Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2026; 23(6):1992-2005. doi:10.7150/ijms.112837 This issue Cite
Research Paper
Therapeutic Hypothermia Ameliorates Apoptosis and Cerebral Injury by Upregulating HECTD1-mediated Ubiquitination and VDAC3 Degradation in a Rat CPR Model
Pei Liu, MD1#, Jiali Wu, MD2#, Lirong Wang, MD1, Jiawei He, MD1, Zhanghuan He, MD1, Lei Wang, MS5, Shen Zhao, MD, PhD1,3,4 ![]()
1. Department of Critical Care Medicine, Beijing Friendship Hospital, Capital Medical University, Beijing, China.
2. Department of Critical Care Medicine, Fuzhou University Affiliated Provincial Hospital, Fujian Provincial Hospital, Fujian Provincial Center for Critical Care Medicine, Fuzhou, China.
3. Fujian Institute of Emergency Research, Fuzhou University Affiliated Provincial Hospital, Fujian Provincial Hospital, Fuzhou, China.
4. Fujian Provincial Key Laboratory of Emergency Medicine, Fuzhou, China.
5. Laboratory Animal Center, Beijing Friendship Hospital, Capital Medical University, Beijing, China.
# The authors contributed equally to this work.
Received 2025-2-25; Accepted 2026-4-10; Published 2026-4-23
Abstract

Background and Objective: Post-cardiac arrest (CA) brain injury is a leading cause of death and disability in patients with return of spontaneous circulation (ROSC). Our previous study showed that hypothermia-induced VDAC3 ubiquitination attenuated apoptosis of microglia cells following simulated ischemia/reperfusion (I/R). This study aimed to further investigate the role of E3 ubiquitin ligase, HECTD1, in the ubiquitination and degradation of VDAC3, which is involved in hypothermic neuroprotection.
Methods: Sprague-Dawley rats were randomized into two groups, namely the normothermic (T37) group and hypothermic (T33) group, which were further divided into three subgroups: PBS, si-NC, and si-Hectd1. All the rats were subjected to 8 min of asphyxia-induced CA and cardiopulmonary resuscitation (CPR). The core temperature in the T33 and T37 groups were maintained at 33 ± 0.5 °C and 37 ± 0.5 °C, respectively, after 5 min following ROSC and maintained for 6 h. The si-Hectd1 subgroups were administered adeno-associated viral vector with siRNA against rat Hectd1 (si-Hectd1). The PBS and si-NC subgroups were administered the same volume of PBS and nonspecific siRNA, respectively, as controls. Survival time was measured for the resuscitated rats. At 72 h after ROSC, the neurological injuries to rats were evaluated using neurological deficit scores, Serum S100B and neuron-specific enolase levels, hematoxylin and eosin staining, and transmission electron microscopy analysis. Apoptosis-related proteins were measured using western blot assay. The interaction and colocalization of HECTD1 and VDAC3 were evaluated by immunoprecipitation and double immunofluorescence assays, respectively.
Results: Survival time and neurological outcomes, as well as ultrastructural damage, were significantly improved in the T33 group, with the T37 + si-Hectd1 subgroup showing the worst outcome at ROSC 72 h. Compared to the T37 group, the T33 group showed reduced expression of VDAC3, cleaved caspase-3, and BAX but increased expression of HECTD1 and BCL-2 at ROSC 72 h. Additionally, compared to the T37 group, the T33 group showed increased VDAC3 and Hectd1 interaction, resulting in increased VDAC3 ubiquitination at ROSC 72 h; Hectd1 knockdown reversed these effects. The PBS and si-NC subgroups showed no significant differences under both targeted temperatures.
Conclusions: Hypothermia treatment contributed to greater antiapoptotic neuroprotection in the rat CA/CPR model, which may be attributed to the promotion of HECTD1-mediated ubiquitination and degradation of VDAC3.
Keywords: cardiac arrest, cardiopulmonary resuscitation, therapeutic hypothermia, apoptosis, ubiquitination
Introduction
Hypoxic-ischemic brain injury (HIBI) is a severe sequela directly induced by global ischemia/reperfusion (I/R) following cardiac arrest (CA). HIBI survivors usually progress to brain death or persistent vegetative state after a stable return of spontaneous circulation (ROSC) [1, 2]. Current guidelines and recent reviews recommend continuous monitoring of core temperature and active temperature management of post-ROSC patients for a better outcome [3-6]. However, the optimal targeted temperature for ROSC patients has not yet been determined. Recent large-scale clinical trials showed no significant positive outcome for hypothermia (33°C) therapy compared to normothermia for patients with ROSC [7, 8]. Moreover, the neuroprotective mechanisms of hypothermia are not yet fully understood. Consequently, further preclinical studies are required to clarify the underlying mechanisms of therapeutic hypothermia for patients with ROSC.
Hypoxic-ischemic injury leads to mitochondrial impairment, which results in the release of proapoptotic proteins, followed by cellular apoptosis [9, 10]. Voltage-dependent anion channel (VDAC), a porin protein in the outer mitochondrial membrane (OMM), regulates ion and metabolite fluxes, modulates reactive oxygen species production, and is associated with mitochondrial-mediated apoptosis [11, 12]. Our previous study demonstrated that therapeutic hypothermia upregulated VDAC3 ubiquitination, thereby protecting murine BV2 microglia from oxygen-glucose deprivation/reoxygenation-induced apoptosis [13]. However, the neuroprotective mechanisms of VDAC3 degradation require further investigation.
Hectd1, a member of homologous to the E6-AP C-terminal (HECT) domain-containing E3 ubiquitin ligase family, is widely expressed in both human and murine primary cells, including neuronal cells and fibroblasts [14, 15]. Hectd1 may play varying roles in the cerebral ubiquitin-proteasome system based on its target proteins [16, 17]. A recent study showed that I/R significantly decreases Hectd1 expression, thereby affecting cell viability and apoptosis [18]. Furthermore, bioinformatics analysis revealed that circular RNA (circRNA) Hectd1 is a promising biomarker for acute ischemic stroke [19]. However, studies investigating the effect of hypothermia on Hectd1 expression are limited. Preliminary findings from our proteomics dataset revealed that HECTD1 rose significantly in hypothermia group when compared with normothermia group at 72 h post-ROSC in a rodent cardiac arrest model (see Supplementary Figure S1). In this study, we hypothesized that therapeutic hypothermia may upregulate Hectd1 expression, resulting in VDAC3 degradation. Thus, we investigated the interaction between Hectd1 and VDAC3 using conditional Hectd1 knockdown rats in an experimental model of asphyxial CA and cardiopulmonary resuscitation (CPR).
Materials and Methods
Ethics statement
This study was approved by the Animal Welfare and Ethics Committee of Beijing Friendship Hospital, Capital Medical University, Beijing, China (license no. 23-1005). All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health [20, 21].
Animals and grouping
Healthy adult male specific-pathogen free Sprague-Dawley rats (weight: 400-450 g) were purchased from the Experimental Animal Center of Capital Medical University (license number: SYXK 2022-0022). All rats were adaptively reared at 22-25 °C and 60-80% relative humidity and subjected to a 12 h/12 h light-dark cycle for 1 week. Rats were fasted overnight but had ad libitum access to water before the CPR experiment. Animals were randomized into two groups (n = 24/group): normothermia (T37) group and hypothermia (T33) group. The core temperature of the T33 rats was rapidly reduced to 33 ± 0.5 °C using ice packs, an electrical fan, and a cooling blanket at 5 min after ROSC; this temperature was maintained for 6 h using a temperature-controlled blanket (Boruida, Chongqing, China). Subsequently, the T33 rats were rewarmed at a rate of 1.0 °C/h, until their core temperature reached 37 ± 0.5 °C. Furthermore, the core temperature of the T37 rats was maintained at 37 ± 0.5 °C. Both the T37 and T33 rats were then randomly divided into three subgroups (n = 8/group) and administered adeno-associated viral vector (AAV) carrying small interfering RNA (siRNA) against rat Hectd1 (si-Hectd1; si-Hectd1 group), phosphate-buffered saline (PBS; PBS group), or nonspecific negative control (NC) siRNA (si-NC group) via lateral ventricle injection 2 weeks before CPR.
siRNA preparation and screening
Specific siRNAs were selected to knockdown Hectd1 expression in the rat brain. Three rat Hectd1 genes, including Hectd1-Rat-265, Hectd1-Rat-2716, and Hectd1-Rat-504, were designed and synthesized as interference targets by Genepharma (Shanghai, China). The specificity of the designed siRNAs was verified by transfecting them into NRK-49F cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The mRNA expression levels of Hectd1 were examined using quantitative polymerase chain reaction (qPCR) (see Supplementary Figure S2). Based on qPCR results, Hectd1-rat-2716-siRNA (5'-GGTGGTAACACTCAGAAAT-3') was selected for the design of si-Hectd1.
Preparation of recombinant adenovirus particles
Rat Hectd1 (Gene ID: 362736) knockdown was achieved by RNA interference using the AAV-based siRNA approach. The generation of recombinant AAV expressing si-Hectd1 (AAV-GP-7-Hectd1-Rat-2716) and si-NCs (AAV-GP-7-NC), as well as viral titer testing, was performed by Genepharma (Shanghai, China). The AAV-293T cells were co-transfected with the RNAi-Mate transfection reagent. After infection, the titers of AAV-GP-7-Hectd1-Rat-2716 and AAV-GP-7-NC were 1.35 × 1013 and 2.72 × 1013 V.G/ml, respectively.
Experimental preparation
The animals were anesthetized using an intraperitoneal injection of pentobarbital (45 mg/kg). Orotracheal intubation was completed with a 14 G cannula (Sirui Teaching experiment instrument, Chongqing, China). End-tidal CO2 was continuously monitored with a CO2 analyzer (Capnostream™ 20; Medtronic, CO, USA). Electrocardiogram lead II was recorded. Mean arterial pressure (MAP) and medicine delivery were monitored by inserting a polyethylene catheter (PE-50; Becton-Dickinson, Franklin Lakes, NJ) from the right femoral artery into the descending aorta. Rectal temperature was maintained at 37 ± 0.5 °C using a temperature-controlled blanket. All data were recorded using a four-channel data acquisition system (Taimeng BL-420N, Chengdu, China).
Establishment of asphyxial CA and CPR
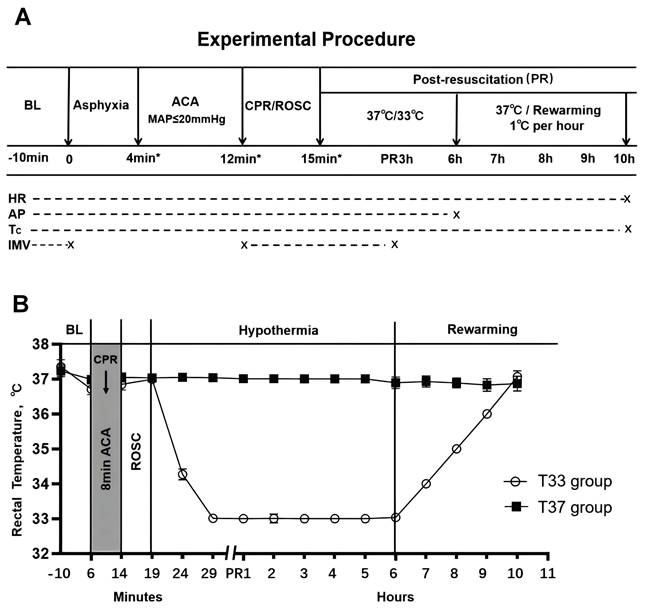
Baseline measurements were recorded and mechanical ventilation (tidal volume: 0.55 mL/100 g body weight, frequency: 100 breaths/minute, and inspired O2 fraction: 0.21) was initiated 10 min prior to asphyxia induction. Asphyxia was induced by a combination of transarterial rocuronium (1 mg/kg; Merk Sharp & Dohme B.V., Netherlands) administration and endotracheal tube clamping. CA was defined as MAP ≤ 20 mmHg which occurred approximately 4 min following asphyxia. Precordial compression (PC) was initiated 8 min after CA onset to provide a 2:1 compression/ventilation ratio with equal compression and relaxation [22, 23]. The compression depth and position were adjusted to maintain a MAP ≥ 20 mmHg. Epinephrine (0.02 mg/kg) was administrated via the femoral artery catheter after 1 min of CPR. In the case of ventricular fibrillation during PC, defibrillation was attempted with a 3-J countershock. ROSC was defined as the return of a supraventricular rhythm with a MAP ≥ 60 mmHg for at least 5 min. If ROSC was not achieved, an additional dose of epinephrine was administered after 3 min of CPR. CPR was halted if ROSC was not achieved after 5 min. Mechanical ventilation with 100% inspired O2 was maintained for 1 h following ROSC, and normoxic ventilation was maintained thereafter until the recovery of autonomous respiration. Ventilation weaning and tracheal extubation were performed, followed by sputum suction. The arterial catheter was removed prior to rewarming. Each resuscitated animal was returned to a separate cage, and all the animals were monitored closely for 72 h. The experimental outline and temperature monitoring are summarized in Figure 1.
(A) Experimental outline. (B) Core temperature (°C) of the T37 and T33 groups during the experiment. The T37 group includes T37 + PBS, T37 + si-NC, and T37 + si-Hectd1 subgroups, while the T33 group includes T33 + PBS, T33 + si-NC, and T33 + si-Hectd1 subgroups. *Values are expressed as means. BL, baseline; ACA, asphyxial cardiac arrest; CPR, cardiopulmonary resuscitation; ROSC, return of spontaneous circulation; PR, post resuscitation; HR, heart rate; AP, arterial pressure; Tc, core temperature; and IMV, invasive mechanical ventilation.
Measurements
Assessment of neurological function
Neurologic function was evaluated based on the neurological deficit score (NDS) method by two investigators who were blinded to the protocol. NDS, ranging between from 0 (no observed neurologic deficit) to 500 (death or brain death), was recorded at 24-h intervals for 72 h [24].
Specimen collection
The animals were euthanized with an overdose of pentobarbital (150 mg/kg, intraperitoneally) after NDS testing at 72 h post resuscitation (PR). Then blood samples were obtained for the measurement of serum biomarkers. For histological evaluation, the animals were sacrificed and subjected to cardiac perfusion with 200 ml of normal saline. Rat brains were obtained and kept on the ice after decapitation. The right frontal cortex of each rat was collected for transmission electron microscopy (TEM) analysis. The right parietal cortex was fixed in 10% formalin. It was then processed through routine dehydration, transparency, embedding, and slicing. Finally, hematoxylin and eosin (HE) staining and immunofluorescence (IF) staining analysis were performed. The left cortex of each rat was quickly separated and stored at -80°C in a refrigerator for CO-immunoprecipitation (CO-IP) and western blot (WB) analyses.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were centrifuged at 1000×g for 15 min, and the obtained serum was stored at -80 °C until further analysis. Serum concentrations of neuron-specific enolase (NSE) and S100B were measured using commercially available ELISA kits (E07963r and E08066r, respectively; CUSABIO), according to the manufacturer's instructions. The optical density was determined within 5 min using a microplate reader set to 450 nm.
Hematoxylin-eosin (HE) staining
The wax block was sliced into coronal sections (4-μm), dewaxed, debenzolized, and HE stained. Then, the histomorphological changes in the cortex were observed under a microscope. Then HE-stained sections were semi-quantitatively assessed using a histopathological injury scale ranging from 0 to 4[25].
TEM analysis
The tissue sample was rapidly cut into 1 × 1 × 1 mm blocks, fixed in 2.5% glutaraldehyde, and stored at 4 °C. Thereafter, the sample was rinsed with PBS and fixed in 1% osmic acid for 2 h at room temperature. The samples were then dehydrated using an ethanol and acetone gradient and embedded in 812 resin. The embedded tissue blocks were sectioned and observed under a TEM (HT7700, Hitachi) to evaluate the ultrastructural changes in the cortex neurons. TEM images were further Semi-quantitative analysed using Mitochondrial damage index (MDI) ranging from 0 to 3 [26].
WB analysis
Protein concentrations were measured using a Bradford protein assay kit (Bio-Rad Laboratories). The protein samples (30 µg/lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories). The membrane was blocked with 5% skim milk in Tris-buffered saline containing Tween 20 (TBST) and incubated overnight at 4°C with the following primary antibodies: anti-HECTD1 (sc-517169; Santa Cruz Biotechnology, USA), anti-VDAC3 (55260-1-AP; Proteintech Group, China), anti-BAX (YT0455; ImmunoWay Biotechnology, USA), anti-BCL-2 (YM3041; ImmunoWay Biotechnology), anti-cleaved caspase-3 (CC3; YC0006; ImmunoWay Biotechnology), or β-actin (YT0099; ImmunoWay Biotechnology). The membrane was then washed with TBST and incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies, goat anti-mouse IgG or goat anti-rabbit IgG (RS0001 and RS0002, respectively; ImmunoWay Biotechnology). β-Actin was used as an internal control to normalize the relative expression of HECTD1, VDAC3, BAX, BCL-2, and CC3. The optical densities of the protein bands were analyzed using ImageJ software.
Double IF staining
Histologic localization of HECTD1 and VDAC3 was determined by IF staining. The brain sections were dewaxed, rehydrated, rinsed with PBS, and incubated with 5% BSA for 2 h at room temperature. Thereafter, the samples were incubated overnight at 4 °C in a wet box with rabbit anti-HECTD1 and anti-VDAC3 polyclonal antibodies. The samples were rewarmed at room temperature for 30 min and then incubated for 2 h with the corresponding fluorescent secondary antibodies at 4 °C in a dark box. Subsequently, the samples were washed thrice with PBS, sealed with an anti-quenching sealing agent containing DAPI, and observed under a fluorescence microscope. Red, green, and blue fluorescence indicated VDAC3, HECTD1, and DAPI expression, respectively. For each slice, five visual fields were randomly captured at 200× magnification using an inverted fluorescence microscope (Leica DMi8, Germany). The fluorescence intensities in each visual field were measured using the Image J software. For the quantification analysis, the captured images were analyzed using the co-localization tools incorporated within the Coloc 2 plugin to obtain a scatter plot. The Pearson colocalization coefficients (PCC) of HECTD1 and VDAC3 were displayed in the colocalized volume, wherein 1, 0, and -1 indicated perfect correlation, no correlation, and perfect inverse correlation, respectively.
CO-immunoprecipitation (CO-IP) and immunoblotting (IB) assays
The interaction between HECTD1 and VDAC3 was verified by CO-IP assay. For this, fresh tissue sample (20 mg) was used to prepare 100-200 μL of cortex lysate. The supernatant was collected after ultrasonic homogenization and centrifugation (12000 rpm, 4 °C, and 15 min), and the total protein concentration of the sample was determined using a bicinchoninic acid assay kit (P0010S; Beyotime Biotechnology, China). IP was performed using Co-IP kits (P2179M; Beyotime Biotechnology, China) according to the manufacturer's instructions. Briefly, the protein samples were divided into three parts, which were then treated with 1 SDS-PAGE loading buffer and boiled for 10 min (positive control; input); incubated overnight with IgG antibodies (negative control); or incubated overnight with rabbit anti-VDAC3 primary antibodies at 4 °C with shaking. The antibody-antigen complexes were mixed with magnetic beads and incubated overnight at 4 °C. After three washes with PBS, the immunoprecipitated proteins were eluted and subjected to SDS-PAGE, followed by immunoblotting with an anti-HECTD1 antibody (Santa Cruz Biotechnology) to detect co-precipitated HECTD1. To further validate this interaction, a reciprocal Co-IP was performed. Cortical lysates were incubated overnight at 4°C with a rabbit anti-HECTD1 antibody. Immune complexes were collected using magnetic beads, washed thoroughly with PBS, and analyzed by immunoblotting with an anti-VDAC3 antibody.
SDS-PAGE loading buffer and boiled for 10 min (positive control; input); incubated overnight with IgG antibodies (negative control); or incubated overnight with rabbit anti-VDAC3 primary antibodies at 4 °C with shaking. The antibody-antigen complexes were mixed with magnetic beads and incubated overnight at 4 °C. After three washes with PBS, the immunoprecipitated proteins were eluted and subjected to SDS-PAGE, followed by immunoblotting with an anti-HECTD1 antibody (Santa Cruz Biotechnology) to detect co-precipitated HECTD1. To further validate this interaction, a reciprocal Co-IP was performed. Cortical lysates were incubated overnight at 4°C with a rabbit anti-HECTD1 antibody. Immune complexes were collected using magnetic beads, washed thoroughly with PBS, and analyzed by immunoblotting with an anti-VDAC3 antibody.
Statistical analysis
Normal and abnormal distribution data were reported as mean ± standard deviation ( ± SD) and median (25% and 75% quartile), respectively. Normal distribution was confirmed with the Kolmogorov-Smirnov test. For normally distributed data, the variables between subgroups were compared using a one-way analysis of variance with Bonferroni's correction. The NDS was compared using the Mann-Whitney U test. The survival rates were compared using Fisher's exact test. Survival time was compared using Kaplan-Meier survival analysis. Comparisons of HE staining and TEM findings between the normothermia and hypothermia groups were conducted using the Kruskal-Wallis test. The SPSS 22.0 software (IBM, New York, NY, USA) was used for statistical analysis. P < 0.05 indicated statistically significant differences.
± SD) and median (25% and 75% quartile), respectively. Normal distribution was confirmed with the Kolmogorov-Smirnov test. For normally distributed data, the variables between subgroups were compared using a one-way analysis of variance with Bonferroni's correction. The NDS was compared using the Mann-Whitney U test. The survival rates were compared using Fisher's exact test. Survival time was compared using Kaplan-Meier survival analysis. Comparisons of HE staining and TEM findings between the normothermia and hypothermia groups were conducted using the Kruskal-Wallis test. The SPSS 22.0 software (IBM, New York, NY, USA) was used for statistical analysis. P < 0.05 indicated statistically significant differences.
Results
Characteristics at baseline and during CPR
Of the 136 rats analyzed in this study, 110 were resuscitated successfully. There were no significant differences in the initial ROSC rate, baseline physiological parameters, asphyxia time, PC duration, total epinephrine dosages, defibrillation numbers, and 72 h-survival rates among the six subgroups (all P > 0.05) (Table 1).
Baseline and outcomes of resuscitation
| Variables | T37+PBS | T37+si-NC | T37+si-Hectd1 | T33+PBS | T33+si-NC | T33+si-Hectd1 | P value |
|---|---|---|---|---|---|---|---|
| Body weight, g | 431±15 | 433±13 | 440±10 | 435±12 | 428±15 | 430±16 | 0.92 |
| Rectal temperature, ℃ | 37.1±0.4 | 37.0±0.2 | 37.3±0.5 | 37.0±0.3 | 37.0±0.6 | 37.1±0.5 | 0.80 |
| ETCO2, mmHg | 51±4 | 53±3 | 52±5 | 51±4 | 53±5 | 52±4 | 0.87 |
| Heart rate, bpm | 451±39 | 457±29 | 442±24 | 456±25 | 449±27 | 463±21 | 0.74 |
| MAP, mmHg | 129±6 | 129±7 | 130±7 | 133±8 | 134±9 | 135±11 | 0.52 |
| Asphyxia time, s | 252±28 | 244±17 | 238±17 | 236±11 | 238±21 | 238±29 | 0.68 |
| PC time, s | 172±75 | 184±69 | 162±68 | 148±58 | 187±63 | 187±77 | 0.82 |
| Total epinephrine dosages, ml | 0.15±0.1 | 0.13±0.2 | 0.14±0.1 | 0.15±0.1 | 0.13±0.1 | 0.15±0.1 | 0.93 |
| Defibrillation numbers | 0.6±0.7 | 0.5±0.5 | 0.5±0.5 | 0.6±0.7 | 0.5±0.5 | 0.6±0.5 | 1.00 |
| ROSC/total CPR | 17/21 | 18/22 | 23/28 | 16/20 | 17/21 | 19/24 | 1.00 |
| 72-hour survival/ROSC | 8/17 | 8/18 | 8/23 | 8/16 | 8/17 | 8/19 | 0.95 |
Note: Values are expressed as means ± SD. ETCO2, End-tidal carbon dioxide; MAP, Mean aortic pressure; Asphyxia time, time from start of asphyxia to MAP≤20mmHg; PC, precordial compression; ROSC, return of spontaneous circulation; CPR, cardiopulmonary resuscitation
Hypothermia improved survival time and neurological outcome in rats following CA/CPR
Despite no significant difference in the 72-h-survival rate, the T33 group demonstrated an overall improved median survival time, NDS, and brain injury biomarkers compared to the T37 group (all P < 0.05). Additionally, at both targeted temperatures, si-Hectd1 subgroups exhibited significantly decreased survival time and neurological function compared to the PBS and si-NC subgroups (all P < 0.05). Among these, the T37 + si-Hectd1 subgroup showed the worst survival time (P = 0.03), NDS (P = 0.04), and brain injury biomarkers, including NSE (P = 0.02) and S100B (P = 0.01) (Figures 2A-D).
(A) Kaplan-Meier survival curves of T37 + PBS, T37 + si-NC, T37 + si-Hectd1, T33 + PBS, T33 + si-NC, and T33 + si-Hectd1 subgroups. Median survival times of the six subgroups (n = 8), as determined by the Breslow test. (B) NDS of the six subgroups PR (n = 8). (C, D) The serum NSE and S100B levels in the six subgroups (n = 8). (E) Representative HE-stained images (magnification: 200×) of the cerebral cortex samples of the six subgroups (n = 8) at 72-h PR. Black arrows indicate pyknosis, while red arrows indicate vacuolar degeneration. (F) Semi-quantitative analyses for the HE images in the six subgroups (n = 8). ROSC, return of spontaneous circulation; NDS, neurological deficit score; NSE, neuron-specific enolase; HE, hematoxylin and eosin; and PR, post resuscitation. *P < 0.05 vs. T37 group and #P < 0.05 vs. PBS or si-NC subgroup.
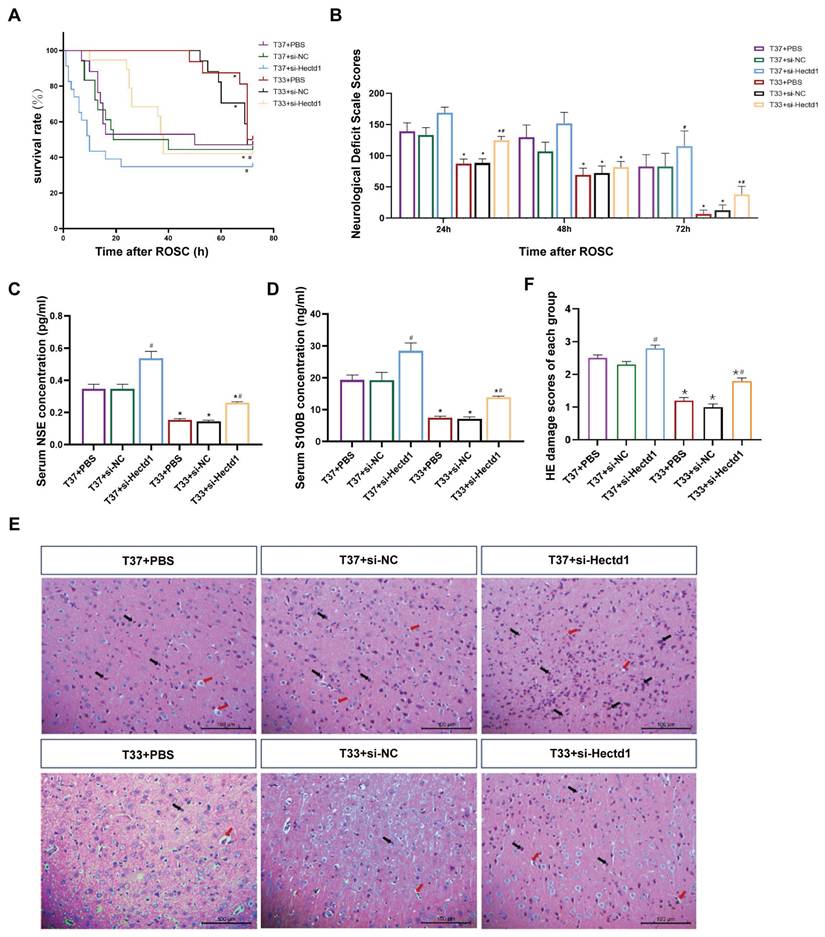
Furthermore, HE staining revealed that the cerebral cortex samples of the T37 group showed deeply stained nuclei, pyknotic changes with vacuolated cytoplasm, and interstitial loosening (Figure 2E). However, the T33 group showed significantly improved cell morphology compared to the T37 group (P < 0.01). Additionally, our results revealed that at both targeted temperatures, si-Hectd1 subgroups showed significantly aggravated pathomorphologic changes compared to the PBS and si-NC subgroups (all P < 0.05) (Figure 2F). Among these, the T37 + si-Hectd1 subgroup showed the worst cerebral pathology (P < 0.05 vs. T37 + si-NC group or T37 + PBS). These results indicate that HECTD1 may contribute to hypothermia-induced neurological protection following asphyxial CA and resuscitation.
Hypothermia induced alterations in the mitochondrial morphology in rats following CA/CPR
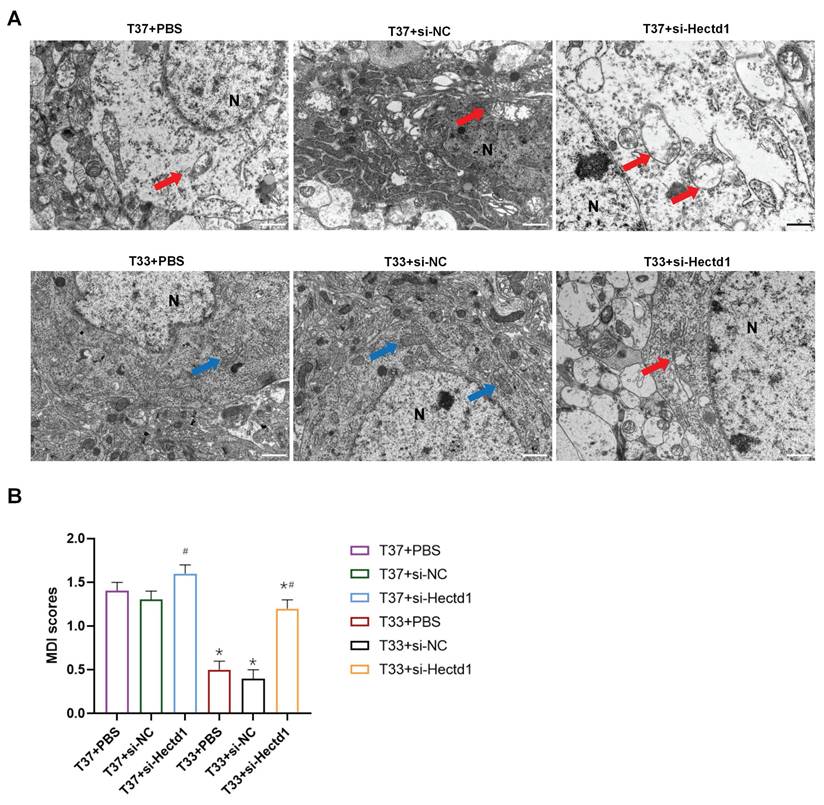
To demonstrate the effect of hypothermia on mitochondria after CA/CPR, we detected their ultrastructure in the cortex tissues with TEM scanning. TEM images revealed that the T37 group showed severe mitochondrial swelling with disrupted bilayer membrane, vacuolation, and few cristae. However, hypothermia alleviated these morphological alterations in the cortex tissues of the T33 group (P < 0.01). The si-Hectd1 subgroups showed significantly altered mitochondrial morphology compared to the PBS and si-NC subgroups at both targeted temperatures (all P < 0.01). The T37 + si-Hectd1 subgroup exhibited the most severe mitochondrial damage (P < 0.05 vs. T37 + si-NC group or T37 + PBS) (Figure 3). These results indicate that HECTD1 may be involved in the hypothermia-mediated alleviation of alterations in mitochondrial morphology.
Hypothermia reduced the mitochondrial ultrastructure damage in the cerebral cortex of the CA/CPR rat model. (A) Representative TEM images (Scale bar = 1 μm) of mitochondria of each group. N indicates nucleus, the blue arrow indicates relatively intact mitochondria, and the red arrow indicates swollen mitochondria with fractured cristae. (B) Semi-quantitative analyses for the TEM images in the six subgroups (n = 8). *P < 0.05 vs. T37 group and #P < 0.05 vs. PBS or si-NC subgroup.
Hypothermia reduced the expression of apoptosis-associated proteins in rats following CA/CPR
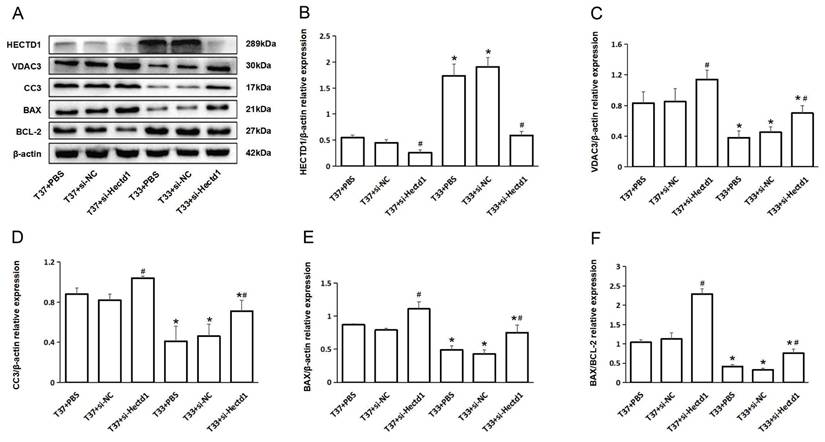
To further analyze the effect of hypothermia on the expression of apoptosis-associated proteins, we conducted WB analysis of HECTD1, VDAC3, CC3, BAX, and Bcl-2. Compared to the T37 group, the T33 group showed significantly increased HECTD1 expression (P < 0.05). In contrast, the T33 group showed significantly reduced VDAC3, CC3, and BAX expression as well as BAX/BCL-2 ratio compared to the T37 group (all P < 0.05), consistent with the results of our previous study. However, the si-Hectd1 subgroups showed significantly increased levels of the aboving markers at both target temperatures compared to the PBS and si-NC subgroups (all P < 0.05). And the T37 + si-Hectd1 subgroup exhibited the highest level of pro-apoptosis-associated protein expression (P < 0.05 vs. T37 + si-NC group or T37 + PBS) (Figure 4). These results suggest that hypothermia may increase HECTD1 expression, resulting in the promotion of VDAC3 degradation in the PR period.
Hypothermia reduced the expression of apoptosis-associated proteins in rats following CA/CPR. (A) WB analysis of HECTD1, VDAC3, CC3, BAX, and BCL-2 expression at 72-h PR (n = 4). (B-F) Quantitative analysis of HECTD1, VDAC3, CC3, BAX, and BCL-2. Values are represented as mean ± SD. *P < 0.05 vs. T37 group and #P < 0.05 vs. PBS or si-NC subgroup.
Hypothermia enhanced Hectd1 and VDAC3 interaction in rats following CA/CPR
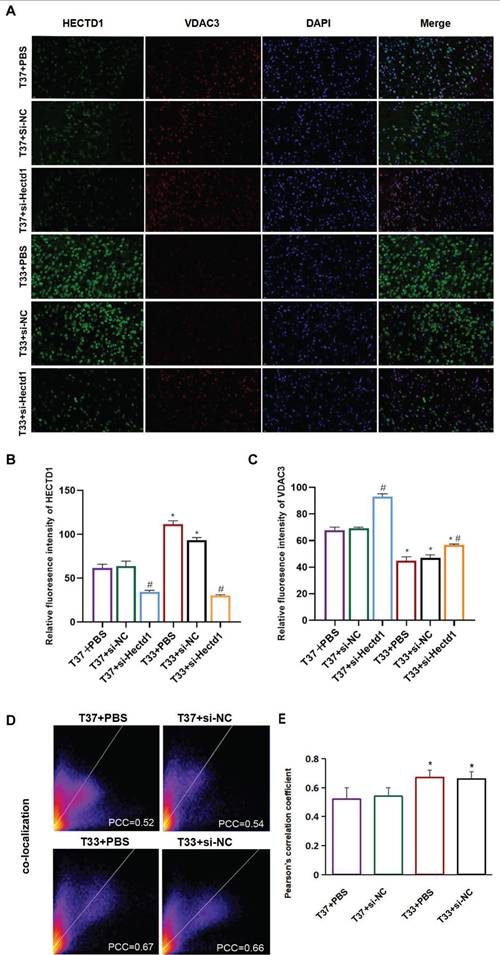
The results of double IF staining using anti-HECTD1 (green) and anti-VDAC3 (red) antibodies revealed that VDAC3 (an OMM marker) was colocalized with HECTD1 in the cerebral cortex of rats following resuscitation (Figure 5A). The fluorescence intensities of both HECTD1 and VDAC3 were consistent with the trends observed with the IB assay (Figures 5B, C). As indicated in Figure 5 D, the distribution scatter plot showed a representative image derived from the PCC obtained using the Coloc 2 plugin. Compared to the T37 group, the T33 group showed a significantly increased fluorescence intensity of HECTD1, resulting in a higher PCC (P < 0.01) (Figure 5 E). Interestingly, fluorescence intensity of VDAC3 was significantly higher in the T37 + si-Hectd1 group than in the T33 + si-Hectd1 group, suggesting that Hectd1 may partly interpret the decrease in VDAC3 expression (P < 0.01) (Figures 5A, C). These results indicate that hypothermia may enhance HECTD1 expression and its colocalization with VDAC3.
Hypothermia enhanced Hectd1 and VDAC3 co-localization in rats following CA/CPR. (A) IF images (magnification: 200×) showing the density and distribution of neurons labeled with anti-HECTD1 (green) and anti-VDAC3 (red) antibodies. Blue fluorescence indicates DAPI-labeled nuclei. (B, C) Statistical analysis of HECTD1 or VDAC levels between T33 and T37 groups. (D) Pixel distribution scatter plot: yellow pixels along the diagonal represent colocalized intensity; PCC, Pearson's correlation coefficient. (E) PCC values were analyzed for at least 3 different fields for each group. Values are represented as the mean ± SD (n = 8). *P < 0.05 vs. T37 group.
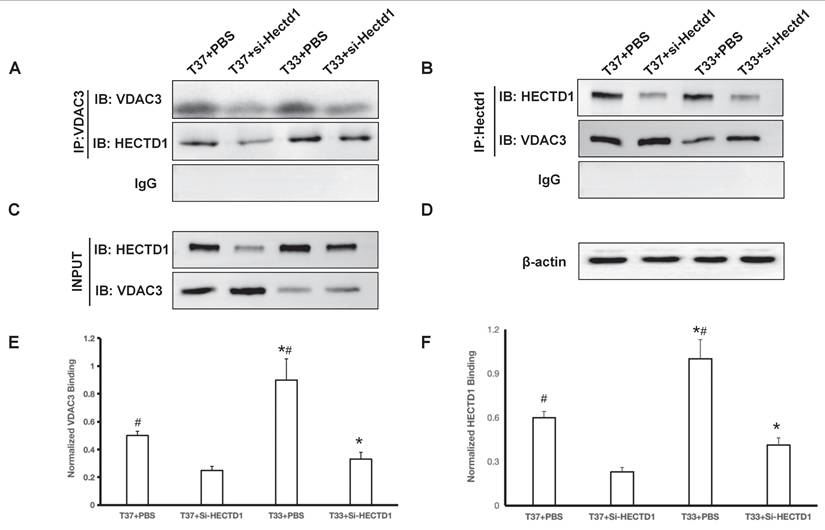
Co-immunoprecipitation assays confirmed a direct interaction between HECTD1 and VDAC3. Lysate from cortex tissues was immunoprecipitated with an anti-HECTD1 antibody. Subsequent IB assay with an anti-VDAC3 antibody revealed a clear band specifically in the HECTD1 immunoprecipitate, but not in the control IgG precipitate (P < 0.01 vs. si-Hectd1 group) (Figure 6A). Reciprocal co-IP using an anti-VDAC3 antibody for pull-down also successfully co-precipitated HECTD1, further validating this interaction (P < 0.01 vs. si-Hectd1 group) (Figure 6B). Hypothermia treatment led to a marked upregulation of HECTD1 expression, as shown in the input lysates (P < 0.01 vs. T 37 group) (Figure 6C). This also reflects a change in binding affinity by normalizing the co-IP signal to the respective input levels (Figure 6E-F). The results suggest that exposure to hypothermia treatment upregulated HECTD1 expression and potentiated its interaction with VDAC3, thereby resulting in enhanced HECTD1-mediated ubiquitination of VDAC3.
Hypothermia enhanced HECTD1 interacted with VDAC3 in rats following CA/CPR. (A-B) Co-IP assays demonstrate reciprocal binding in the PBS and si-Hectd1 subgroups of T37 and T33 groups. Lysates were immunoprecipitated with antibody against HECTD1 (IB: VDAC3) and VDAC3 (IB: HECTD1) as shown. (C) Input controls of VDAC3 and HECTD1. (D) Loading control β-actin in the lysates. (E-F) Quantitative analysis of protein interaction. The relative binding affinity between HECTD1 and VDAC3 was quantified using the formula: (VDAC3 intensity in Co-IP / HECTD1 intensity in Co-IP) ÷ (VDAC3 intensity in Input / Loading control intensity in Input) and (HECTD1 intensity in Co-IP / VDAC3 intensity in Co-IP) ÷ (HECTD1 intensity in Input / Loading control intensity in Input). Data are presented as mean ± SEM from four independent experiments. Values are presented as the mean ± SD. *P < 0.05 vs. T37 group and #P < 0.05 vs. si-Hectd1 subgroup.
Proposed mechanism underlying the neuroprotective effect of therapeutic hypothermia after cardiac arrest. Therapeutic hypothermia (TH) increases the expression of the E3 ubiquitin ligase HECTD1, which promotes ubiquitination-dependent degradation of the mitochondrial channel protein VDAC3. The reduction of VDAC3 is associated with decreased mitochondrial apoptotic signaling and lower levels of cleaved caspase-3, ultimately attenuating neuronal apoptosis and improving neurological outcomes.
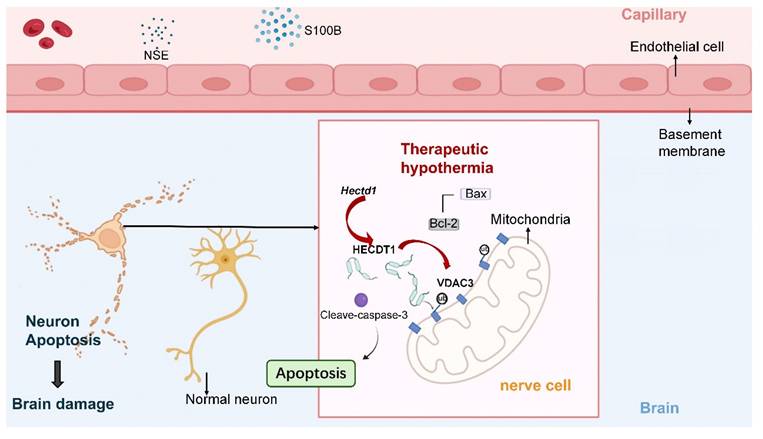
Discussion
This study demonstrated that mild hypothermia following ROSC alleviated neurological dysfunction and apoptosis in the CA/CPR rat model. Additionally, this study revealed that the beneficial effect of hypothermia was associated with VDAC3 ubiquitination in vivo, consistent with our previous findings [13]. Furthermore, this study indicated that, compared to the normothermic treatment, hypothermic treatment increased the interaction between the E3 ubiquitin ligase HECTD1 and VDAC3, which retained the improved neurologic outcomes and survival time in the CA/CPR rat model.
VDACs are considered highly conserved proteins of the OMM and are known to be involved in many cellular processes, including regulation of energy metabolism/ion transport and mitochondria-mediated apoptosis, autophagy, or ferroptosis, by interacting with cytosolic and mitochondrial proteins [27-29]. Many studies have shown that mitochondrial permeability transition pore formation, dependent on VDACs and other component proteins, induces apoptosis during I/R [30, 31]. Electrophysiological and immunocytochemical observations showed an increase in VDAC expression or activation in neurons during apoptosis and/or oxidative damage [32, 33]. Furthermore, studies have shown that post-translational modifications of VDACs, including phosphorylation, helix docking, or ubiquitination, affect neuronal function [34-36]. Increasing evidence indicates that the stability, aggregation, and physiological properties of VDACs are highly dependent on the temperature conditions [37, 38]. However, only a few studies have been conducted on the association between hypothermia-induced VDAC ubiquitination and organ protection during I/R. In this study, we demonstrated that the increase in the level of VDAC3, a temperature-sensitive protein, is associated with neurological dysfunction during PR. However, VDAC3 homeostasis induced by therapeutic hypothermia contributed to the attenuation of mitochondria-mediated apoptosis and neuroprotection in the CA/CPR rat model.
Ankyrin repeat (AR) domains can identify and interact with a variety of intracellular substrates, which serve a critical role in biological processes, including the ubiquitination signaling pathway [39]. HECTD1 is a key AR-containing E3 ubiquitin ligase, and it regulates neuroinflammation and neuron cell apoptosis by ubiquitinating its target proteins [14, 40]. Furthermore, the downregulation of HECTD1 expression is associated with stress-induced apoptosis and endothelial injury during I/R [16, 18, 41]. Several studies found that circRNA Hectd1, derived from the Hectd1 gene, is a reliable biomarker for acute ischemic stroke [42, 43]; However, the downstream pathways of circRNA Hectd1-mediated regulation of neurological deficit development have not been fully explored. Further studies should explore the protein-protein interaction networks associated with HECTD1 to determine its target proteins for ubiquitination. In this study, we found that hypothermia enhanced HECTD1 expression and promoted its interaction with VDAC3 during PR. Therefore, our results revealed that VDAC3 could serve as a substrate for HECTD1-mediated ubiquitination, contributing to mitochondrial apoptosis in the CA/CPR rat model.
We observed that the T37 + si-Hectd1 subgroup showed higher VDAC3 expression than the T33 + si-Hectd1 subgroup. This explains the complex regulation of VDAC3 expression, including HECTD1-mediated VDAC3 degradation under hypothermic conditions. Additionally, this result indicates the efficiency of siRNA delivery by AAV. Hectd1 knockout using transgenic techniques, such as clustered regularly interspaced short palindromic repeats/Cas9, may enable more efficient gene targeting to produce desired phenotypes in rats [44].
In this study, there was no statistical difference in NDS between the subgroups of the T37 group within 48 h. This finding may be attributed to severe myocardial dysfunction induced by normothermia during the early post-resuscitation period. As post-resuscitation myocardial function subsequently improved, the elevated expression of HECTD1 demonstrated more pronounced neuroprotective effects by 72 hours post-resuscitation. Accordingly, the 72-hour timepoint after ROSC was selected for the current investigation, as it aligns with critical outcomes including neurological function, survival, and definitive histological evidence of cellular injury. Future studies examining the temporal dynamics of HECTD1 and VDAC3 expression may further elucidate the neuroprotective mechanisms underlying therapeutic hypothermia.
This study has some limitations. First, we used polyclonal antibodies against VDAC3 for the CO-IP experiment, which could not effectively prevent persistent nonspecific interactions between the antibody complex and HECTD1. This limitation could affect the comprehensiveness of our mechanistic interpretation, though our controlled experimental conditions maintain internal validity. Second, the cellular origin of the observed HECTD1 and VDAC3 expression changes remains partially unresolved. Without co-localization studies using specific markers for neurons and microglia, neuroprotective process cannot be exclusively attributed to a particular cell type. This represents an important avenue for future investigation. Finally, while the animal protocol used in this study was well controlled, the application in clinical practice might be limited due to the unavailability of target protein detection methods. The technical limitation, combined with the current lack of validated clinical-grade assays for real-time protein quantification, necessitate cautious interpretation of these preclinical results until further methodological validation studies are conducted. Nevertheless, we observed the protein expression patterns following the CA/CPR and targeted temperature management protocols, which could provide insights into the mechanisms underlying the neuroprotective benefits of therapeutic hypothermia.
Conclusion
Our study demonstrated that therapeutic hypothermia improved neurological dysfunction and survival outcomes in a rat model of asphyxial CA and resuscitation. This may be attributed to increased Hectd1-induced VDAC3 degradation, which results in the attenuation of mitochondria-mediated apoptosis of neurons.
Abbreviations
CA: cardiac arrest; ROSC: return of spontaneous circulation; I/R: ischemia/reperfusion; T37: normothermic; T33: hypothermic; CPR: cardiopulmonary resuscitation; si-Hectd1: siRNA against rat Hectd1; HIBI: hypoxic-ischemic brain injury; VDAC: voltage-dependent anion channel; OMM: outer mitochondrial membrane; HECT: homologous to the E6-AP C-terminal; circRNA: circular RNA; AAV: adeno-associated viral vector; siRNA: small interfering RNA; PBS: phosphate-buffered saline; NC: negative control; qPCR: quantitative polymerase chain reaction; PC: precordial compression; NDS: neurological deficit score; PR: post resuscitation; TEM: transmission electron microscopy; HE: hematoxylin and eosin; IF: immunofluorescence; WB: western blot; IP: immunoprecipitation; ELISA: enzyme-linked immunosorbent assay; NSE: neuron-specific enolase; HE: hematoxylin-eosin; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TBST: Tris-buffered saline containing Tween 20; PCC: Pearson colocalization coefficients; IB: immunoblotting.
Supplementary Material
Supplementary figures.
Acknowledgements
Funding
This project was funded by National Natural Science Foundation of China (82272242); Medical innovation project of Fujian Province, China (2020CXA003); Program for Young and Middle-aged Health Professionals of Fujian Province, China (2016-ZQN-1).
Author contributions
Conceptualization: Shen Zhao; Data curation: Pei Liu, Jiali Wu and Shen Zhao; Formal analysis: Pei Liu, Jiali Wu; Funding acquisition: Shen Zhao; Investigation: Pei Liu, Jiali Wu, Jiawei He, Zhanghuan He and Lei Wang; Methodology: Pei Liu, Jiali Wu and Shen Zhao; Project administration: Shen Zhao; Supervision: Shen Zhao; Writing-original draft: Pei Liu, Jiali Wu and Shen Zhao; Writing-review and editing: Pei Liu, Jiali Wu, Lirong Wang and Shen Zhao.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sandroni C, D'Arrigo S, Cacciola S. et al. Prediction of poor neurological outcome in comatose survivors of cardiac arrest: a systematic review. Intensive Care Med. 2020Oct;46(10):1803-1851
2. Sandroni C, D'Arrigo S, Nolan JP. Prognostication after cardiac arrest. Crit Care. 2018Jun5;22(1):150
3. Elmer J, Callaway CW. Temperature control after cardiac arrest. Resuscitation. 2023Aug;189:109882
4. Howes D, Gray SH, Brooks SC. et al. Canadian Guidelines for the use of targeted temperature management (therapeutic hypothermia) after cardiac arrest: A joint statement from The Canadian Critical Care Society (CCCS), Canadian Neurocritical Care Society (CNCCS), and the Canadian Critical Care Trials Group (CCCTG). Resuscitation. 2016Jan;98:48-63
5. Callaway CW, Donnino MW, Fink EL. et al. Part 8: Post-Cardiac Arrest Care: 2015 American Heart Association Guidelines Update for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2015 Nov 3;132(18 Suppl 2): S465-82. Erratum in: Circulation. 2017Sep5;136(10):e197
6. Nolan JP, Soar J, Cariou A. et al. European Resuscitation Council and European Society of Intensive Care Medicine Guidelines for Post-resuscitation Care 2015: Section 5 of the European Resuscitation Council Guidelines for Resuscitation 2015. Resuscitation. 2015Oct;95:202-22
7. Nielsen N, Wetterslev J, Cronberg T. et al. Targeted temperature management at 33°C versus 36°C after cardiac arrest. N Engl J Med. 2013Dec5;369(23):2197-206
8. Dankiewicz J, Cronberg T, Lilja G. et al. Hypothermia versus Normothermia after Out-of-Hospital Cardiac Arrest. N Engl J Med. 2021Jun17;384(24):2283-2294
9. Robertson CL, Scafidi S, McKenna MC. et al. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol. 2009Aug;218(2):371-80
10. Bakthavachalam P, Shanmugam PST. Mitochondrial dysfunction - Silent killer in cerebral ischemia. J Neurol Sci. 2017Apr15;375:417-423
11. Park E, Lee GJ, Choi S. et al. The role of glutamate release on voltage-dependent anion channels (VDAC)-mediated apoptosis in an eleven vessel occlusion model in rats. PLoS One. 2010Dec21;5(12):e15192
12. Su L, Zhang J, Gomez H. et al. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy. 2023Feb;19(2):401-414
13. Zhao S, Xiao P, Cui H. et al. Hypothermia-Induced Ubiquitination of Voltage-Dependent Anion Channel 3 Protects BV2 Microglia Cells From Cytotoxicity Following Oxygen-Glucose Deprivation/Recovery. Front Mol Neurosci. 2020Jun5;13:100
14. Chen Y, Cao P. Circ-Bnc2 alleviates neuroinflammation in LPS-stimulated microglial cells to inhibit neuron cell apoptosis through regulating miR-497a-5p/HECTD1 axis. Brain Behav. 2023May;13(5):e2935
15. Chu H, Wang W, Luo W. et al. CircHECTD1 mediates pulmonary fibroblast activation via HECTD1. Ther Adv Chronic Dis. 2019Nov27;10:2040622319891558
16. Zhuang JH, Chen HX, Gao N. et al. CircUCK2 regulates HECTD1-mediated endothelial-mesenchymal transition inhibition by interacting with FUS and protects the blood-brain barrier in ischemic stroke. Kaohsiung J Med Sci. 2023Jan;39(1):40-51
17. Rui L, Weiyi L, Yu M. et al. The serine/threonine protein kinase of Streptococcus suis serotype 2 affects the ability of the pathogen to penetrate the blood-brain barrier. Cell Microbiol. 2018Oct;20(10):e12862
18. Chen L, Luo W, Zhang W. et al. circDLPAG4/HECTD1 mediates ischaemia/reperfusion injury in endothelial cells via ER stress. RNA Biol. 2020Feb;17(2):240-253
19. Zhang X, Wan M, Min X. et al. Circular RNA as biomarkers for acute ischemic stroke: A systematic review and meta-analysis. CNS Neurosci Ther. 2023Aug;29(8):2086-2100
20. National Society for Medical Research. Guide for the Care and Use of Laboratory Animals. Washington DC: National Academic Press. 1996
21. Institute of Laboratory Animal Resources. Guide for the Care and Use of Laboratory Animals. Atlanta, GA: National Institutes of Health. 1985 NIH publication No. 86-32
22. Lin QM, Zhao S, Zhou LL. et al. Mesenchymal stem cells transplantation suppresses inflammatory responses in global cerebral ischemia: contribution of TNF-α-induced protein 6. Acta Pharmacol Sin. 2013Jun;34(6):784-92
23. Yang S, Yu C, Yang Z. et al. DL-3-n-butylphthalide-induced neuroprotection in rat models of asphyxia-induced cardiac arrest followed by cardiopulmonary resuscitation. J Cell Physiol. 2021Nov;236(11):7464-7472
24. Hendrickx HH, Rao GR, Safar P. et al. Asphyxia, cardiac arrest and resuscitation in rats. I. Short term recovery. Resuscitation. 1984Jul;12(2):97-116
25. Shi L, Yang AC, Meng DW. et al. Pathological alterations and stress responses near DBS electrodes after MRI scans at 7.0T, 3.0T and 1.5T: an in vivo comparative study. PLoS One. 2014Jul2;9(7):e101624
26. Sheng YY, Xiang J, Lu JL. et al. Protective effects of gallocatechin gallate against ultraviolet B induced skin damages in hairless mice. Sci Rep. 2022Jan25;12(1):1310
27. Caterino M, Ruoppolo M, Mandola A. et al. Protein-protein interaction networks as a new perspective to evaluate distinct functional roles of voltage-dependent anion channel isoforms. Mol Biosyst. 2017Nov21;13(12):2466-2476
28. Lemasters JJ. Modulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolism. J Gastroenterol Hepatol. 2007Jun;22(Suppl 1):S31-7
29. Fu X, Qu L, Xu H. et al. Ndfip1 protected dopaminergic neurons via regulating mitochondrial function and ferroptosis in Parkinson's disease. Exp Neurol. 2024May;375:114724
30. Koushi M, Aoyama Y, Kamei Y. et al. Bisindolylpyrrole triggers transient mitochondrial permeability transitions to cause apoptosis in a VDAC1/2 and cyclophilin D-dependent manner via the ANT-associated pore. Sci Rep. 2020Oct12;10(1):16751
31. Zheng Y, Shi Y, Tian C. et al. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene. 2004Feb12;23(6):1239-47
32. Akanda N, Tofighi R, Brask J. et al. Voltage-dependent anion channels (VDAC) in the plasma membrane play a critical role in apoptosis in differentiated hippocampal neurons but not in neural stem cells. Cell Cycle. 2008Oct;7(20):3225-34
33. Tian T, Pang H, Li X. et al. The role of DRP1 mediated mitophagy in HT22 cells apoptosis induced by silica nanoparticles. Ecotoxicol Environ Saf. 2024Mar1;272:116050
34. Gupta R, Ghosh S. Phosphorylation of voltage-dependent anion channel by c-Jun N-terminal Kinase-3 leads to closure of the channel. Biochem Biophys Res Commun. 2015Mar27;459(1):100-6
35. Liu Y, Zhang H, Liu Y. et al. Hypoxia-induced GPCPD1 depalmitoylation triggers mitophagy via regulating PRKN-mediated ubiquitination of VDAC1. Autophagy. 2023Sep;19(9):2443-2463
36. Gupta A, Mahalakshmi R. Helix-strand interaction regulates stability and aggregation of the human mitochondrial membrane protein channel VDAC3. J Gen Physiol. 2019Apr1;151(4):489-504
37. Chowdhury S, Jarecki BW, Chanda B. A molecular framework for temperature-dependent gating of ion channels. Cell. 2014Aug28;158(5):1148-1158
38. Queralt-Martín M, Hoogerheide DP, Noskov SY. et al. VDAC Gating Thermodynamics, but Not Gating Kinetics, Are Virtually Temperature Independent. Biophys J. 2020Dec15;119(12):2584-2592
39. Kane EI, Spratt DE. Structural Insights into Ankyrin Repeat-Containing Proteins and Their Influence in Ubiquitylation. Int J Mol Sci. 2021Jan9;22(2):609
40. Zhang Y, Du L, Bai Y. et al. CircDYM ameliorates depressive-like behavior by targeting miR-9 to regulate microglial activation via HSP90 ubiquitination. Mol Psychiatry. 2020Jun;25(6):1175-1190
41. Sun X, Dai M, Liu X. et al. Hsa_circ_0090002 regulates miR-186-5p/HECTD1 axis to mediate brain microvascular endothelial cell dysfunction. Brain Res Bull. 2022Jan;178:97-107
42. Peng X, Jing P, Chen J. et al. The role of circular RNA HECTD1 expression in disease risk, disease severity, inflammation, and recurrence of acute ischemic stroke. J Clin Lab Anal. 2019Sep;33(7):e22954
43. Han B, Zhang Y, Zhang Y. et al. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142-TIPARP: implications for cerebral ischemic stroke. Autophagy. 2018;14(7):1164-1184
44. Filipiak WE, Hughes ED, Gavrilina GB. et al. Next Generation Transgenic Rat Model Production. Methods Mol Biol. 2019;2018:97-114
Author contact
![]() Corresponding author: Shen Zhao, MD, PhD, Department of Critical Care Medicine, Beijing Friendship Hospital, Capital Medical University. No. 95 Yong'an Road, Xicheng District, Beijing 100050, China. Tel: 010-63138814; Mail: jzkzhaoshencom.
Corresponding author: Shen Zhao, MD, PhD, Department of Critical Care Medicine, Beijing Friendship Hospital, Capital Medical University. No. 95 Yong'an Road, Xicheng District, Beijing 100050, China. Tel: 010-63138814; Mail: jzkzhaoshencom.