Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2026; 23(7):2197-2208. doi:10.7150/ijms.133077 This issue Cite
Research Paper
Genetic Evidence for the Benefits and Risks of Glucose-Lowering Drugs on Cardiovascular-Kidney-Metabolic Syndrome: A Drug-Target Mendelian Randomization Study
Jian Lu1*, Shuaigang Sun1,2*, Xinru Shang1,3, Shimin Jiang1, Zekai Deng4, Shunwei Wang5, Chenping Wei4, Jiaqi Hu6,7 ![]() , Wenge Li1,2
, Wenge Li1,2 ![]()
1. Department of Nephrology, China-Japan Friendship Hospital, Beijing, China.
2. Department of Nephrology, Capital Medical University China-Japan Friendship School of Clinical Medicine, Beijing, China.
3. Department of Nephrology, Peking University China-Japan Friendship School of Clinical Medicine, Beijing, China.
4. School of Basic Medical Sciences, Capital Medical University, Beijing, China.
5. School of Public Health, Capital Medical University, Beijing, China.
6. School of Healthcare Management, Tsinghua Medicine, Tsinghua University, Beijing, China.
7. Institute for Hospital Management, Tsinghua University, Beijing, China.
* These authors contributed equally.
Received 2026-2-12; Accepted 2026-4-20; Published 2026-5-11
Abstract

Background: To explore the potential benefits and risks of glucose-lowering drugs on cardiovascular-kidney-metabolic (CKM) syndrome outcomes using drug-target Mendelian randomization (MR).
Methods: Genetic instruments for eight glucose-lowering drugs were identified from variants associated with glycated hemoglobin (HbA1c) and drug target gene expression. We employed two-sample MR and meta-analysis to estimate associations with nine CKM syndrome-related diseases, including coronary artery disease (CAD), myocardial infarction (MI), stroke, heart failure (HF), atrial fibrillation (AF), venous thromboembolism (VTE), peripheral artery disease (PAD), chronic kidney disease (CKD), and metabolic syndrome (MetS), using data from UK Biobank, FinnGen, and other large GWAS consortia. Supplementary analyses included summary-data-based MR (SMR) and colocalization.
Results: Genetic proxies for sodium—glucose cotransporter 2 inhibitors (SGLT2i) were associated with reduced risks of CAD (OR 0.85, 95% CI 0.73—0.98), HF (OR 0.66, 95% CI 0.44—0.98), and MetS (OR 0.64, 95% CI 0.50—0.82). GLP-1 receptor agonists (GLP-1RA) were linked to lower risks of CAD (OR 0.90, 95% CI 0.84—0.96), HF (OR 0.92, 95% CI 0.86—0.99), and CKD (OR 0.87, 95% CI 0.79—0.95). Metformin showed protective association with CAD (OR 0.51, 95% CI 0.40—0.66) and PAD (OR 0.99, 95% CI 0.99—1.00). Sulfonylureas showed a modest association with reduced CAD (OR 0.98, 95% CI 0.96—1.00). Conversely, insulin was associated with a higher risk of MetS (OR 1.07, 95% CI 1.02—1.13), and thiazolidinediones (TZDs) with an increased risk of HF (OR 1.12, 95% CI 1.05—1.20). SMR provided additional evidence for protective roles of GLP1R and VEGFA for CAD, KCNJ11 for HF, ABCC8 for HF and AF, and PRKAB1 for multiple cardiovascular outcomes. The detrimental effects of INSR on MetS and SERPINE1 on CKD and MetS were also verified.
Conclusions: This study provides genetic evidence revealing the potential benefits and risks of certain glucose-lowering drugs in the management of CKM syndrome. These findings may inform target validation, drug repurposing, and personalized therapies for CKM syndrome.
Keywords: cardiovascular-kidney-metabolic syndrome, Mendelian randomization, glucose-lowering drugs, SGLT2 inhibitors, GLP-1 receptor agonists
Introduction
Growing recognition has been given to the complex pathological links among metabolic dysfunction, cardiovascular disease (CVD), and chronic kidney disease (CKD). These interconnected mechanisms—insulin resistance, advanced glycation, dyslipidemia, chronic inflammation, and endothelial dysfunction—contribute to multi-organ impairment and increase the risk of adverse cardiovascular and renal outcomes [1]. In 2023, the American Heart Association (AHA) formally defined this cluster as cardiovascular-kidney-metabolic (CKM) syndrome and further staged its progression from 0 to 4 [2]. Epidemiological evidence indicates that nearly 90% of U.S. adults meet criteria for stage 1 or higher, with approximately 15% in advanced stages [3]. Similar burdens are observed among middle-aged and older Chinese populations [4]. Patients with CKM syndrome face markedly increased premature mortality [5], and CKM status is now recognized as a key determinant of all-cause mortality [6]. Together, these findings highlight CKM syndrome as a pressing global health challenge and underscore the urgent need for therapeutic strategies that provide broad protection across metabolic, cardiovascular, and renal domains.
Diabetes is a major driver of CKM syndrome [7]. In 2021, the International Diabetes Federation estimated that approximately 537 million adults worldwide have diabetes, accounting for 11% of the global population [8]. A cohort study of 1.2 million type 2 diabetes (T2D) patients across six countries found that, among those without pre-existing CVD or CKD, 24% of initial events were heart failure (HF), stroke (16%), myocardial infarction (MI) (14%), peripheral artery disease (PAD) (10%), and CKD (36%) [9]. Additionally, a study of 530,747 U.S. adults with T2D found that isolated T2D without other CKM conditions was rare (6.4%), with around 51% having three or more additional CKM-related conditions [10]. These findings suggest that glucose-lowering drugs may have consequences far beyond glycemic control, shaping outcomes across the CKM spectrum.
In recent years, antidiabetic agents have been recognized not only for glucose-lowering efficacy but also for cardiovascular and renal benefits. Landmark randomized controlled trials (RCTs) [11-21] have demonstrated that sodium-glucose cotransporter-2 inhibitors (SGLT2i) and glucagon-like peptide-1 receptor agonists (GLP-1RA) substantially reduce risks of major adverse cardiovascular events, heart failure hospitalization, and kidney disease progression—providing “triple protection” across metabolic, cardiovascular, and renal axes. These findings have shifted clinical practice guidelines and positioned SGLT2i and GLP-1RA as cornerstone therapies in T2D and atherosclerotic cardiovascular disease (ASCVD) [22-23]. Nevertheless, the underlying mechanisms remain incompletely understood, and evidence for other glucose-lowering drug classes such as insulin and its analogues, metformin, sulfonylureas, α-glucosidase inhibitors (AGIs), thiazolidinediones (TZDs), dipeptidyl peptidase-4 inhibitors (DPP4i) remains inconsistent and even harmful in certain contexts [24-27]. Moreover, traditional clinical trials are limited by selective enrollment and potential confounding, leaving knowledge gaps about long-term efficacy and safety.
Mendelian randomization (MR) offers a powerful complementary approach by leveraging genetic variants as instrumental variables to infer causal effects while minimizing bias from confounding and reverse causation [28]. Extending this framework, drug-target MR utilizes genetic proxies of therapeutic targets to evaluate the likely effects of pharmacological modulation [29]. In this study, we applied a two-sample drug-target MR design across large-scale GWAS datasets of European ancestry to systematically evaluate the associations of eight major glucose-lowering drug classes on nine CKM syndrome-related outcomes. We further integrated summary-data-based MR (SMR) and colocalization analyses to validate gene-trait expression and distinguish pleiotropy from shared genetic causation. This integrative framework aims to refine the understanding of drug mechanisms and provide genetic evidence for potential therapeutic and repurposing strategies in the context of CKM syndrome.
Methods
Identification and validation of glucose-lowering drug targets
The overall workflow of this study is illustrated in Figure 1. Briefly, we conducted the analysis in two stages: (i) a two-sample MR analysis to assess the causal effects of genetic proxies for glucose-lowering drug targets on a spectrum of CKM syndrome outcomes, and (ii) SMR and colocalization analyses to evaluate whether any detected drug-disease associations were mediated through gene expression, thereby serving as complementary validation.
Overall workflow of this study.
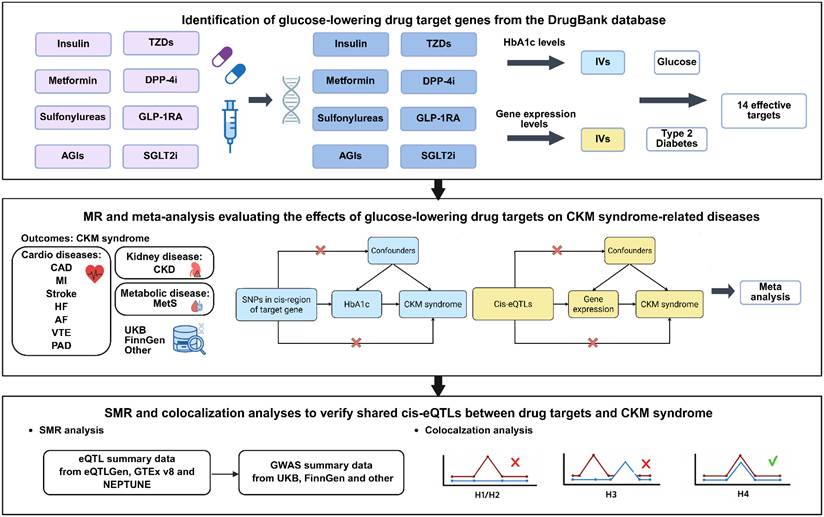
We comprehensively identified genetic targets of eight commonly prescribed glucose-lowering drug classes using the DrugBank pharmacogenomics database (https://go.drugbank.com/) (Table S1): insulin and its analogues, metformin, sulfonylureas, AGIs, TZDs, DPP4i, GLP-1 RAs, and SGLT2i. The pharmacological mechanisms and chromosomal locations of these targets are presented in Table S2.
Instrumental variables (IVs) were extracted from the GWAS of glycated hemoglobin (HbA1c) in the UK Biobank population [30], using the thresholds of P < 5 × 10⁻⁸ and r² < 0.2 [31]. For two adjacent drug targets located within overlapping cis regions and exerting effects in the same mechanistic direction, we combined them and denoted them with a slash (e.g., ABCC8/KCNJ11 and ABCC9/KCNJ8). Furthermore, eQTLs for drug-target genes were retrieved from the eQTLGen Consortium (https://www.eqtlgen.org), with only cis-eQTLs located within 500 kb of the target gene included as IVs [32].
To ensure consistency with pharmacological mechanisms, we adjusted the directionality of effect estimates for all genetic instruments at the individual target gene level prior to downstream analyses. Specifically, for genetic proxies corresponding to agonists or mechanisms of uncertain directionality (e.g., modulators, agonists, activators, substrates, or unknown/other mechanisms), we retained the original β coefficients. Conversely, for inhibitors (e.g., blockers, inhibitors, antagonists, or inverse agonists), we applied the negative β values. Then, all validated and direction-aligned SNPs corresponding to the various targets of a specific drug class (e.g., pooling SLC5A1 and SLC5A2 for SGLT2i) were combined into a single composite IV set in order to reflect the composite action of drug classes rather than isolated targets. To minimize weak IV bias, we calculated F-statistics using the formula F = R² × (N - 2) / (1 - R²), where R² = β² / (β² + SE² × (N - 1)), excluding IVs with F-statistics < 10 (Table S3) [33]. Finally, to validate the robustness of the selected IVs, we employed four independent GWAS datasets related to blood glucose levels and T2D as positive controls [31,33]. Given the established glucose-lowering effects of these drugs, only drug-gene target pairs showing significant associations consistent with reduced glycemia (β < 0, P < 0.05) were advanced to subsequent analyses.
Outcome data of the CKM syndrome
We focused on nine major clinical outcomes that represent the core components of the CKM syndrome, including coronary artery disease (CAD), MI, stroke, HF, atrial fibrillation (AF), venous thromboembolism (VTE), PAD, CKD, and metabolic syndrome (MetS). For each outcome, we obtained summary-level GWAS statistics from three independent data sources to enhance robustness and reduce dataset-specific bias. The primary GWAS datasets were obtained from the UK Biobank, FinnGen, and other large disease-specific international consortia, such as CKDGen for CKD and GIGASTROKE for stroke, or GWAS meta-analyses. However, due to the unavailability of FinnGen GWAS data for MetS, only two GWAS datasets were used. All data were derived from previously published GWAS studies, and detailed information is provided in Table S4.
Evaluation of sample overlap bias
Given that both the HbA1c exposure and several outcome studies included participants from the UK Biobank, potential bias and Type 1 error rate inflation associated with varying levels of sample overlap rates were estimated using the mrSampleOverlap R package (https://www.levin-lab.org/mrSampleOverlap/), which strictly implements the analytic formulae derived by Burgess et al. for two-sample MR [34].
Two-sample MR analysis and meta-analysis
Two-sample MR was applied to evaluate the causal effect of genetically proxied glucose-lowering drug targets on outcomes, rather than the effects of blood glucose changes. As illustrated in Figure 1, MR design relies on three core assumptions: (i) IVs are strongly associated with the exposure; (ii) IVs are independent of potential confounders; and (iii) IVs affect the outcome only through the exposure of interest.
The inverse variance-weighted (IVW) method was used as the primary MR approach, under either a random-effects or fixed-effects model depending on the results of heterogeneity testing [35]. To assess the robustness of the findings, we additionally performed sensitivity analyses using MR-Egger regression, weighted median, simple mode, and weighted mode methods. For drug targets with fewer than three valid SNP instruments, causal estimates were obtained using the Wald ratio method. Heterogeneity was evaluated using Cochran's Q statistic derived from both IVW and MR-Egger models. Potential horizontal pleiotropy was examined by testing the intercept term from MR-Egger regression. All MR analyses were performed using the TwoSampleMR package in R (https://mrcieu.github.io/TwoSampleMR/) with default settings.
To obtain an overall causal effect estimate, we performed a meta-analysis of the causal estimates derived from the primary MR method (IVW for ≥ 3 SNPs or the Wald ratio for < 3 SNPs) for each drug target-outcome pair. Heterogeneity was evaluated using Cochran's Q statistic and the I² index. A fixed-effects model was applied when no significant heterogeneity was observed (P ≥ 0.05 and I² ≤ 50%), whereas a random-effects model was used when significant heterogeneity was present (P < 0.05 and I² > 50%). The pooled estimates from the meta-analysis were utilized to quantify the magnitude of the final causal effects. However, if only one reliable MR result was available, the causal inference was based on that single estimate. Given the broadly exploratory nature of this study, the false discovery rate (FDR) was applied for multiple testing correction. Specifically, an FDR < 0.05 indicated a significant causal relationship between the exposure and the outcome, while a nominal P < 0.05 with an FDR ≥ 0.05 was considered suggestive.
Summary-data-based MR
To further examine the associations between gene expression of eight glucose-lowering drug targets and the risk of CKM syndrome, we applied the SMR method, using the most significantly associated cis-eQTL SNP as the primary IV, and effect estimates were expressed as OR for disease risk per-SD increase in genetically predicted expression of each drug target gene. To complement the whole-blood cis-eQTL from eQTLGen, we additionally integrated tissue-specific cis-eQTLs from cardiac tissues (left ventricle and atrial appendage) from genotype-tissue expression (GTEx) version 8 [36], and renal tissues (glomeruli and tubulointerstitium) from NEPTUNE [37]. The HEIDI (heterogeneity in dependent instruments) test was conducted to distinguish true causal associations from those potentially driven by linkage disequilibrium (LD) between the eQTL SNP and another causal variant. A HEIDI test P-value < 0.05 was considered indicative that the observed association might be attributable to LD rather than gene expression itself. SMR analyses were performed using the default parameters (https://yanglab.westlake.edu.cn/software/smr/#SMR&HEIDIanalysis).
Colocalization analysis
To explore whether the associations between specific gene expression and CKM syndrome outcomes were attributable to a shared causal genetic variant, we performed a Bayesian colocalization analysis using the coloc R package (http://cran.r-project.org/web/packages/coloc) with default prior settings (P1 = 1 × 10⁻⁴, P2 = 1 × 10⁻⁴, P12 = 1 × 10⁻⁵) [38]. Evidence for colocalization was considered strong to indicate a high probability of a shared causal variant when the posterior probability for hypothesis 4 (PPH4) exceeded 0.8.
Results
Identification and validation of glucose-lowering drug targets
Based on HbA1c levels, we identified 18 potential target genes across eight classes of glucose-lowering drugs: Insulin (LRP2), Metformin (GPD1), Sulfonylureas (ABCC8/KCNJ11, ABCC9/KCNJ8, ABCB11, CPT1A, VEGFA, INS, KCNJ1), AGIs (GANC), TZDs (PPARG, ESRRA, SERPINE1, SLC29A1, RXRB), GLP-1RA (GLP1R), and SGLT2i (SLC5A2, SLC5A1). Bias due to sample overlap from the UK Biobank participants included in both the HbA1c exposure and several outcome GWASs was mathematically negligible across a wide range of assumed observational effect sizes: for instance, in the analysis of LRP2 on AF, at 80.5% sample overlap, bias was estimated to be 0.001 for an observational OR of 1.3, and 0.002 for an OR of 1.6 (Table S5). Among these, nine targets were significantly associated with reduced glycemia or reduced risk of T2D and were retained for further analyses (Table S6). It should be noted that no valid IVs meeting our stringent selection criteria were available for DPP4i, therefore, this drug class was not included in the analyses.
Based on gene expression levels, we initially identified 26 potential target genes from the eQTLGen Consortium: Insulin (INSR, IGF1R, IGFBP7), Metformin (ETFDH, PRKAB1, ACACB), Sulfonylureas (KCNJ11, ABCA1, CPT1A, TRPM4, VEGFA), AGIs (MGAM, GAA, GANAB, GANC), TZDs (PPARG, ESRRA, PPARD, PPARA, GSTP1, SERPINE1, SLC29A1, RXRA), DPP4i (DPP4), GLP-1RA (GLP1R), and SGLT2i (SLC5A2). However, only five target genes passed positive control analyses.
Subsequently, after adjusting effect directions according to drug-target mechanisms, we combined all validated IVs within each drug class to construct the overall genetic proxies for drug effects: Insulin (INSR), Metformin (GPD1), Sulfonylureas (ABCC8/KCNJ11 + ABCB11 + CPT1A + KCNJ11 + VEGFA), TZDs (ESRRA + SERPINE1 + SLC29A1 + PPARD), GLP-1RA (GLP1R), and SGLT2i (SLC5A2 + SLC5A1). All drug-level instruments were validated by positive control analyses.
Effects of genetic variation in glucose-lowering drug targets on CKM syndrome risk by MR and meta-analysis
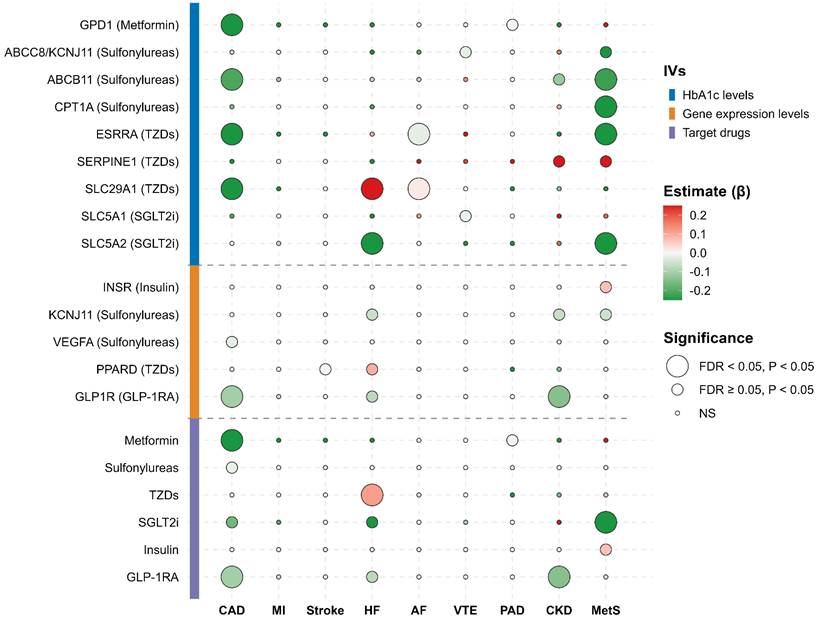
The associations of genetically proxied glucose-lowering drug targets with CKM syndrome-related diseases across three independent GWAS datasets for each trait were evaluated by MR (Table S7) and the pooled estimates from meta-analysis were regarded as the final causal effect estimates (Table S8, Figure S1-S14).
Based on HbA1c-derived IVs, GPD1 (Metformin) significantly reduced CAD risk (OR = 0.51, 95% CI 0.40-0.66, P < 0.001, FDR < 0.001), alongside a potential benefit for PAD (OR = 0.99, 95% CI 0.99-1.00, P = 0.035). For Sulfonylureas, ABCC8/KCNJ11 showed suggestive protective associations against VTE (OR = 0.98, 95% CI 0.96-1.00, P = 0.039) and MetS (OR = 0.58, 95% CI 0.38-0.88, P = 0.011). ABCB11 demonstrated significant protective effects against CAD (OR = 0.81, 95% CI 0.76-0.86, P < 0.001, FDR < 0.001) and MetS (OR = 0.80, 95% CI 0.74-0.85, P < 0.001, FDR < 0.001), with a suggestive benefit for CKD (OR = 0.89, 95% CI 0.81-0.97, P = 0.009). CPT1A was significantly associated with a lower risk of MetS (OR = 0.18, 95% CI 0.09-0.37, P < 0.001, FDR < 0.001). Among TZD targets, ESRRA conferred significant reductions in the risks of CAD (OR = 0.30, 95% CI 0.21-0.41, P < 0.001, FDR < 0.001), AF (OR = 0.98, 95% CI 0.96-0.99, P < 0.001, FDR = 0.002), and MetS (OR = 0.15, 95% CI 0.10-0.24, P < 0.001, FDR < 0.001), whereas SERPINE1 showed suggestive associations with increased risks of CKD (OR = 3.91, 95% CI 1.08-14.11, P = 0.037) and MetS (OR = 3.63, 95% CI 1.19-11.07, P = 0.023). SLC29A1 was significantly protective against CAD (OR = 0.41, 95% CI 0.27-0.63, P < 0.001, FDR < 0.001) but increased the risks of HF (OR = 1.48, 95% CI 1.15-1.91, P = 0.002, FDR = 0.016) and AF (OR = 1.02, 95% CI 1.01-1.02, P < 0.001, FDR = 0.002). For SGLT2i, SLC5A1 showed a suggestive association with lower VTE risk (OR = 0.98, 95% CI 0.97-1.00, P = 0.019), whereas SLC5A2 demonstrated significant protection against HF (OR = 0.70, 95% CI 0.57-0.86, P = 0.001, FDR = 0.005) and MetS (OR = 0.44, 95% CI 0.32-0.60, P < 0.001, FDR < 0.001).
Gene expression-based analyses further supported these findings. INSR (Insulin) was suggestively associated with increased MetS risk (OR = 1.07, 95% CI 1.02-1.13, P = 0.013). For Sulfonylureas, KCNJ11 showed suggestive protective associations with HF (OR = 0.94, 95% CI 0.90-0.99, P = 0.010), CKD (OR = 0.94, 95% CI 0.88-1.00, P = 0.048), and MetS (OR = 0.94, 95% CI 0.90-0.99, P = 0.031), while VEGFA was suggestively associated with reduced CAD risk (OR = 0.98, 95% CI 0.96-1.00, P = 0.025). Among TZDs, PPARD demonstrated a suggestive association with stroke (OR = 0.99, 95% CI 0.99-1.00, P = 0.045) and an increased HF risk (OR = 1.10, 95% CI 1.03-1.18, P = 0.006). For GLP-1RA, GLP1R exhibited robust protective associations with CAD (OR = 0.90, 95% CI 0.84-0.96, P = 0.002, FDR = 0.043) and CKD (OR = 0.87, 95% CI 0.79-0.95, P = 0.002, FDR = 0.043), alongside a suggestive protective effect against HF (OR = 0.92, 95% CI 0.86-0.99, P = 0.020).
At the drug overall level, pooled analyses revealed that Metformin significantly reduced the risk of CAD (OR = 0.51, 95% CI 0.40-0.66, P < 0.001, FDR < 0.001) and suggestively reduced PAD risk (OR = 0.99, 95% CI 0.99-1.00, P = 0.035). Sulfonylureas were suggestively associated with lower CAD risk (OR = 0.98, 95% CI 0.96-1.00, P = 0.024). TZDs significantly increased HF risk (OR = 1.12, 95% CI 1.05-1.20, P < 0.001, FDR = 0.007). SGLT2i significantly reduced the risk of MetS (OR = 0.64, 95% CI 0.50-0.82, P < 0.001, FDR = 0.007), with suggestive reductions in CAD (OR = 0.85, 95% CI 0.73-0.98, P = 0.027) and HF (OR = 0.66, 95% CI 0.44-0.98, P = 0.040). Insulin suggestively increased the risk of MetS (OR = 1.07, 95% CI 1.02-1.13, P = 0.013). Finally, GLP-1RA conferred significant protection against CAD (OR = 0.90, 95% CI 0.84-0.96, P = 0.002, FDR = 0.021) and CKD (OR = 0.87, 95% CI 0.79-0.95, P = 0.002, FDR = 0.021), and suggestive protection against HF (OR = 0.92, 95% CI 0.86-0.99, P = 0.020). These results are summarized in Figure 2.
MR and meta-analysis of glucose-lowering drug targets and CKM syndrome-related diseases. The bubble heatmap illustrates the association between genetically proxied drug targets and nine CKM syndrome outcomes. Targets are categorized by IV sources: HbA1c levels, gene expression levels, and overall target drugs. A red bubble indicates a positive association, and a green bubble indicates a negative association. Bubble size corresponds to the level of statistical significance (large for FDR < 0.05, medium for P < 0.05, and small for non-significant results).
Summary-based MR
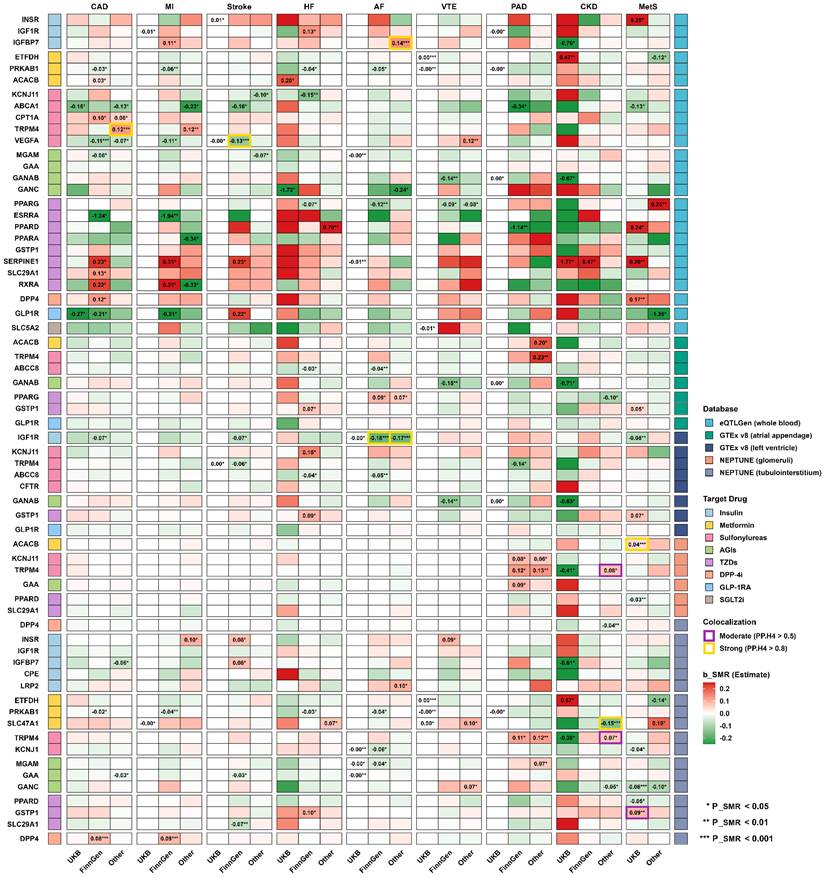
In the SMR analyses, several drug targets previously identified in our MR analyses were further validated (Figure 3, Table S9). Specifically, in whole blood, genetically proxied activation of GLP1R was validated as protective against CAD (OR = 0.81, 95% CI 0.66-0.99, P_SMR = 0.034, P_HEIDI = 0.151). Similarly, for Sulfonylureas, the genetically proxied inhibition of KCNJ11 was confirmed to reduce HF risk (OR = 0.86, 95% CI 0.77-0.96, P_SMR = 0.009, P_HEIDI = 0.754), and VEGFA was validated as protective against CAD (OR = 0.90, 95% CI 0.84-0.96, P_SMR = 0.001, P_HEIDI = 0.086). In contrast, INSR (Insulin) was verified to increase MetS risk (OR = 1.28, 95% CI 1.06-1.55, P_SMR = 0.011, P_HEIDI = 0.146). Among TZD targets, genetically proxied inhibition of SERPINE1 showed consistent harmful associations, with increased risks of CKD across two independent datasets (OR = 5.88, 95% CI 1.09-31.67, P_SMR = 0.039, P_HEIDI = 0.497; and OR = 1.60, 95% CI 1.08-2.39, P_SMR = 0.021, P_HEIDI = 0.321), as well as an increased MetS risk (OR = 1.33, 95% CI 1.10-1.61, P_SMR = 0.003, P_HEIDI = 0.767). Furthermore, SMR analyses utilizing eQTLs from cardiac and renal tissues provided complementary insights. Notably, for Sulfonylureas, the genetically proxied inhibition of ABCC8 in the atrial appendage and left ventricle was associated with lower risks of AF and HF. Additionally, the genetically proxied activation of PRKAB1 in whole blood and renal tubulointerstitium exhibited broad protective effects against multiple cardiovascular diseases, such as CAD, AF, HF, MI, PAD, and VTE (all P_SMR < 0.05, P_HEIDI > 0.05).
SMR and colocalization analysis of glucose-lowering drug targets and CKM syndrome-related diseases. The matrix heatmap illustrates the association between the genetically predicted expression of drug target genes across five eQTL databases and nine CKM syndrome outcomes. A red region indicates a positive association, a green region indicates a negative association, and the β_SMR estimates are displayed if the results are significant (P_SMR < 0.05). Asterisks denote the level of significance (* P_SMR < 0.05, ** P_SMR < 0.01, *** P_SMR < 0.001). Additionally, highlighted cell borders denote the posterior probability of colocalization, with gold and purple borders representing strong (PP.H4 > 0.8) and moderate (PP.H4 > 0.5) evidence, respectively.
Colocalization analysis
Consistent with SMR analyses (Figure 3), strong evidence for a shared causal variant was observed for left ventricle IGF1R eQTL and AF across two independent datasets (PPH4 = 0.990 and 0.968) (Table S10). High posterior probabilities were also identified for whole blood TRPM4, IGFBP7, VEGFA eQTL with CAD (PPH4 = 0.932), AF (PPH4 = 0.825) and stroke (PPH4 = 0.812), respectively. Meanwhile, moderate evidence of colocalization was observed for GSTP1 eQTL and MetS risk in tubulointerstitium (PPH4 = 0.591). However, no strong evidence for colocalization was observed for the causal associations identified through the two-sample MR analyses.
Discussion
This is the first and most comprehensive MR study to date investigating the potential benefits and risks of genetic variation in glucose-lowering drug targets on CKM syndrome-related diseases. Using multi-dataset two-sample MR and meta-analysis, genetically proxied SGLT2i and GLP-1RA demonstrated prominent and consistent protective associations across three comorbid conditions, while metformin and sulfonylureas showed moderate benefits in one to two comorbid conditions. In contrast, insulin and specific targets of TZDs were associated with adverse outcomes. Additionally, supplementary validation through SMR identified several key target genes mediating these drug-disease associations. From a genetic perspective, these findings offer new insights into the therapeutic potential of glucose-lowering drugs in the context of CKM syndrome comorbidities.
SGLT2i lower blood glucose levels by reducing the reabsorption of sodium and glucose in the proximal tubules of the kidney. They also exert multiple beneficial effects, including weight loss and improved lipid metabolism, and have been shown to significantly improve various aspects of MetS, such as obesity, dyslipidemia, and insulin resistance [39-40]. In recent years, SGLT2i have garnered widespread attention due to their ability to reduce cardiovascular mortality, heart failure hospitalization rates, and the incidence of adverse kidney outcomes [41-43]. These benefits extend far beyond glucose control and may be associated with antioxidant effects, reduced inflammation and fibrosis, decreased circulatory load, enhanced erythropoiesis, modulation of mitochondrial function, and improved cardiac energy metabolism and vascular function [44-46]. Notably, the favorable effects on the heart and kidneys appear to be independent of glucose-lowering effects, as similar phenomena have been observed in individuals without diabetes [47-48]. Our MR study, focusing on the target-specific perturbation of SGLT2i, rather than generalized glucose-lowering effects, aligns with prior evidence, suggesting that SGLT2i are associated with a reduced risk of CAD, HF, and MetS. However, we did not observe a potential benefit of SGLT2i on CKD, which may be attributed to the limitations of the data used, which did not adequately cover the diversity of different CKD subtypes or patient populations. The benefits of SGLT2i may be more relevant to specific CKD subtypes [e.g., diabetic kidney disease]. Additionally, we noted that SGLT2i may have a protective effect on VTE through the SLC5A1 gene, which could provide new perspectives for the drug's indications. Indirect evidence suggests that SGLT2i might reduce the risk of ischemic stroke in patients with AF and diabetes, with its potential mechanism of preventing thrombosis related to the improvement of inflammatory states in AF patients [49]. A nationwide retrospective cohort study from China indicated that, compared to DPP-4 inhibitors, SGLT2i are associated with a lower risk of VTE, and this finding was further supported by a subsequent meta-analysis [50].
GLP-1RA were initially developed as glucose-lowering drugs by stimulating insulin secretion, inhibiting glucagon release, and slowing gastric emptying [51]. However, large RCTs have revealed an unexpected benefit: GLP-1RA acts as a systemic metabolic modulator, providing cardiovascular and renal benefits far beyond glucose control and weight loss. A recent meta-analysis, which included 67,769 diabetic patients across 11 RCTs, showed that, compared to a placebo, GLP-1RA reduced the risk of composite renal adverse events by 18%, the risk of renal failure by 16%, and the risk of major adverse cardiovascular events by 13% [52]. Similarly, the ERA Diabesity working group reviewed 9 high-quality RCTs and 4 real-world cohorts (with a total sample size of over 170,000 and a maximum follow-up of 8.5 years), reporting a 14% reduction in the overall risk of MACE, a 31% reduction in heart failure hospitalization rates in high-risk subgroups, and a 22% reduction in the risk of adverse renal outcomes [53]. Based on GLP1R expression-based MR analysis independent of HbA1c, clear protective associations of genetically proxied GLP-1RA with CAD, HF, and CKD were observed, with the effect on CAD further validated in SMR analysis. Mechanistically, the beneficial effects of GLP-1RA on the heart and kidneys may be related to its suppression of oxidative stress, reduction of inflammation and fibrosis [54-59]. Specifically, activation of GLP1R reduces the production of reactive oxygen species (ROS) through the downregulation of HO-1 and non-receptor-mediated NADPH oxidase, and inhibits the activity of NF-κB, thus reducing the production of pro-inflammatory chemokines, cytokines, adhesion molecules, and pro-fibrotic factors [60]. Studies have shown that the most common causes of death in DKD patients are heart failure and CAD, further highlighting the potential of GLP-1RA as a therapeutic strategy for managing comorbid conditions [61].
Metformin has been used for over 70 years in the treatment of diabetes [62]. It controls blood glucose levels by inhibiting hepatic gluconeogenesis, enhancing peripheral insulin sensitivity, and reducing intestinal glucose absorption. It remains one of the most commonly prescribed drugs for the treatment of T2D. In this study, we observed potential benefits of metformin for CAD and PAD. A substantial body of evidence indicates that metformin significantly reduces the risk of major cardiovascular events and mortality in patients with ASCVD associated with T2D [63-68]. Although previous studies have suggested metformin's significant potential in combating atherosclerosis, its precise mechanisms remain unclear. The largest and longest double-blind placebo-controlled RCT, the REmoval trial (REversing with MetfOrmin Vascular Adverse Lesions), found that metformin significantly slowed the progression of atherosclerosis in patients with type 1 diabetes, as measured by the averaged maximal carotid intima-media thickness (-0.013 mm/year, -0.024 to -0.003; P = 0.0093) [69]. Li et al. further confirmed in a cholesterol-fed rabbit model of atherosclerosis that metformin intervention significantly reduced atherosclerotic plaque formation and inhibited the phosphorylation of IκB and activation of NF-κB in the vascular wall, while lowering serum hs-CRP levels [70]. Similarly, Forouzandeh et al. observed similar results in ApoE-/- C57BL/6J mice, with the protective effects potentially attributed in part to the downregulation of the angiotensin II type 1 receptor [71]. Additional studies on metformin's potential mechanisms in combating atherosclerosis have shown that it may enhance endothelial function through the activation of the AMP-activated protein kinase (AMPK) pathway to promote nitric oxide (NO) production [72-73]. Consistent with this, our SMR analysis confirmed that genetically predicted PRKAB1 (encoding the AMPK β1 subunit) exhibited broad protective effects across multiple cardiovascular outcomes. Furthermore, metformin may inhibit the abnormal migration of vascular smooth muscle cells and delay intimal thickening [74]; regulate triglyceride levels and HDL functionality [75-76]; and suppress the activation of NF-κB in endothelial cells and vascular smooth muscle cells, thereby reducing the secretion of pro-inflammatory cytokines induced by interleukin-1β [77], which weakens the chronic inflammation process that contributes to atherosclerosis [78]. Other studies have reported that metformin can prevent local complications in PAD patients [79] and improve survival rates [80]. Additionally, the ongoing MOBILE IC trial (NCT05132439) aims to evaluate the ability of metformin to improve function in patients with intermittent claudication and delay the progression of PAD, which could provide robust evidence for positioning metformin as a novel therapeutic strategy for PAD management [81].
As insulin secretagogues, sulfonylureas have traditionally been used for blood glucose control, but their role in cardioprotective and nephroprotective outcomes remains a subject of ongoing debate [82-85]. In recent years, with the emergence of newer glucose-lowering drugs such as SGLT2i and GLP-1RA, research on the cardiovascular and renal safety of sulfonylureas often seems to take a backseat, with conflicting conclusions. A large-scale meta-analysis (47 RCTs, 37,650 patients) found that sulfonylureas did not significantly increase the risk of all-cause mortality, cardiovascular mortality, MI, or stroke [86]. However, some high-quality observational studies have shown that sulfonylureas are associated with an increased risk of cardiovascular events and mortality, particularly when compared with drugs like metformin [87]. Despite this, our MR-meta analysis indicates that certain targets of sulfonylureas, such as ABCC8/KCNJ11, ABCB11, and VEGFA, are significantly associated with a reduction in the risk of diseases like CAD, CKD, and MetS. At the drug level, sulfonylureas are also linked to a reduction in CAD risk. However, given the lack of definitive evidence in real-world settings, our conclusions must be interpreted with caution.
We also identified some adverse effects associated with certain drugs. INSR, a key target gene in the insulin signaling pathway, is typically linked to insulin resistance and other manifestations of MetS [88]. However, our study found that the genetic proxy for insulin, which activates INSR, while lowering blood glucose, also increases the risk of MetS. This can be explained by several factors: On one hand, insulin, as a potent anabolic hormone, promotes fat synthesis and storage, leading to obesity [89-90]. On the other hand, excessive insulin use increases the risk of hypoglycemia, triggering the release of counter-regulatory hormones that promote lipolysis, increase free fatty acids, and induce insulin resistance, ultimately exacerbating the metabolic abnormalities associated with MetS [91-92]. It is important to emphasize that MR estimates reflect the lifetime, moderate disruption of the target, and the effect size should not be directly equated to the results observed in short-term clinical trials. While insulin is effective in lowering blood glucose, its long-term use may have potential adverse effects on overall metabolic health. Furthermore, TZDs, as classic insulin sensitizers, improve insulin resistance and effectively lower blood glucose by activating the PPARγ pathway. However, their cardiovascular and metabolic effects have been a subject of debate. In our MR study, the effects of TZD-related targets were complex and inconsistent: for instance, ESRRA was associated with reduced risks of CAD, AF, and MetS, while targets such as SERPINE1, SLC29A1, and PPARD were linked to adverse outcomes, including HF, AF, CKD, and MetS. Overall, the drug-level results indicated a significant increase in the risk of HF associated with TZDs, a finding well-supported by numerous real-world studies [93-97]. The main mechanisms for this are likely fluid retention, weight gain, and enhanced sodium and water reabsorption, leading to increased cardiac preload and deterioration of heart function [98-99].
This study has several limitations. First, the genetic data used in this research is solely derived from individuals of European descent, which may limit the generalizability of our findings. Second, our genetic instruments capture only highly specific targeted effects, without accounting for potential off-target effects. Third, MR estimates reflect lifelong, low-intensity genetic modulation of drug targets, whereas clinical pharmacotherapy typically involves short-term, high-intensity drug exposure in middle-aged or elderly patients. Therefore, the absolute effect sizes should not be directly equated with the anticipated magnitude of benefit in real-world clinical settings, nor should they be interpreted as clinical recommendations for patient management. Fourth, although we employed various methods to ensure the robustness of the analysis, the inherent potential for pleiotropy and bias in MR analyses remains unavoidable. Despite selecting genetic variants from the cis-regions of each diabetes-related target gene, some variants may still influence disease risk through other pathways. Specifically, results lacking strong colocalization support should be interpreted with caution, as they may merely represent pleiotropic or proxy associations rather than definitive drug-target effects. Further multi-exposure MR analysis could help deepen the validation of these causal relationships. Fifth, as this study fundamentally relies on robust SNPs derived from large-scale GWAS and tissue-specific eQTL data, not all drug targets possessed available IVs to be comprehensively explored. Additionally, we did not explore the effects in specific disease subtypes, such as the treatment responses in patients with DKD. Finally, while MR provides strong causal inference evidence, the results still need to be validated through larger-scale clinical and mechanistic trials.
This is the first and most comprehensive systematic evaluation of the genetic variations in glucose-lowering drug targets and their potential benefits and risks in diseases related to CKM syndrome. From a genetic perspective, our findings provide mechanistic evidence supporting the broad cardiorenal benefits of SGLT2i and GLP-1R targets, while highlighting potential genetic liabilities associated with insulin and specific TZD targets. While not serving as direct clinical guidelines, these findings have significant implications for future target validation, drug repurposing, and the development of personalized precision therapies.
Supplementary Material
Supplementary figures.
Supplementary tables.
Acknowledgements
We acknowledge the essential efforts of research groups that have shared GWAS summary statistics publicly.
Funding
This study was supported by National High Level Hospital Clinical Research Funding 2025-NHLHCRF-JBGS-A-WZ-05 (W.G.L), the National Natural Science Foundation of China 82500909 (J.L), and the National Natural Science Foundation of China 82300815 (S.M.J).
Availability of data and materials
All data used in this study are publicly available as described in Table S4. Summary statistics can be accessed via specified repositories.
Author contributions
J.L. and S.S. conceived and designed the study. S.S. performed the statistical analysis and drafted the manuscript. J.H. and W.L. provided a critical review of the manuscript. X.S., S.J., Z.D., S.W., and C.W. advised on statistical aspects and performed interpretation of the results. All authors agreed on the final version of the manuscript and take responsibility for its content.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sebastian SA, Padda I, Johal G. Cardiovascular-Kidney-Metabolic (CKM) syndrome: A state-of-the-art review. Curr Probl Cardiol. 2024;49(2):102344
2. Ndumele CE, Rangaswami J, Chow SL. et al. Cardiovascular-Kidney-Metabolic Health: A Presidential Advisory from the American Heart Association. Circulation. 2023;148(20):1606-1635
3. Aggarwal R, Ostrominski JW, Vaduganathan M. Prevalence of Cardiovascular-Kidney-Metabolic Syndrome Stages in US Adults, 2011-2020. JAMA. 2024;331(21):1858-1860
4. Zhang H, Hu Z, Wu J. et al. Prevalence of Cardiovascular-Kidney-Metabolic Syndrome Stages Among Middle-Aged and Older Adults in China. JACC Asia. 2025;5(3 Pt 1):393-395
5. Cotton A, Salerno PRVO, Deo SV. et al. The association between county-level premature cardiovascular mortality related to cardio-kidney-metabolic disease and the social determinants of health in the US. Sci Rep. 2024;14(1):24984
6. Li N, Li Y, Cui L. et al. Association between different stages of cardiovascular-kidney-metabolic syndrome and the risk of all-cause mortality. Atherosclerosis. 2024;397:118585
7. Xu Z, Yang S, Tan Y. et al. Inflammation in cardiovascular-kidney-metabolic syndrome: key roles and underlying mechanisms-a comprehensive review. Mol Cell Biochem. 2025;480(12):6039-6075
8. Sun H, Saeedi P, Karuranga S. et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119
9. Olufade T, Jiang L, Israni R. et al. Cardiovascular and renal disease manifestation and healthcare resource utilization in patients on first-line oral therapy for type 2 diabetes: A claims-based observational cohort study. Diabetes Obes Metab. 2021;23(12):2741-2751
10. Ostrominski JW, Thierer J, Claggett BL. et al. Cardio-Renal-Metabolic Overlap, Outcomes, and Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. JACC Heart Fail. 2023;11(11):1491-1503
11. Marso SP, Daniels GH, Brown-Frandsen K. et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2016;375(4):311-322
12. Hernandez AF, Green JB, Janmohamed S. et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392(10157):1519-1529
13. Gerstein HC, Colhoun HM, Dagenais GR. et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394(10193):121-130
14. Gerstein HC, Sattar N, Rosenstock J. et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N Engl J Med. 2021;385(10):896-907
15. Marso SP, Bain SC, Consoli A. et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375(19):1834-1844
16. Perkovic V, Tuttle KR, Rossing P. et al. Effects of Semaglutide on Chronic Kidney Disease in Patients with Type 2 Diabetes. N Engl J Med. 2024;391(2):109-121
17. Zinman B, Wanner C, Lachin JM. et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373(22):2117-2128
18. Neal B, Perkovic V, Mahaffey KW. et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med. 2017;377(7):644-657
19. Mahaffey KW, Jardine MJ, Bompoint S. et al. Canagliflozin and Cardiovascular and Renal Outcomes in Type 2 Diabetes Mellitus and Chronic Kidney Disease in Primary and Secondary Cardiovascular Prevention Groups. Circulation. 2019;140(9):739-750
20. Wiviott SD, Raz I, Bonaca MP. et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2019;380(4):347-357
21. The EMPA-KIDNEY Collaborative Group, Herrington WG, Staplin N. et al. Empagliflozin in Patients with Chronic Kidney Disease. N Engl J Med. 2023;388(2):117-127
22. Marx N, Federici M, Schütt K. et al. 2023 ESC Guidelines for the management of cardiovascular disease in patients with diabetes. Eur Heart J. 2023;44(39):4043-4140
23. American Diabetes Association Professional Practice Committee. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2022. Diabetes Care. 2022;45(Suppl 1):S125-S143
24. Yandrapalli S, Jolly G, Horblitt A. et al. Cardiovascular benefits and safety of non-insulin medications used in the treatment of type 2 diabetes mellitus. Postgrad Med. 2017;129(8):811-821
25. Lexis CP, van der Horst IC. Metformin for cardiovascular disease: promise still unproven. Lancet Diabetes Endocrinol. 2014;2(2):94-95
26. Xie Y, Bowe B, Xian H. et al. Comparative effectiveness of SGLT2 inhibitors, GLP-1 receptor agonists, DPP-4 inhibitors, and sulfonylureas on risk of major adverse cardiovascular events: emulation of a randomised target trial using electronic health records. Lancet Diabetes Endocrinol. 2023;11(9):644-656
27. Yen FS, Wei JC, Chiu LT. et al. Thiazolidinediones were associated with higher risk of cardiovascular events in patients with type 2 diabetes and cirrhosis. Liver Int. 2021;41(1):110-122
28. Sanderson E, Glymour MM, Holmes MV. et al. Mendelian randomization. Nat Rev Methods Primers. 2022;2:6
29. Schmidt AF, Finan C, Gordillo-Marañón M. et al. Genetic drug target validation using Mendelian randomisation. Nat Commun. 2020;11(1):3255
30. Mbatchou J, Barnard L, Backman J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat Genet. 2021;53(7):1097-1103
31. Fu K, Si S, Jin X. et al. Exploring antidiabetic drug targets as potential disease-modifying agents in osteoarthritis. EBioMedicine. 2024;107:105285
32. Võsa U, Claringbould A, Westra HJ. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300-1310
33. Li Y, Zhou T, Liu Z. et al. Protective effect of antidiabetic drugs against male infertility: evidence from Mendelian randomization. Diabetol Metab Syndr. 2025;17(1):140
34. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40(7):597-608
35. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658-665
36. GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318-1330
37. Han SK, McNulty MT, Benway CJ. et al. Mapping genomic regulation of kidney disease and traits through high-resolution and interpretable eQTLs. Nat Commun. 2023;14(1):2229
38. Foley CN, Staley JR, Breen PG. et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun. 2021;12(1):764
39. Olagunju A, Yamani N, Kenny D. et al. Potential for sodium-glucose cotransporter-2 inhibitors in the management of metabolic syndrome: A systematic review and meta-analysis. World J Cardiol. 2022;14(11):599-616
40. Cortés-Camacho F, Zambrano-Vásquez OR, Aréchaga-Ocampo E. et al. Sodium-Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome. Antioxidants (Basel). 2024;13(7):768
41. Ali MU, Mancini GBJ, Fitzpatrick-Lewis D. et al. The effectiveness of sodium-glucose co-transporter 2 inhibitors on cardiorenal outcomes: an updated systematic review and meta-analysis. Cardiovasc Diabetol. 2024;23(1):72
42. Scheen AJ. Sodium-glucose cotransporter type 2 inhibitors for the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2020;16(10):556-577
43. Li HL, Lip GYH, Feng Q. et al. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: a systematic review and meta-analysis. Cardiovasc Diabetol. 2021;20(1):100
44. Guerrero-Mauvecin J, Villar-Gómez N, Miño-Izquierdo L. et al. Antioxidant Effects of SGLT2 Inhibitors on Cardiovascular-Kidney-Metabolic (CKM) Syndrome. Antioxidants (Basel). 2025;14(6):701
45. Lopaschuk GD, Verma S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl Sci. 2020;5(6):632-644
46. Salvatore T, Galiero R, Caturano A. et al. An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors. Int J Mol Sci. 2022;23(7):3651
47. Nyström T. Key results from observational studies and real-world evidence of sodium-glucose cotransporter-2 inhibitor effectiveness and safety in reducing cardio-renal risk. Diabetes Obes Metab. 2024;26(Suppl 5):35-57
48. Heerspink HJL, Stefánsson BV, Correa-Rotter R. et al. Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med. 2020;383(15):1436-1446
49. Chang SN, Chen JJ, Huang PS. et al. Sodium-Glucose Cotransporter-2 Inhibitor Prevents Stroke in Patients with Diabetes and Atrial Fibrillation. J Am Heart Assoc. 2023;12(10):e027764
50. Tsai HR, Lin YJ, Yeh JI. et al. Sodium-glucose co-transporter-2 inhibitors and the risk of venous thromboembolism: A nationwide population-based study and meta-analysis. Diabetes Metab Res Rev. 2024;40(2):e3739
51. Alicic RZ, Cox EJ, Neumiller JJ. et al. Incretin drugs in diabetic kidney disease: biological mechanisms and clinical evidence. Nat Rev Nephrol. 2021;17(4):227-244
52. Badve SV, Bilal A, Lee MMY. et al. Effects of GLP-1 receptor agonists on kidney and cardiovascular disease outcomes: a meta-analysis of randomised controlled trials. Lancet Diabetes Endocrinol. 2025;13(1):15-28
53. Trillini M, Jenssen TG, Martin WP. et al. The future of glucagon-like peptide-1 receptor agonists in cardiovascular-kidney-metabolic diseases considerations from the ERA Diabesity Working Group. Nephrol Dial Transplant. 2025;40(6):1069-1076
54. Zhao YY, Chen LH, Huang L. et al. Cardiovascular protective effects of GLP-1: a focus on the MAPK signaling pathway. Biochem Cell Biol. 2022;100(1):9-16
55. Drucker DJ. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metab. 2016;24(1):15-30
56. Hendarto H, Inoguchi T, Maeda Y. et al. GLP-1 analog liraglutide protects against oxidative stress and albuminuria in streptozotocin-induced diabetic rats via protein kinase A-mediated inhibition of renal NAD(P)H oxidases. Metabolism. 2012;61(10):1422-1434
57. Sancar-Bas S, Gezginci-Oktayoglu S, Bolkent S. Exendin-4 attenuates renal tubular injury by decreasing oxidative stress and inflammation in streptozotocin-induced diabetic mice. Growth Factors. 2015;33(5-6):419-429
58. Kodera R, Shikata K, Kataoka HU. et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54(4):965-978
59. Park CW, Kim HW, Ko SH. et al. Long-term treatment of glucagon-like peptide-1 analog exendin-4 ameliorates diabetic nephropathy through improving metabolic anomalies in db/db mice. J Am Soc Nephrol. 2007;18(4):1227-1238
60. Alicic RZ, Neumiller JJ, Tuttle KR. Combination therapy: an upcoming paradigm to improve kidney and cardiovascular outcomes in chronic kidney disease. Nephrol Dial Transplant. 2025;40(Suppl 1):i3-i17
61. Tuttle KR, Wong L, St Peter W. et al. Moving from Evidence to Implementation of Breakthrough Therapies for Diabetic Kidney Disease. Clin J Am Soc Nephrol. 2022;17(7):1092-1103
62. Rena G, Lang CC. Repurposing Metformin for Cardiovascular Disease. Circulation. 2018;137(5):422-424
63. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352(9131):854-865
64. Hong J, Zhang Y, Lai S. et al. Effects of metformin versus glipizide on cardiovascular outcomes in patients with type 2 diabetes and coronary artery disease. Diabetes Care. 2013;36(5):1304-1311
65. Holman RR, Paul SK, Bethel MA. et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577-1589
66. Kooy A, de Jager J, Lehert P. et al. Long-term effects of metformin on metabolism and microvascular and macrovascular disease in patients with type 2 diabetes mellitus. Arch Intern Med. 2009;169(6):616-625
67. Katakami N, Yamasaki Y, Hayaishi-Okano R. et al. Metformin or gliclazide, rather than glibenclamide, attenuate progression of carotid intima-media thickness in subjects with type 2 diabetes. Diabetologia. 2004;47(11):1906-1913
68. Lamanna C, Monami M, Marchionni N. et al. Effect of metformin on cardiovascular events and mortality: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2011;13(3):221-228
69. Petrie JR, Chaturvedi N, Ford I. et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017;5(8):597-609
70. Li SN, Wang X, Zeng QT. et al. Metformin inhibits nuclear factor kappaB activation and decreases serum high-sensitivity C-reactive protein level in experimental atherogenesis of rabbits. Heart Vessels. 2009;24(6):446-453
71. Forouzandeh F, Salazar G, Patrushev N. et al. Metformin beyond diabetes: pleiotropic benefits of metformin in attenuation of atherosclerosis. J Am Heart Assoc. 2014;3(6):e001202
72. O'Hora TR, Markos F, Wiernsperger NF. et al. Metformin causes nitric oxide-mediated dilatation in a shorter time than insulin in the iliac artery of the anesthetized pig. J Cardiovasc Pharmacol. 2012;59(2):182-187
73. Davis BJ, Xie Z, Viollet B. et al. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55(2):496-505
74. Ding Y, Zhang M, Zhang W. et al. AMP-Activated Protein Kinase Alpha 2 Deletion Induces VSMC Phenotypic Switching and Reduces Features of Atherosclerotic Plaque Stability. Circ Res. 2016;119(6):718-730
75. Goldberg RB, Temprosa M, Mele L. et al. Change in adiponectin explains most of the change in HDL particles induced by lifestyle intervention but not metformin treatment in the Diabetes Prevention Program. Metabolism. 2016;65(5):764-775
76. Matsuki K, Tamasawa N, Yamashita M. et al. Metformin restores impaired HDL-mediated cholesterol efflux due to glycation. Atherosclerosis. 2009;206(2):434-438
77. Isoda K, Young JL, Zirlik A. et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26(3):611-617
78. Legein B, Temmerman L, Biessen EA. et al. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci. 2013;70(20):3847-3869
79. Tan S, Goudot G, Arnoux A. et al. Occurrence of Major Local Lower Limb Events in Type 2 Diabetic Patients with Lower Extremity Arterial Disease: Impact of Metformin. Ann Vasc Surg. 2023;90:153-161
80. Khan SZ, Rivero M, Nader ND. et al. Metformin Is Associated with Improved Survival and Decreased Cardiac Events with No Impact on Patency and Limb Salvage after Revascularization for Peripheral Arterial Disease. Ann Vasc Surg. 2019;55:63-77
81. Reitz KM, Althouse AD, Forman DE. et al. MetfOrmin BenefIts Lower Extremities with Intermittent Claudication (MOBILE IC): randomized clinical trial protocol. BMC Cardiovasc Disord. 2023;23(1):38
82. Neumiller JJ, Herrin J, Swarna KS. et al. Kidney Outcomes with Glucagon-Like Peptide-1 Receptor Agonists, Sodium-Glucose Cotransporter 2 Inhibitors, Dipeptidyl Peptidase-4 Inhibitors, and Sulfonylureas in Type 2 Diabetes and Moderate Cardiovascular Risk. Clin J Am Soc Nephrol. 2024;20:206-217
83. Richardson TL Jr, Hackstadt AJ, Hung AM. et al. Hospitalization for Heart Failure Among Patients with Diabetes Mellitus and Reduced Kidney Function Treated with Metformin Versus Sulfonylureas: A Retrospective Cohort Study. J Am Heart Assoc. 2021;10:e019211
84. Roumie CL, Chipman J, Min JY. et al. Association of Treatment with Metformin vs Sulfonylurea with Major Adverse Cardiovascular Events Among Patients with Diabetes and Reduced Kidney Function. JAMA. 2019;322(12):1167-1177
85. Whitlock RH, Hougen I, Komenda P. et al. A Safety Comparison of Metformin vs Sulfonylurea Initiation in Patients with Type 2 Diabetes and Chronic Kidney Disease: A Retrospective Cohort Study. Mayo Clin Proc. 2020;95(1):90-100
86. Varvaki Rados D, Catani Pinto L, Reck Remonti L. et al. The Association between Sulfonylurea Use and All-Cause and Cardiovascular Mortality: A Meta-Analysis with Trial Sequential Analysis of Randomized Clinical Trials. PLoS Med. 2016;13(4):e1001992
87. Azoulay L, Suissa S. Sulfonylureas and the Risks of Cardiovascular Events and Death: A Methodological Meta-Regression Analysis of the Observational Studies. Diabetes Care. 2017;40(5):706-714
88. Taylor SI, Cama A, Accili D. et al. Mutations in the insulin receptor gene. Endocr Rev. 1992;13(3):566-595
89. Kolb H, Stumvoll M, Kramer W. et al. Insulin translates unfavourable lifestyle into obesity. BMC Med. 2018;16(1):232
90. van Vliet S, Koh HE, Patterson BW. et al. Obesity Is Associated with Increased Basal and Postprandial β-Cell Insulin Secretion Even in the Absence of Insulin Resistance. Diabetes. 2020;69(10):2112-2119
91. Cryer PE. Hypoglycemia, functional brain failure, and brain death. J Clin Invest. 2007;117(4):868-870
92. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. 2008;37(3):635-646
93. Delea TE, Edelsberg JS, Hagiwara M. et al. Use of thiazolidinediones and risk of heart failure in people with type 2 diabetes: a retrospective cohort study. Diabetes Care. 2003;26(11):2983-2989
94. Page RL 2nd, Gozansky WS, Ruscin JM. Possible heart failure exacerbation associated with rosiglitazone: case report and literature review. Pharmacotherapy. 2003;23(7):945-954
95. Lipscombe LL, Gomes T, Lévesque LE. et al. Thiazolidinediones and cardiovascular outcomes in older patients with diabetes. JAMA. 2007;298(22):2634-2643
96. Singh S, Loke YK, Furberg CD. Thiazolidinediones and heart failure: a teleo-analysis. Diabetes Care. 2007;30(8):2148-2153
97. Lu CJ, Sun Y, Muo CH. et al. Risk of stroke with thiazolidinediones: a ten-year nationwide population-based cohort study. Cerebrovasc Dis. 2013;36(2):145-151
98. Giles TD. The patient with diabetes mellitus and heart failure: at-risk issues. Am J Med. 2003;115(Suppl 8A):107S-110S
99. Hollenberg NK. Considerations for management of fluid dynamic issues associated with thiazolidinediones. Am J Med. 2003;115(Suppl 8A):111S-115S
Author contact
![]() Corresponding authors: Jiaqi Hu, Ph.D., School of Healthcare Management, Tsinghua Medicine, Tsinghua University, Beijing, China; Institute for Hospital Management, Tsinghua University, Beijing, China; 30 Shuangqing Rd, Haidian District, Beijing, China 100084; Email: hujiaqiedu.cn; ORCID: 0000-0002-4082-4755; Wenge Li, M.D., Department of Nephrology, China-Japan Friendship Hospital, No. 2 East Yinghuayuan Street, Chaoyang District, Beijing, China 100029; Email: liwengeedu.cn; ORCID: 0000-0002-4749-1009.
Corresponding authors: Jiaqi Hu, Ph.D., School of Healthcare Management, Tsinghua Medicine, Tsinghua University, Beijing, China; Institute for Hospital Management, Tsinghua University, Beijing, China; 30 Shuangqing Rd, Haidian District, Beijing, China 100084; Email: hujiaqiedu.cn; ORCID: 0000-0002-4082-4755; Wenge Li, M.D., Department of Nephrology, China-Japan Friendship Hospital, No. 2 East Yinghuayuan Street, Chaoyang District, Beijing, China 100029; Email: liwengeedu.cn; ORCID: 0000-0002-4749-1009.