Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
UIP Pattern: Overlap and...
Concluding remarks and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2026; 23(6):1908-1920. doi:10.7150/ijms.129394 This issue Cite
Review
Biomarkers on Fibrotic Lung Diseases Associated with Usual Interstitial Pneumonia
Yuhang Cheng1#, Qing Chang2#, Feng Li2 ![]()
1. Department of Basic Medical Sciences, Shanghai Jiao Tong University School of Medicine, No1, Banxia Road, Pudong new area, Shanghai, 201318, P.R. China.
2. Department of Pulmonary and Critical Care Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University School of Medicine, No.241, West Huaihai Road, Xuhui, Shanghai, 200030, P.R. China.
# Both authors contributed equally to the manuscript.
Received 2025-12-3; Accepted 2026-4-1; Published 2026-4-16
Abstract

Usual interstitial pneumonia (UIP) is a common pattern in fibrotic lung diseases characterized by distinctive radiological and histopathological features. UIP is associated with various underlying causes including idiopathic UIP, i.e. idiopathic pulmonary fibrosis (IPF), and secondary UIP, e.g. connective tissue disease-associated interstitial lung diseases (CTD-ILDs) and fibrotic hypersensitivity pneumonia (fHP). Previous work suggests radiological UIP patterns have strong correlations with overall poor prognosis. This review synthesizes current knowledge on the diverse entities of fibrotic lung diseases presenting with the UIP pattern. This review also highlights previous studies in biomarker searching of IPF and secondary UIP. A number of biomarkers associated with disease diagnosis are summarized, providing new insights for clinicians in disease differentiation. Finally, this review emphasizes the future directions of differential diagnosis through radiology- and blood- biomarkers with the integration of artificial intelligence (AI).
Keywords: usual interstitial pneumonia, diagnosis, biomarkers
Introduction
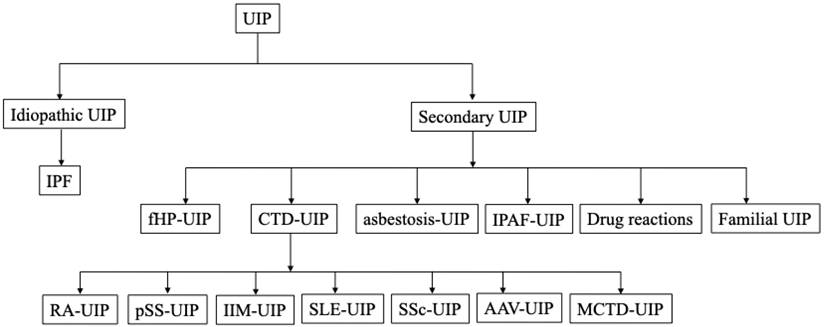
The usual interstitial pneumonia (UIP) pattern is one of the most common radiological and histopathological features observed in fibrotic lung diseases, characterized by heterogeneity in the lung parenchyma, subpleural predominant reticulation and honeycombing [1]. For decades, the UIP has been considered the pathognomonic hallmark of idiopathic pulmonary fibrosis (IPF), while other fibrotic lung diseases, including connective tissue disease-associated interstitial lung diseases (CTD-ILDs), interstitial pneumonia with autoimmune features (IPAF), fibrotic hypersensitivity pneumonitis (fHP), asbestosis, drug reactions, and familial pulmonary fibrosis, can also present UIP patterns (Figure 1, Figure 2). Discriminating idiopathic UIP (IPF) with secondary UIP is clinically significant for its potential therapeutic and prognostic implications. Management for fibrotic lung diseases differs significantly by etiology. While corticosteroids, immunosuppressants, and certain biological agents (e.g., rituximab) have shown efficacy in improving lung functions and slowing disease progression in patients with CTD-ILDs and fHP [2], antifibrotic treatment strategy remains the established standard of care for IPF patients for slowing the decline of FVC. The prognosis of fibrotic lung diseases also differs by etiology. Previous studies have demonstrated that with biopsy- or radiology- proven UIP, patients with IPF had worse survival (median survival 3.5-4.7 years) compared with fHP group (median survival 4.8 years), CTD-ILDs group (median survival 5.5-7.1 years) [3-6]. Therefore, the differential diagnosis for the UIP pattern becomes particularly important in clinical care.
Underlying diseases associated with a radiologic UIP pattern. The tree diagram shows the spectrum of clinical entities that may manifest as a UIP pattern. Abbreviations: UIP, Usual interstitial pneumonia; IPF, Idiopathic pulmonary fibrosis; fHP, fibrotic hypersensitivity pneumonitis; CTD-UIP, Connective tissue disease-related UIP; IPAF-UIP, Interstitial pneumonia with autoimmune features related UIP; RA, Rheumatoid arthritis; pSS, primary sjögren's syndrome; IIM, Idiopathic inflammatory myopathy; SLE, Systemic lupus erythematosus; SSc, Systemic sclerosis; AAV, Anti-neutrophil cytoplasmic antibody associated vasculitis; MCTD, Mixed connective tissue disease; UCTD, Undifferentiated connective tissue disease
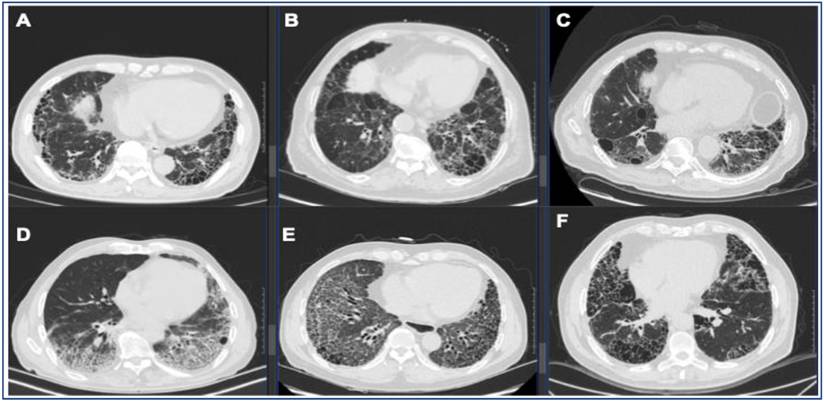
Representative UIP pattern on HRCT in IPF and secondary UIP. (A) IPF, (B) fHP, (C) RA-ILD, (D) pSS-ILD, (E) SSc-ILD, (F) AAV-ILD. Radiologic UIP pattern is defined as basilar, subpleural distribution of reticulation and traction bronchiectasis with honeycombing and without features incompatible with UIP.
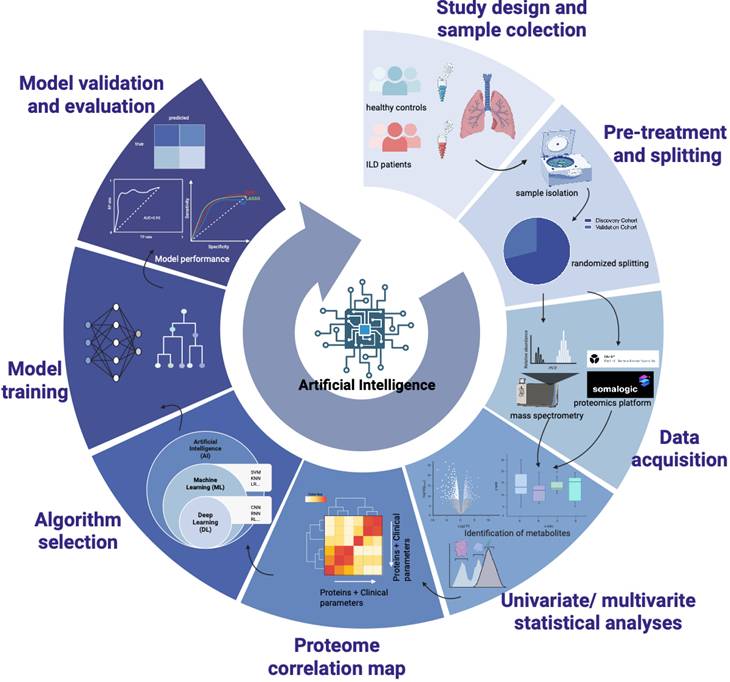
AI-driven workflow for proteomic biomarker discovery and model validation in ILDs. This integrated pipeline encompasses seven key stages of development. First, study design and sample collection involve cohorts of healthy controls and ILDs patients. Second, pre-treatment and splitting are performed to divide samples into discovery and validation cohorts. Third, data acquisition utilizes high-throughput technologies such as mass spectrometry and the Somalogic proteomics platform. Fourth, univariate and multivariate statistical analyses are applied for metabolite identification and data processing. Fifth, a proteome correlation map is generated to integrate protein data with clinical parameters. Sixth, algorithm selection involves choosing between machine learning and deep learning. Finally, model training is followed by rigorous model validation and evaluation using performance metrics such as AUC and sensitivity-specificity curves. Created in BioRender.com, with permission.
The underlying causes of the UIP pattern primarily encompass five clinical entities. IPF accounts for the largest proportion (54.15%-85.15%) [3-5, 7-9], followed by CTD-ILDs (10.46%-38.75%) [3, 5, 7-9], fHP (2.37%-6.60%) [5, 7, 8], asbestosis (about 4.74%) [5], and other (about 6.67%) [7], of which CTD can be subdivided into rheumatoid arthritis (RA) (22.71%-41.27%) [3-5, 9], primary sjögren's syndrome (pSS) (11.11%-31.00%) [4, 5, 9], anti-neutrophil cytoplasmic antibody associated vasculitis (AAV) (14.29%-25.33%) [4, 5, 9], systemic lupus erythematosus (SLE) (19.05%-19.64%) [4], and other CTD (14.28%-57.14%) including mixed CTD(mCTD), unclassifiable CTD (uCTD), and polymyalgia rheumatica etc [3-5, 9]. Such broad ranges in proportional of underlying causes reflect significant heterogeneity across studies, primarily driven by population variance (e.g., geographic and environmental exposure differences), evolving diagnostic criteria and inherent referral bias or selection bias among different study cohorts (e.g., specialized interstitial lung disease centers versus general clinics).
Although distinctive CT signs have been proposed to differentiate UIP pattern of IPF from CTD-ILDs, i.e. anterior upper lobe sign, exuberant honeycombing, and straight edge sign [10], precisely diagnosing the underlying causes of UIP still remains challenging, especially at primary hospitals, albeit with growing understanding of IPF and secondary UIP. The effective diagnostic standard for IPF and secondary UIP is lacking, while current guidelines suggest multidisciplinary discussion (MDD) as a surrogate standard and ultimate choice [1], which can be unavailable at inexperienced centers.
Biomarker is measured as an indicator of normal biological processes, pathogenic processes or responses to an exposure or intervention. Biomarkers may exhibit promising applications in disease differentiation, monitoring and prognosis. This review focuses on the radiological UIP pattern and we aim to provide an evidence-informed overview of the risk factors and diagnostic biomarkers for UIP subtypes. Finally, we tend to discuss future research directions in artificial intelligence (AI) integrating with multimodal learning to realize precise diagnosis.
UIP Pattern: Overlap and Distinction
IPF
IPF is one of the most common fibrosing ILDs characterized by fatal lung function loss and progressive tissue remodeling. Radiological features of UIP or probable UIP, as defined by the guideline, is the hallmark of IPF [11]. Diagnosis of IPF requires the exclusion of all other risk factors or exposures that are known to cause secondary UIP [1]. The prevalence of IPF ranges from 14-27.9 cases to 42.7-63 cases per 100,000 people, depending on statistical methods and selection criteria [12]. Risk factors include age, male sex, smoking, wood/ metal dust, sand/ silica, agricultural exposures and gastroesophageal reflux [13-15]. Genomic risk factors include MUC5B promoter polymorphism rs35705950 [16], TOLLIP [17], short telomere length [18], TLR3, IL1RN, IL-8, IL-4, TGFB1 [19], DSP rs2076295 [16] and AKAP13 [16]. FUT3, ADAM15 and USP28 are found to be possible genetic-informed proteomic risk factors [20]. Recent evidence suggests that specific autoantibodies offer a distinct perspective for understanding IPF through immune mechanisms. Anti-periplakin autoantibodies target epithelial desmosomes and impair wound repair, while anti-HSPA2 antibodies may play a protective role [19, 21, 22]. Diagnostic delay is widely observed in IPF, with median time being 2.1 years [23].
fHP
HP is an immune-mediated, inflammatory parenchymal lung disease after inhalational exposure to an inciting antigen or mixture of antigens [24-26]. It is estimated that the incidence of HP is 0.3-0.9 cases per 100,000 population, and the annual prevalence of HP is 1.67-2.71 cases per 100,000 individuals in the US [12]. Current guideline propose a phenotype-based approach in definition, i.e. non-fHP and fHP [25]. Definite or probable UIP pattern are observed in fHP patients, taking up 17% of fHP patients, and UIP pattern, whether histologically or radiologically proven, are often linked to worse survival similar to IPF [27, 28]. MUC5B promoter polymorphism rs35705950, as a genomic risk factor in IPF patients, is also associated with HP incidence, UIP histopathology, and radiological fibrosis, explaining why fHP and IPF share certain common risk factors and clinical manifestations [27].
RA-ILD
RA affects nearly 1% of the population [29], whereas RA-ILD is highly prevalent in RA cohorts (up to 60% [30]). UIP or probable UIP pattern on CT are correlated with unfavorable outcomes similar to IPF, compared with other RA-ILD patients with alternative CT patterns [31-33] .Anti-cyclic citrullinated peptide (anti-CCP) positivity and titre are found to be strongly associated with RA-ILD susceptibility [34], while male sex, age at onset, cigarette smoking and rheumatoid factor titre are all risk factors independently associated with RA-ILD [35, 36]. Higher levels of c-reactive protein, erythrocyte sedimentation rate (ESR), and tumor markers especially CA125 levels are also risk factors [37, 38]. A recent study indicates that MUC5B promoter variant rs35705950 is associated with increased risk (at least 3-fold increased) of pulmonary involvement [39]. A risk score system was developed and upgraded with MUC5B rs35705950 to improve model precision for subclinical RA-ILD [40]. Genetic susceptibility to RA-ILD is significantly linked to variants in TOLLIP and HLA-DRB1 [19]. Several genomic characteristics observed in RA-ILD patients such as familial pulmonary fibrosis (FPF) linked genes (TERT, RTEL1, PARN or SFTPC) mutations and short telomeres that were previously linked with FPF susceptibility [41].
SSc-ILD
SSc is characterized by vasculopathy and fibrosis in skin and multi-internal organs with a prevalence of 1 in 100,000 individuals, whereas ILD is observed in 25%-30% of SSc patients and acts as a major cause of mortality [42, 43]. Older age, male sex, diffuse cutaneous SSc and cardio involvement are well-established risk factors for SSc-ILD [43, 44], while there is an uncertain association between cigarette smoking and susceptibility of SSc-ILD. UIP pattern accounts for less than 10% of patients in a retrospective cohort study but is linked with relatively faster lung function deterioration [45]. Specific genetic variants, including STAT4 and IRF5, have been identified as key risk factors predisposing patients to SSc-ILD [19]. MUC5B promoter polymorphism rs35705950 was not associated with SSc-ILD, suggesting its different pathophysiology with IPF [46].
pSS-ILD
pSS is a common systemic autoimmune disease with a prevalence of 0.3-1 per 1000 individuals and a female predominance [47]. Pulmonary involvement is a frequent manifestation of pSS and ILD is observed in 17% of pSS patients in a recent meta-analysis [48]. Male sex remains one of the important risk factors for pSS-ILD [49, 50]. Other identified risk factors include older age at onset and longer disease duration [48]. Anti-SSA/Ro and anti-SSB/La antibodies are critical for diagnosing pSS-ILD for their specificity in pSS patients [51]. However, current understanding on pSS-ILD is still limited. A particular diagnostic challenge arises when ILD precedes the formal diagnosis of pSS in individuals with subclinical disease, especially for those with a definite or probable UIP pattern on HRCT [52]. This condition is challenging for clinicians and often results in misdiagnosis as unclassifiable ILD or IPF, with the correct diagnosis rate being only 4% [53]. From a clinical standpoint, female sex, younger age, systemic manifestations and serological abnormalities are important discriminators between pSS-ILD and IPF.
Microscopic polyangiitis (MPA)-ILD
MPA, a major subtype of AAV, is a pauci-immune necrotizing vasculitis with a prevalence of 3-10 cases per 1,000,000 individuals in the USA and Europe [54]. ILD is observed in 2.7% to 47.4% of patients with MPA and often leads to worse survival [55]. Age, male sex and smoking history are risk factors for MPA-ILD, and interestingly, TERT rs2736100A and DSP rs2076295G, both genomic risk factors for IPF, are associated with MPA-ILD, indicating that IPF and MAP may share some common pathogenic mechanisms [56, 57]. UIP is the most frequent radiologic pattern in MAP-ILD patients, and is observed in more than 50% of MPA-ILD patients [55, 56, 58, 59]. Serological test for MPO-ANCA is critical for diagnosis, which is found in over 80% MPA-ILD patients and shows high specificity in differential diagnosis [60, 61]. However, pulmonary manifestations can precede the onset of systemic vasculitis symptoms, resulting in an initial misdiagnosis as IPF and delaying the treatment for MPA [56]. The clinical features of idiopathic UIP and common secondary UIP diseases are summarized in Table 1.
Clinic features of Idiopathic UIP (IPF) and Secondary UIP.
| Idiopathic UIP | Secondary UIP | |||||
|---|---|---|---|---|---|---|
| IPF | fHP | CTD-UIP | ||||
| RA-ILD | SSc-ILD | pSS-ILD | MPA-ILD | |||
| Age | Most patients diagnosed with IPF are over 60 years old [84] | The average age of onset is 52 years, and higher risk for individuals over 65 [85] | Most diagnosed at sixth decade of life, and increased mortality risk after 64.7 | Median age of 60.7 years old [86] | Peak incidence at 55-61 [47, 53] | Most diagnosed over 65 years [58] |
| Sex | Males represent ∼70% of all patients with IPF [6, 87] | Males represent fewer than half of the cases [85] | Males are affected twice as frequently as females [88] | Female predominance in absolute numbers (over 75% female patients) [86] | Female predominance in absolute numbers (about 90% female patients) [47] | Higher frequency of men [56] |
| Cigarette smoking | Ever-smokers account for more than 50% of IPF patients [14, 89] | Less frequent in ever-smokers than non-smokers, and smoking is associated with lower survival rates. [90] | Smoking over 25 pack years have 2.4 times greater chances of demonstrating lung erosions [34] | Unknown association | Ever-smokers account for about 20% of pSS-ILD patients [53] | Higher frequency of smokers, and smoking is associated with higher mortality [56] |
| UIP pattern | Hallmark of diagnosis [1] | Observed in 17% of fHP patients [27] | Over 50% of RA-ILD patients [91, 92] | Less than 10% of patients [45] | About 10% of patients [53] | More than 50% of patients [55, 56, 58, 59] |
| Gastro-esophageal reflux | Associated risk factor [13] | Unknown association | Unknown association | Present in 80% SSc-ILD patients [93] | Present in 50% pSS-ILD patients [53] | Unknown association |
| Exposures | Associated with environmental factors such as agriculture, farming, livestock, wood dust, and metal dust [94] | Antigen exposures are essential components in diagnosis, including microbes, proteins, enzymes, chemicals and medication [25] | Associated with inhalational exposures, include silica, stone dust, rock drilling or stone crushing [95] | Unknown association | Unknown association | Unknown association |
| Obstructive sleep apnea | Highly prevalent in IPF [96, 97] | Unknown association | Unknown association | Highly prevalent [98] | OSA observed in about 30% of pSS patients [99] | Unknown association |
| Acute exacerbation (AE) | AE occurs, with 1-year, 2-year and 3- year incidence of 14.2%, 18.8% and 20.7% respectively [100] | AE occurs, with 1-year and 3-year incidence of 6.0%, 13.6% and 22.8% respectively [101] | AE occurs, with 1-year incidence of 2.8% [102] | AE occurs, but the incidence not fully studied | AE occurs, but the incidence not fully studied [103] | AE occurs, with 1-year incidence of 7.5% [56] |
| Prognosis | Median survival 3.5-4.7 years [4, 6] | Median survival 2.76 years [28] | Median survival 5.5 years [3] | Median survival 13.5 years [104] | 5-year survival rate 84% [53] | 5-year survival rate 29%-60% [58] |
| Immune suppression based treatment strategy | Proven to associated with shorter survival and high mortality [2] | Immunosuppressants is associated with increased mortality [105]. Corticosteroid has no influence on long-term results [106] | Leflunomide is associated with better prognosis [32] | Mycophenolate mofetil, cyclophosphamide, and tocilizumab [107] | Azathioprine and prednisone [108] | Glucocorticoid and cyclophosphamide [109] |
| Anti-fibrotic treatment strategy | Slow lung function decline [110, 111] | Evaluation of efficiency and safety is still in need. | Slow lung function decline [112] | Slow lung function decline [113, 114] | May be effective, but not proven [115] | May be effective, but not proven [56] |
Diagnostic biomarkers
IPF
Genetic variants influence IPF susceptibility, progression and prognosis. The MUC5B polymorphism rs35705950 is known to be the strongest genetic risk factor with a 14-fold increased risk for IPF. However, its prognostic value remains a subject of debate, with conflicting evidence observed across different studies. Previous studies have reported that carriers of the MUC5B risk allele exhibit slower disease progression and improved survival compared to non-carriers [27, 62]. Conversely, recent studies demonstrate conflicting results, indicating that this genetic variant may not always consistently correlate with better survival outcomes [63-65]. These disparate findings suggest that the prognostic value of MUC5B remains a matter of controversy influenced by many reasons. In contrast, TOLLIP variant (rs5743890 minor allele) accelerates the progression of lung fibrosis and leads to worse survival [17]. Telomere length is associated with fibrosis extent, and shortened telomeres (lengths less than the 10th percentile for age) result in cellular senescence, rapid disease progression and reduced survival [27].
Serological biomarkers for IPF diagnosis can be categorized into a few groups based on their association with core pathological mechanisms: alveolar epithelial injury (e.g., SP-D, CA19-9, CA-125), extracellular matrix remodeling (e.g., MMP-1, MMP-7, Osteopontin), and immune/chemokine signaling and fibrotic mediators (e.g., CCL17, BPI, LTBP2). What's more, protein signatures (e.g., PC37, a 5-protein panel) and microRNA profiles demonstrate diagnostic utility in discriminating IPF from healthy controls and other ILDs. Biomarkers associated with IPF are summarized in Table 2.
Biomarkers for diagnosis of IPF.
| Biomarkers | Clinical utility /Results | Reference |
|---|---|---|
| SP-D | SP-D > 31ng/mL differentiate IPF from other-ILD and healthy controls | [116] |
| CA19-9, CA-125 | CA19-9 and CA-125 discriminate IPF and other ILDs | [116] |
| MMP-1, MMP-7 | MMP-7 > 1.75ng/mL distinguish IPF from other-ILD, and MMP-7 > 12.1ng/mL are predictive mortality and TFS | [117, 118] |
| MMP-1 ≥ 1.99ng/mL and MMP-7 ≥ 2.15ng/mL distinguish IPF from other ILDs | ||
| Osteopontin | Osteopontin > 6ng/mL distinguish IPF and other-ILDs | [117] |
| GT1, vWF, CCL17, BPI | Circulating proteins (GT1, vWF, CCL17 and BPI) differentiate IPF and other healthy controls | [119] |
| LTBP2 | Higher levels in IPF than seen in CTD-ILDs | [117] |
| LTBP2 > 33.75 ng/mL discriminate IPF and CTD-UIP (AUC 0.682) | ||
| LTBP2 > 38.33 ng/mL discriminate IPF and RA-UIP (AUC 0.681) | ||
| cCCK18 | Serum cCK-18 levels are elevated in IPF patients and may be clinically informative as a diagnostic marker of IPF | [120] |
| LDHA, CCT6A | LDHA and CCT6A differentiate IPF from healthy controls | [15] |
| Protein signature | A combinatorial signature of five proteins (MMP7, MMP1, MMP8, IGFBP1, and TNFRSF1A) discriminates IPF from healthy controls with a sensitivity of 98.6% and specificity of 98.1% | [121] |
| PC37 | A classifier with 37 proteins consistently distinguishes CTD-ILDs from IPF | [78] |
| eNose | eNose technology can be a novel biomarker for its potential to discriminate IPF and other ILDs | [66] |
| microRNAs | Downregulated micro RNAs (miR-29, let-7d) and upregulated micro RNAs (miR-21, miR-154) are shown to have diagnostic capacity in IPF | [122] |
Abbreviations: LTBP2, Latent transforming growth factor-beta binding protein-2; SP-D, Surfactant protein D; MMP-7, Matrix metalloproteinase-7; PC37, Proteomic classifier 37; TFS, Transplant free survival; LDHA, Lactate dehydrogenase A; CCT6A, Chaperonin containing TCP1 subunit 6A; GT1, Glycoproteins thrombospondin 1; vWF, von Willebrand factor; CCL17/18, C-C motif chemokine ligand 17/18; BPI, Bactericidal permeability-increasing protein; IGFBP1, insulin-like growth factor binding protein1; TNFRSF1A, tumor necrosis factor receptor superfamily member, 1A.
Novel biomarkers such as electronic Nose (eNose) and genome UIP (gUIP) have shown adequate specificity and sensitivity in differentiating ILD subtypes. Exhaled breath analysis-based eNose technology is a promising non-invasive biomarker to discriminate healthy individuals from various ILD subgroups by analyzing different chemical compounds in volatile organic compounds (VOCs) in exhaled breath [66]. Derived from transbronchial biopsies, gUIP has been validated in predicting histopathological UIP and increasing diagnostic confidence for IPF, and positive gUIP classification is associated with reduced transplant free survival (TFS) [67]. Biomarkers for diagnosis of IPF (Table 2).
fHP
A large-scale study of single-cell immune profiles revealed novel immune perturbations in fHP. Both fHP and IPF share enriched S100Aʰⁱ and CCL3ʰⁱ/CCL4ʰⁱ classical monocytes whereas cytotoxic GZMhi lymphocytes are enriched specifically in fHP and may contribute to disease discrimination. The study also found variance in cell compositions in both blood and BAL fluid between fHP and IPF patients [68]. Compared to IPF, fHP shows increased classical monocytes and platelets but decreased memory B cells, Tregs, MAIT cells, and CD56ʰⁱ NK cells in blood and fewer alveolar macrophages but more T cells in BAL fluid [68]. Previous studies have verified SP-D as an alveolar epithelial injury marker for HP diagnosis. New evidence identifies YKL-40 and KL-6 as viable candidates with adequate specificity and sensitivity to discriminate HP from healthy controls and IPF patients. Differences in microbiome patterns in fHP and IPF also contribute to discrimination [69]. Biomarkers associated with fHP are summarized in Table 3.
Biomarkers for diagnosis of fHP.
| Biomarkers | Clinical utility /Results | Reference |
|---|---|---|
| S100hi monocytes, CCL3hi/CCL4hi monocytes | S100hi monocytes and CCL3hi/CCL4hi monocytes are both highly enriched in fHP patients with the potential to discriminate fHP and healthy individuals. | [68] |
| GZMhi lymphocytes | Cytotoxic GZMhi lymphocytes are increased in fHP compared with healthy controls and IPF patients with the potential for discrimination between fHP and others. | [68] |
| SP-D | Serum SP-D level in HP are higher than in IPF, CTD-ILDs and sarcoidosis and SP-D is useful for diagnosis. | [123] |
| YKL-40, KL-6 | Serum and sputum YKL-40 and KL-6 levels in fHP are higher than healthy individuals while lower than IPF, with adequate specificity and sensitivity for diagnosis. | [124] |
| Microbiome | Differences in bacterial community composition and diversity contribute to discrimination of fHP and IPF. | [69] |
Abbreviations: CCL3/4, C-C motif chemokine ligand3/4; GZM, Granzyme; SP-D, Surfactant protein D; YKL-40(CHI3L1), Chitinase 3-like protein 1; KL-6, Krebs von den lungen-6
CTD-ILDs
The biomarker landscape for CTD-ILDs is rapidly evolving, encompassing markers of genetic risk (MUC5B [39]), autoimmunity (ACPA, Citrullinated HSP90), inflammation (chemokines), cellular senescence (telomere length), epithelial injury (KL-6, SP), and matrix remodeling (MMP-7, LOXL2). A recent study suggests Reticulocalbin 3 (Rcn3), an endoplasmic reticulum lumen protein concerning alveolar epithelial injury, holds promise in discriminating CTD-ILDs from IPF, for serum Rcn3 concentration was significantly higher in patients with CTD-ILDs than in those with IPF; yet, further studies are still needed [70].
A few biomarkers contribute to pSS-ILD diagnosis. A machine learning based study identified age, disease duration, and serum levels of KL-6 and TNFα as highly discriminating biomarkers for pSS-ILD and pSS-non-ILD [71]. A recent study investigated compositional and functional similarities and differences of the gut microbiota in pSS and SLE patients, suggesting the potential role of microbiome profiling in CTD-ILDs diagnosis. [72]
Current research on biomarkers for MPA-ILD remains limited. MPO-ANCA, while being a cornerstone serological marker for MPA and demonstrating high specificity in distinguishing MPA-ILD from other ILDs, is shared across AAV (GPA, MPA, EGPA included) and does not specifically reflect pulmonary involvement or disease progression [61, 73]. Significantly elevated serum CCL2 levels in MPA-ILD patients enable discrimination between MPA-ILD patients, MPA-non-ILD patients and healthy individuals [74]. Biomarkers correlating to alveolar epithelial injury, including KL-6 and SP, reflect lung-specific activity and are useful in monitoring pulmonary lesions, but their utility in diagnosing and monitoring MPA-ILD are limited [75]. Biomarkers associated with CTD-ILDs are summarized in Table 4.
Biomarkers for diagnosis of CTD-ILDs.
| Biomarkers | Clinical utility /Results | Reference | |
|---|---|---|---|
| RA-ILD | Uric Acid | Elevated serum and BAL fluid uric acid levels may serve as a diagnostic biomarker for RA-ILD, particularly RA-UIP. | [125] |
| Citrullinated Hsp 90α/β | Citrullinated Hsp 90α/β can differentiate RA-ILD from RA-non-ILD, IPF and MCTD. | [126] | |
| ACPA | ACPA differentiates RA-ILD from RA-non-ILD, and associates with worse survival. | [36] | |
| LOXL2 | Serum LOXL2 levels are higher in RA-ILD patients than healthy individuals, with potential utility in diagnosis. | [127] | |
| MMP-3, MMP-7, MMP-9, MMP-10, TIMP-1 | MMPs and TIMPs found as potential candidates for the discrimination of RA-ILD and IPF. | [128] | |
| pSS-ILD | KL-6, TNF-α | Age, KL-6, and TNFα effectively differentiated pSS-ILD from pSS-non-ILD with high sensitivity and specificity. | [71] |
| CA153 | Serum CA153 levels are higher in pSS-ILD patients than pSS-non-ILD. | [129] | |
| Angptl2 | Serum Angptl2 level are highly elevated in pSS-ILD patients compared with healthy controls and pSS-non-ILD patients. | [130] | |
| CYSLTR1, SIGLEC10 | Differential expression of CYSLTR1 and SIGLEC10 in pSS-ILD and healthy controls revealed their diagnostic potential for pSS-ILD | [131] | |
| MPA-ILD | CCL2 | Initial serum CCL2 levels were significantly higher in MPA-ILD patients than MPA-non-ILD patients. | [132] |
| KL-6 | Higher serum KL-6 were in MPA-ILD compared with those without ILD, and serum KL-6 > 513 U/mL differentiates MPA-ILD with other AAV. | [133] |
Abbreviations: ACPA, Anticitrullinated protein antibody; LOXL2, Lysyl oxidase-like 2; MMPs, Matrix metalloproteinases; TIMPs, Tissue inhibitor of metalloproteinases; KL-6, Krebs von den lungen-6; CCL2, C-C motif chemokine ligand 2; CA153, Cancer antigen 153; TNF-α, Tumor necrosis factor-alpha; Angptl2, Angiopoietin-like protein 2; CYSLTR1, Cysteinyl Leukotriene Receptor 1; SIGLEC10, Sialic acid binding Ig like lectin 10
Artificial intelligence (AI)-driven biomarker discovery for diagnosis
The application of AI and, more specifically, machine learning (ML) has revolutionized the discovery of biomarkers for ILDs. By integrating multi-omics data and dealing with large-scale biomedical data (such as genomic sequencing and proteomics) simultaneously, AI-assisted analytical tools enable a systems-level, data-driven paradigm that integrates multimodal information [76].
ML-based techniques have been applied in plasma and serum proteomics to identify novel circulating biomarkers in ILDs, where they now outperform the classic and established biomarkers. AI-driven ILD biomarkers have been proven efficient in some real-world case studies. Bowman et al. applied LASSO (least absolute shrinkage and selection operator) logistic regression and ten-fold cross-validation to identify a set of biomarkers predictive of progressive fibrosing ILDs. A semi-quantitative proteomic signature comprising twelve biomarkers achieved a sensitivity of 0.90 in discriminating progressive and non-progressive ILD patients [77]. Huang et al. performed SVM, LASSO and RF to develop and validate a proteomics classifier with 37 proteins (PC37) associated with bronchiole development and immune responses, demonstrating 82.9% accuracy in discriminating CTD-ILDs and IPF. By iteratively classifying single samples and applying composite scoring methods across all four machine learning models, they established a single-patient diagnosis model mimicking clinical practice settings [78]. Zhou et al. developed a pulmonary fibrosis model using RF and SVM. Three key genes including TMEM52B, PHACTR1 and BLVRB were selected using RF and SVM found that the model achieved an accuracy of 0.786 when 15 genes were incorporated, providing a new solution for early diagnosis of pulmonary fibrosis [79]. Oldham et al. identified 140 circulating plasma proteins associated with differential transplant-free survival (TFS) in IPF cohorts. LASSO was applied to build a proteomic signature of TFS and the model performance was estimated using decision curve analysis. The model significantly outperformed a clinical prediction model on discriminating TFS, showing its potential in clinical decision-making and disease monitoring [80].
The identification of novel biomarkers with AI assistance involves a multi-step, rigorous process. Well-designed clinical cohorts have to be recruited and participants have to be categorized or phenotyped, discovery cohort and validation cohort have to be randomly assigned after sample preprocessing. Either mass spectrometry (MS) or high-throughput proteomics platforms can be applied in data acquisition. Omic data and/ or clinical data are appropriately collected and stored integrated and pre-processed. The processed data are divided into a training and a testing dataset. Having obtained such extensive data sets, the next step is univariate and/ or multivariate statistical analyses to quantify the proteome differences between cases and controls, often visualized through volcano plots and heatmaps. We hereby select candidate differentially accumulated metabolites (DAMs) and conduct cross validation. Then we train the ML models (LASSO, RF, SVM-RFE, LR, XGBoost, etc) to obtain optimal parameters and tune hyperparameters. The learning approach (supervised, semi-supervised, unsupervised) is steered by the ultimate goals and the availability of collected data. Evaluation of biomarkers found by ML and ML models should be done in the following steps, such as with confusion matrices and ROC curves, which quantify the sensitivity and specificity of candidate biomarkers and facilitate their potential translation into clinical settings. The predictive power and explainability of the biomarker are validated on external datasets to assess its robustness and generalizability on unseen data (usually from another medical center).
To ensure the robustness and reproducibility of the AI-driven workflow, we recommend a few technical details as indicated by Mann et al., which adheres to the best practices for machine learning in proteomics [76]. Good ML models should be configured with explicit hyperparameter settings, such as learning rate or specific coefficients to control model complexity. We suggest a strict splitting of training and evaluation data, where the evaluation data are never employed in training. Usually a training set (~70%), an internal validation set (~15%), and an independent external testing cohort (~15%) is ideal. Of note, the accurate quantification of the biomarkers is even more critical than the identification of as many as possible of them, and when samples are really restricting, overfitting prevention methods such as cross-validation are essential for robust evaluation of model performance. K-fold cross-validation and regularization techniques (i.e., Ridge regression) are also procedures for preventing overfitting and confusion matrices and ROC curves are often applied for biomarker evaluation [76].
The AI models and biomarker discovery also have limitations. First, the explainability of DL and ML models is crucial, as ML and DL models often contain thousands of nodes and often lose interpretability; hence, ML and DL models are often treated as black boxes with no clinical significance. Beyond that, it would also be troublesome when considering discordance between the AI classifier and MDD [78], thus it is important to address these challenges through explainable AI (XAI) and more in-depth model evaluation to determine how well the AI classifier correlates with real-world patient diagnosis.
Concluding remarks and future directions
UIP is a well-established terminology back to 1969. The emphasis and usage of the term UIP has shifted over decades, but with obvious limitations. The designation of UIP remains ambiguous in a series of conditions [81]. Changes in guidelines (from 2011 to 2018 and 2022 updates) aim to reduce heterogeneity but still result in inconsistent classification. The classification of UIP into “typical,” “probable,” “indeterminate,” or “alternative diagnosis” relies heavily on personal experience and expertise. In a study concerning a group of thoracic radiologists, significant interobserver variance and disagreement exist, with only moderate consensus achieved, irrespective of their expertise or experience [82]. Current guidelines clarify UIP as a pathologic fibrotic pattern, not synonymous with IPF, but terms like “UIP pattern” or “IPF” are often used interchangeably, causing confusion and misleading outcomes. Shifting diagnostic focus and developing new classifications are still needed.
This review summarizes underlying causes, risk factors, clinical features and diagnostic biomarkers of idiopathic UIP and secondary UIP subtypes. UIP pattern can be found in a series of conditions, but recently, it has been proposed that UIP be defined as a distinct disease entity, due to similarities in disease behaviors and pathogenic mechanisms [83]. However, we have to recognize that the clinical trials are based on large patient cohorts, not individuals. Therefore, when dealing with patients with histological or radiological UIP pattern, the primary task is to separate idiopathic UIP (IPF) from secondary UIP, mainly fHP and CTD-ILDs.
The biomarkers we described here are obvious candidate biomarkers based on our current understanding of diseases. Biomarkers are less invasive and more objective indicators, crucial for early diagnosis, monitoring treatment response, and prognostication. Exploring biomarkers may just be the key we need in unlocking the complex nature of UIP, allowing for early-stage interventions that could change patients' lives.
Despite the fact that considerable advances have been made and substantial biomarkers have been observed and advanced techniques such as single-cell sequencing and multi-omics data integration offer unprecedented insights, their application into routine clinical practice is currently hindered by infrastructure, technical standardization and reproducibility, population heterogeneity and generalizability, high costs and translational challenges, especially in primary clinical settings. To bridge this gap, a biomarker must be measurable, accurate, reproducible, clinically actionable, and cost-effective. Beyond that, the selection and combination of biomarkers should be guided by a clear framework: prioritizing widely available serum markers for longitudinal monitoring, while reserving high-specificity proteomic or metabonomic signatures for complex differential diagnosis. Balancing feasibility, diagnostic validity, and the specific clinical context will be essential for improving patient quality of life and survival substantially.
These studies on biomarkers also have limitations and deficiencies. The variance on disease definition and clinical diagnosis criteria is the primary reason. What's more, statistical manipulation and overfitting also occur. Inappropriate statistical methods can cause bias and misleading results. Although many studies used a multivariate Cox proportional hazards model to adjust for potential confounders, others relied on univariate analyses. The application of univariate methods is a significant limitation, for their defects in capturing the data's complex, multi-dimensional patterns. Thus, it is essential for biomarker studies to employ multivariate analysis (e.g., Cox proportional hazards models or logistic regression) to adjust for key clinical variables including age, sex, BMI, smoking history, medication use and comorbidities to ensure that the identified biomarkers are independent predictors of disease progression and outcomes rather than reflections of underlying clinical characteristics. Insufficient sample sizes and retrospective bias also contribute to it. Future studies should improve data standardization, reproducibility, cross-platform validation, and prioritize AI integration in multi-omics data to provide new insights in novel biomarkers.
Abbreviations
UIP: Usual interstitial pneumonia; CTDs: Connective tissue diseases; IPF: Idiopathic pulmonary fibrosis; fHP: fibrotic hypersensitivity pneumonitis; CTD-UIP: Connective tissue disease associated UIP; IPAF-UIP: Interstitial pneumonia with autoimmune features associated UIP; RA: Rheumatoid arthritis; pSS: primary Sjogren's syndrome; IIM: Idiopathic inflammatory myopathy; SLE: Systemic lupus erythematosus; SSc: Systemic sclerosis; AAV: Anti-neutrophil cytoplasmic antibody associated vasculitis; MCTD: Mixed connective tissue disease; UCTD: Undifferentiated connective tissue disease; LTBP2: Latent transforming growth factor-beta binding protein-2; SP-D: Surfactant protein D; MMP-7: Matrix metalloproteinase-7; PC37: Proteomic classifier 37; TFS: Transplant free survival; LDHA: Lactate dehydrogenase A; CCT6A: Chaperonin containing TCP1 subunit 6A; GT1: Glycoproteins thrombospondin 1; vWF: von Willebrand factor; CCL17/18: C-C motif chemokine ligand 17/18; BPI: Bactericidal permeability-increasing protein; IGFBP1: insulin-like growth factor binding protein1; TNFRSF1A: tumor necrosis factor receptor superfamily member: 1A; CCL3/4: C-C motif chemokine ligand3/4; GZM: Granzyme; YKL-40(CHI3L1): Chitinase 3-like protein 1; KL-6: Krebs von den lungen-6; ACPA: Anticitrullinated protein antibody; LOXL2: Lysyl oxidase-like 2; MMPs: Matrix metalloproteinases; TIMPs: Tissue inhibitor of metalloproteinases; CCL2: C-C motif chemokine ligand 2; CA153: Cancer antigen 153; TNF-α: Tumor necrosis factor-alpha; Angptl2: Angiopoietin-like protein 2; CYSLTR1: Cysteinyl Leukotriene Receptor 1; SIGLEC10: Sialic acid binding Ig like lectin 10.
Acknowledgements
Funding
This work was funded by Shanghai Municipal Health Commission (No.20244Z0019), National Key Technologies R & D Program Precision Medicine Research (2024ZD0528901), Physician-scientist project of Shanghai Jiao Tong University School of Medicine (No.20240820), and Shanghai Jiao Tong University Cultivation Platform for Future Cross-Disciplinary Innovative Talents.
Author Contributions
F.L. and Y.C. conceived this review. Y.C. wrote the manuscript. F.L. and Q.C. reviewed and edited the manuscript. All authors contributed to figure plotting, manuscript revision and have reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T. et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205:e18-e47
2. Idiopathic Pulmonary Fibrosis Clinical Research N, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968-77
3. Strand MJ, Sprunger D, Cosgrove GP, Fernandez-Perez ER, Frankel SK, Huie TJ. et al. Pulmonary function and survival in idiopathic vs secondary usual interstitial pneumonia. Chest. 2014;146:775-85
4. Park JH, Kim DS, Park IN, Jang SJ, Kitaichi M, Nicholson AG. et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007;175:705-11
5. Yang S, Wang J, Sun D, Wang Y, Xue C, Ye Q. Disease progression in patients with usual interstitial pneumonia and probable UIP patterns on computed tomography with various underlying etiologies: a retrospective cohort study. Front Med (Lausanne). 2023;10:1246767
6. Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ. et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010;137:129-37
7. Oh AS, Lynch DA, Swigris JJ, Baraghoshi D, Dyer DS, Hale VA. et al. Deep Learning-based Fibrosis Extent on Computed Tomography Predicts Outcome of Fibrosing Interstitial Lung Disease Independent of Visually Assessed Computed Tomography Pattern. Ann Am Thorac Soc. 2024;21:218-27
8. Kim JS, Pugashetti J, Ma SF, Huang Y, Podolanczuk AJ, Lynch DA. et al. Associations of interstitial lung disease subtype and CT pattern with lung function and survival. Thorax. 2025
9. Chung JH, Cox CW, Montner SM, Adegunsoye A, Oldham JM, Husain AN. et al. CT Features of the Usual Interstitial Pneumonia Pattern: Differentiating Connective Tissue Disease-Associated Interstitial Lung Disease From Idiopathic Pulmonary Fibrosis. AJR Am J Roentgenol. 2018;210:307-13
10. Chung JH, Cox CW, Montner SM, Adegunsoye A, Oldham JM, Husain AN. et al. CT Features of the Usual Interstitial Pneumonia Pattern: Differentiating Connective Tissue Disease-Associated Interstitial Lung Disease From Idiopathic Pulmonary Fibrosis. American Journal of Roentgenology. 2017;210:307-13
11. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ. et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198:e44-e68
12. Spagnolo P, Guler SA, Chaudhuri N, Udwadia Z, Sese L, Kaul B. et al. Global epidemiology and burden of interstitial lung disease. Lancet Respir Med. 2025;13:739-55
13. Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ. et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:1390-4
14. Ekstrom M, Gustafson T, Boman K, Nilsson K, Tornling G, Murgia N. et al. Effects of smoking, gender and occupational exposure on the risk of severe pulmonary fibrosis: a population-based case-control study. BMJ Open. 2014;4:e004018
15. Wang L, Zhu M, Li Y, Yan P, Li Z, Chen X. et al. Serum Proteomics Identifies Biomarkers Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis. Mol Cell Proteomics. 2023;22:100524
16. Allen RJ, Porte J, Braybrooke R, Flores C, Fingerlin TE, Oldham JM. et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med. 2017;5:869-80
17. Newton CA, Oldham JM, Ley B, Anand V, Adegunsoye A, Liu G. et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J. 2019 53
18. Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD. et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051-6
19. Kamiya M, Carter H, Espindola MS, Doyle TJ, Lee JS, Merriam LT. et al. Immune mechanisms in fibrotic interstitial lung disease. Cell. 2024;187:3506-30
20. Zhu J, Liu H, Gao R, Gong R, Wang J, Zhou D. et al. Genetic-informed proteome-wide scan reveals potential causal plasma proteins for idiopathic pulmonary fibrosis. Thorax. 2024;79:878-82
21. Mills R, Mathur A, Nicol LM, Walker JJ, Przybylski AA, Mackinnon AC. et al. Intrapulmonary Autoantibodies to HSP72 Are Associated with Improved Outcomes in IPF. Journal of Immunology Research. 2019;2019:1845128
22. Taillé C, Grootenboer-Mignot S, Boursier C, Michel L, Debray M-P, Fagart J. et al. Identification of Periplakin as a New Target for Autoreactivity in Idiopathic Pulmonary Fibrosis. American Journal of Respiratory and Critical Care Medicine. 2011;183:759-66
23. Hoyer N, Prior TS, Bendstrup E, Wilcke T, Shaker SB. Risk factors for diagnostic delay in idiopathic pulmonary fibrosis. Respir Res. 2019;20:103
24. Hamblin M, Prosch H, Vasakova M. Diagnosis, course and management of hypersensitivity pneumonitis. Eur Respir Rev. 2022 31
25. Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M. et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;202:e36-e69
26. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186:314-24
27. Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E. et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5:639-47
28. Salisbury ML, Gu T, Murray S, Gross BH, Chughtai A, Sayyouh M. et al. Hypersensitivity Pneumonitis: Radiologic Phenotypes Are Associated with Distinct Survival Time and Pulmonary Function Trajectory. Chest. 2019;155:699-711
29. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388:2023-38
30. Kawano-Dourado L, Doyle TJ, Bonfiglioli K, Sawamura MVY, Nakagawa RH, Arimura FE. et al. Baseline Characteristics and Progression of a Spectrum of Interstitial Lung Abnormalities and Disease in Rheumatoid Arthritis. Chest. 2020;158:1546-54
31. Yunt ZX, Chung JH, Hobbs S, Fernandez-Perez ER, Olson AL, Huie TJ. et al. High-resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: Relationship to survival. Respir Med. 2017;126:100-4
32. Chen N, Diao CY, Gao J, Zhao DB. Risk factors for the progression of rheumatoid arthritis-related interstitial lung disease: Clinical features, biomarkers, and treatment options. Semin Arthritis Rheum. 2022;55:152004
33. Zamora-Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Progressive Decline of Lung Function in Rheumatoid Arthritis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2017;69:542-9
34. Saag KG, Cerhan JR, Kolluri S, Ohashi K, Hunninghake GW, Schwartz DA. Cigarette smoking and rheumatoid arthritis severity. Ann Rheum Dis. 1997;56:463-9
35. Kelly CA, Saravanan V, Nisar M, Arthanari S, Woodhead FA, Price-Forbes AN. et al. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics-a large multicentre UK study. Rheumatology (Oxford). 2014;53:1676-82
36. Johnson C. Recent advances in the pathogenesis, prediction, and management of rheumatoid arthritis-associated interstitial lung disease. Curr Opin Rheumatol. 2017;29:254-9
37. Joy GM, Arbiv OA, Wong CK, Lok SD, Adderley NA, Dobosz KM. et al. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. Eur Respir Rev. 2023 32
38. Wang T, Zheng XJ, Ji YL, Liang ZA, Liang BM. Tumour markers in rheumatoid arthritis-associated interstitial lung disease. Clin Exp Rheumatol. 2016;34:587-91
39. Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S. et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N Engl J Med. 2018;379:2209-19
40. Juge PA, Granger B, Debray MP, Ebstein E, Louis-Sidney F, Kedra J. et al. A Risk Score to Detect Subclinical Rheumatoid Arthritis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2022;74:1755-65
41. Juge PA, Borie R, Kannengiesser C, Gazal S, Revy P, Wemeau-Stervinou L. et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J. 2017 49
42. Perelas A, Silver RM, Arrossi AV, Highland KB. Systemic sclerosis-associated interstitial lung disease. Lancet Respir Med. 2020;8:304-20
43. Khanna D, Tashkin DP, Denton CP, Renzoni EA, Desai SR, Varga J. Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease. Am J Respir Crit Care Med. 2020;201:650-60
44. Rahaghi FF, Hsu VM, Kaner RJ, Mayes MD, Rosas IO, Saggar R. et al. Expert consensus on the management of systemic sclerosis-associated interstitial lung disease. Respir Res. 2023;24:6
45. Zheng B, Marinescu DC, Hague CJ, Muller NL, Murphy D, Churg A. et al. Lung imaging patterns in connective tissue disease-associated interstitial lung disease impact prognosis and immunosuppression response. Rheumatology (Oxford). 2024;63:2734-40
46. Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C. et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8:e70621
47. Mariette X, Criswell LA. Primary Sjögren's Syndrome. New England Journal of Medicine. 2018;378:931-9
48. Joy GM, Arbiv OA, Wong CK, Lok SD, Adderley NA, Dobosz KM. et al. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. European Respiratory Review. 2023;32:220210
49. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren's syndrome. European Respiratory Review. 25: 110-23.
50. Berardicurti O, Marino A, Genovali I, Navarini L, D'Andrea S, Currado D. et al. Interstitial Lung Disease and Pulmonary Damage in Primary Sjögren's Syndrome: A Systematic Review and Meta-Analysis. J Clin Med. 2023 12
51. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM. et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren's syndrome: A consensus and data-driven methodology involving three international patient cohorts. Annals of the Rheumatic Diseases. 2017;76:9-16
52. Konak HE, Atalar E, Hezer H, Koçak Ulucaköy R, Kayacan Erdoğan E, Babaoğlu H. et al. Interstitial Lung Disease in Primary Sjögren's Syndrome: Risk factors for occurrence and radiographic progression. Sarcoidosis Vasc Diffuse Lung Dis. 2024;41:e2024035
53. Sambataro G, Ferro F, Orlandi M, Sambataro D, Torrisi SE, Quartuccio L. et al. Clinical, morphological features and prognostic factors associated with interstitial lung disease in primary Sjӧgren's syndrome: A systematic review from the Italian Society of Rheumatology. Autoimmunity Reviews. 2020;19:102447
54. Banerjee S, Grayson PC. Vasculitis Around the World: Epidemiologic Insights into Causality and a Need for Global Partnerships. The Journal of Rheumatology. 2017;44:136-9
55. Yamakawa H, Toyoda Y, Baba T, Kishaba T, Fukuda T, Takemura T. et al. Anti-Inflammatory and/or Anti-Fibrotic Treatment of MPO-ANCA-Positive Interstitial Lung Disease: A Short Review. J Clin Med. 2022 11
56. Hozumi H, Kono M, Hasegawa H, Yasui H, Suzuki Y, Karayama M. et al. Clinical Significance of Interstitial Lung Disease and Its Acute Exacerbation in Microscopic Polyangiitis. Chest. 2021;159:2334-45
57. Kawasaki A, Namba N, Sada KE, Hirano F, Kobayashi S, Nagasaka K. et al. Association of TERT and DSP variants with microscopic polyangiitis and myeloperoxidase-ANCA positive vasculitis in a Japanese population: a genetic association study. Arthritis Res Ther. 2020;22:246
58. Alba MA, Flores-Suárez LF, Henderson AG, Xiao H, Hu P, Nachman PH. et al. Interstital lung disease in ANCA vasculitis. Autoimmun Rev. 2017;16:722-9
59. Matsuda S, Kotani T, Okazaki A, Nishioka D, Watanabe R, Gon T. et al. Prediction model for respiratory-related mortality in microscopic polyangiitis with interstitial lung disease: multicentre REVEAL cohort study. Rheumatology. 2024;63:1607-15
60. Villeneuve T, Faguer S, Collot S, Pugnet G, Prévot G. HRCT imaging of pulmonary involvement in granulomatosis with polyangiitis and microscopic polyangiitis at disease onset and during follow-up. Seminars in Arthritis and Rheumatism. 2023;63:152307
61. Fernandes-Serodio J, Prieto-González S, Espígol-Frigolé G, Ríos-Garcés R, Gómez-Caverzaschi V, Araújo O. et al. Significance of clinical-immunological patterns and diagnostic yield of biopsies in microscopic polyangiitis and granulomatosis with polyangiitis. Journal of Internal Medicine. 2024;295:651-67
62. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE. et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503-12
63. van der Vis JJ, Prasse A, Renzoni EA, Stock CJW, Caliskan C, Maher TM. et al. MUC5B rs35705950 minor allele associates with older age and better survival in idiopathic pulmonary fibrosis. Respirology. 2023;28:455-64
64. Mota PC, Soares ML, Vasconcelos CD, Ferreira AC, Lima BA, Manduchi E. et al. Predictive value of common genetic variants in idiopathic pulmonary fibrosis survival. J Mol Med (Berl). 2022;100:1341-53
65. Liu J, Yi Z, Chen T, Ying Y, Hu Y. An Overview of the Role of Genetic factors in Idiopathic Pulmonary Fibrosis: Insights from Epidemiology to Prognosis. Int J Med Sci. 2025;22:2992-3006
66. Moor CC, Oppenheimer JC, Nakshbandi G, Aerts J, Brinkman P, Maitland-van der Zee AH. et al. Exhaled breath analysis by use of eNose technology: a novel diagnostic tool for interstitial lung disease. Eur Respir J. 2021 57
67. Chaudhary S, Weigt SS, Ribeiro Neto ML, Benn BS, Pugashetti JV, Keith R. et al. Interstitial lung disease progression after genomic usual interstitial pneumonia testing. Eur Respir J. 2023 61
68. Zhao AY, Unterman A, Abu Hussein NS, Sharma P, Nikola F, Flint J. et al. Single-Cell Analysis Reveals Novel Immune Perturbations in Fibrotic Hypersensitivity Pneumonitis. Am J Respir Crit Care Med. 2024;210:1252-66
69. Invernizzi R, Wu BG, Barnett J, Ghai P, Kingston S, Hewitt RJ. et al. The Respiratory Microbiome in Chronic Hypersensitivity Pneumonitis Is Distinct from That of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2021;203:339-47
70. Ding F, Yang L, Wang Y, Wang J, Ma Y, Jin J. Serum Rcn3 level is a potential diagnostic biomarker for connective tissue disease-associated interstitial lung disease and reflects the severity of pulmonary function. BMC Pulmonary Medicine. 2023;23:68
71. Weng L, Chen Y, Liang T, Lin Y, Liu D, Yu C. et al. Biomarkers of interstitial lung disease associated with primary Sjögren's syndrome. Eur J Med Res. 2022;27:199
72. Jia X-m, Wu B-x, Chen B-d, Li K-t, Liu Y-d, Xu Y. et al. Compositional and functional aberrance of the gut microbiota in treatment-naïve patients with primary Sjögren's syndrome. Journal of Autoimmunity. 2023;141:103050
73. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W. et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68:310-7
74. Matsuda S, Kotani T, Kuwabara H, Suzuka T, Kiboshi T, Fukui K. et al. CCL2 produced by CD68+/CD163+ macrophages as a promising clinical biomarker of microscopic polyangiitis-interstitial lung disease. Rheumatology (Oxford). 2021;60:4643-53
75. Matsuda S, Kotani T, Okazaki A, Nishioka D, Masuda Y, Shiomi M. et al. Poor prognostic factors for relapse of interstitial lung disease in microscopic polyangiitis: the Japanese multicentre REVEAL cohort study. Arthritis Res Ther. 2024;26:221
76. Mann M, Kumar C, Zeng WF, Strauss MT. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021;12:759-70
77. Bowman WS, Newton CA, Linderholm AL, Neely ML, Pugashetti JV, Kaul B. et al. Proteomic biomarkers of progressive fibrosing interstitial lung disease: a multicentre cohort analysis. Lancet Respir Med. 2022;10:593-602
78. Huang Y, Ma SF, Oldham JM, Adegunsoye A, Zhu D, Murray S. et al. Machine Learning of Plasma Proteomics Classifies Diagnosis of Interstitial Lung Disease. Am J Respir Crit Care Med. 2024;210:444-54
79. Zhou Y, Tong Z, Zhu X, Wu C, Zhou Y, Dong Z. Deciphering the cellular and molecular landscape of pulmonary fibrosis through single-cell sequencing and machine learning. J Transl Med. 2025;23:3
80. Oldham JM, Huang Y, Bose S, Ma SF, Kim JS, Schwab A. et al. Proteomic Biomarkers of Survival in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2024;209:1111-20
81. Larsen BT. Usual interstitial pneumonia: a clinically significant pattern, but not the final word. Mod Pathol. 2022;35:589-93
82. Antin-Ozerkis D, Kolb M. Interstitial lung disease: time to rethink the snapshot diagnosis? Thorax. 2016;71:5-7
83. Selman M, Pardo A, Wells AU. Usual interstitial pneumonia as a stand-alone diagnostic entity: the case for a paradigm shift? Lancet Respir Med. 2023;11:188-96
84. Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. American Journal of Respiratory and Critical Care Medicine. 2006;174:810-6
85. Fernandez Perez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of Hypersensitivity Pneumonitis among an Insured Population in the United States: A Claims-based Cohort Analysis. Ann Am Thorac Soc. 2018;15:460-9
86. Nasser M, Larrieu S, Boussel L, Si-Mohamed S, Bazin F, Marque S. et al. Prevalence and mortality of systemic sclerosis-associated interstitial lung disease (SSc-ILD) using the French national health insurance system (SNDS) database in France. European Respiratory Journal. 56: 805.
87. Podolanczuk AJ, Thomson CC, Remy-Jardin M, Richeldi L, Martinez FJ, Kolb M. et al. Idiopathic pulmonary fibrosis: state of the art for 2023. Eur Respir J. 2023 61
88. Gauhar UA, Gaffo AL, Alarcón GS. Pulmonary manifestations of rheumatoid arthritis. Semin Respir Crit Care Med. 2007;28:430-40
89. Furusawa H, Cardwell JH, Okamoto T, Walts AD, Konigsberg IR, Kurche JS. et al. Chronic Hypersensitivity Pneumonitis, an Interstitial Lung Disease with Distinct Molecular Signatures. Am J Respir Crit Care Med. 2020;202:1430-44
90. Blanchet MR, Israël-Assayag E, Cormier Y. Inhibitory effect of nicotine on experimental hypersensitivity pneumonitis in vivo and in vitro. Am J Respir Crit Care Med. 2004;169:903-9
91. Ekici M, Baytar Y, Kardas RC, Sari A, Akdogan A, Durhan G. et al. Predictors of mortality in rheumatoid arthritis-associated lung disease: A retrospective study on ten years. Joint Bone Spine. 2021;88:105133
92. Assayag D, Elicker BM, Urbania TH, Colby TV, Kang BH, Ryu JH. et al. Rheumatoid arthritis-associated interstitial lung disease: radiologic identification of usual interstitial pneumonia pattern. Radiology. 2014;270:583-8
93. Roth E, Bruni C, Petelytska L, Becker MO, Dobrota R, Jordan S. et al. Gastroesophageal reflux disease is associated with a more severe interstitial lung disease in systemic sclerosis in the EUSTAR cohort. Rheumatology. 2025
94. Kaul B, Lee JS, Zhang N, Vittinghoff E, Sarmiento K, Collard HR. et al. Epidemiology of Idiopathic Pulmonary Fibrosis among U.S. Veterans, 2010-2019. Ann Am Thorac Soc. 2022;19:196-203
95. Sullivan DI, Ascherman DP. Rheumatoid Arthritis-Associated Interstitial Lung Disease (RA-ILD): Update on Prevalence, Risk Factors, Pathogenesis, and Therapy. Curr Rheumatol Rep. 2024;26:431-49
96. Gille T, Didier M, Boubaya M, Moya L, Sutton A, Carton Z. et al. Obstructive sleep apnoea and related comorbidities in incident idiopathic pulmonary fibrosis. Eur Respir J. 2017 49
97. Lancaster LH, Mason WR, Parnell JA, Rice TW, Loyd JE, Milstone AP. et al. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest. 2009;136:772-8
98. Bagheri L, Kavosi H, Shokouhi N, Aghayani S, Haghighi KS, Najafizadeh SR. Sleep disorders and other medical and socio-demographic factors in systemic scleroderma. Eur J Transl Myol. 2024 34
99. Amlani B, Elsayed G, Barvalia U, Kanne JP, Meyer KC, Sandbo N. et al. Treatment of primary sjögren's syndrome-related interstitial lung disease: a retrospective cohort study. Sarcoidosis Vasc Diffuse Lung Dis. 2020;37:136-47
100. Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356-63
101. Kang J, Kim YJ, Choe J, Chae EJ, Song JW. Acute exacerbation of fibrotic hypersensitivity pneumonitis: incidence and outcomes. Respir Res. 2021;22:152
102. Hozumi H, Nakamura Y, Johkoh T, Sumikawa H, Colby TV, Kono M. et al. Acute exacerbation in rheumatoid arthritis-associated interstitial lung disease: a retrospective case control study. BMJ Open. 2013;3:e003132
103. Parambil JG, Myers JL, Lindell RM, Matteson EL, Ryu JH. Interstitial Lung Disease in Primary Sjögren Syndrome. Chest. 2006;130:1489-95
104. Saldana DC, Hague CJ, Murphy D, Coxson HO, Tschirren J, Peterson S. et al. Association of Computed Tomography Densitometry with Disease Severity, Functional Decline, and Survival in Systemic Sclerosis-associated Interstitial Lung Disease. Ann Am Thorac Soc. 2020;17:813-20
105. Adegunsoye A, Oldham JM, Fernández Pérez ER, Hamblin M, Patel N, Tener M. et al. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res. 2017 3
106. Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer's lung. Am Rev Respir Dis. 1992;145:3-5
107. Yan Q, Bruni C, Garaiman A, Mihai C, Jordan S, Becker MO. et al. Post hoc comparison of the effectiveness of tocilizumab, rituximab, mycophenolate mofetil, and cyclophosphamide in patients with SSc-ILD from the EUSTAR database. Annals of the Rheumatic Diseases. 2025;84:620-31
108. Ito I, Nagai S, Kitaichi M, Nicholson AG, Johkoh T, Noma S. et al. Pulmonary Manifestations of Primary Sjögren's Syndrome. American Journal of Respiratory and Critical Care Medicine. 2005;171:632-8
109. Zhang Y, Ding Q, Lv C, Ying Y, Cen Z, Zhou H. et al. Clinical significance of microscopic polyangiitis with interstitial lung disease and bronchiectasis: probability of preexisting comorbidities. Ann Med. 2023;55:2204449
110. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U. et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071-82
111. King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK. et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083-92
112. Solomon JJ, Danoff SK, Woodhead FA, Hurwitz S, Maurer R, Glaspole I. et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: a randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir Med. 2023;11:87-96
113. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD. et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med. 2019;380:2518-28
114. Acharya N, Sharma SK, Mishra D, Dhooria S, Dhir V, Jain S. Efficacy and safety of pirfenidone in systemic sclerosis-related interstitial lung disease—a randomised controlled trial. Rheumatology International. 2020;40:703-10
115. Lee AS, Scofield RH, Hammitt KM, Gupta N, Thomas DE, Moua T. et al. Consensus Guidelines for Evaluation and Management of Pulmonary Disease in Sjögren's. Chest. 2021;159:683-98
116. Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA. et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. 2017;5:946-55
117. Zou M, Hu X, Song W, Gao H, Wu C, Zheng W. et al. Plasma LTBP2 as a potential biomarker in differential diagnosis of connective tissue disease-associated interstitial lung disease and idiopathic pulmonary fibrosis: a pilot study. Clin Exp Med. 2023;23:4809-16
118. Tzouvelekis A, Herazo-Maya JD, Slade M, Chu JH, Deiuliis G, Ryu C. et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology. 2017;22:486-93
119. Todd JL, Neely ML, Overton R, Durham K, Gulati M, Huang H. et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry. Respir Res. 2019;20:227
120. Cha S-I, Ryerson CJ, Lee JS, Kukreja J, Barry SS, Jones KD. et al. Cleaved cytokeratin-18 is a mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respiratory Research. 2012;13:105
121. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE. et al. MMP1 and MMP7 as Potential Peripheral Blood Biomarkers in Idiopathic Pulmonary Fibrosis. PLOS Medicine. 2008;5:e93
122. Karampitsakos T, Juan-Guardela BM, Tzouvelekis A, Herazo-Maya JD. Precision medicine advances in idiopathic pulmonary fibrosis. eBioMedicine. 2023;95:104766
123. Okamoto T, Fujii M, Furusawa H, Tsuchiya K, Miyazaki Y, Inase N. The usefulness of KL-6 and SP-D for the diagnosis and management of chronic hypersensitivity pneumonitis. Respir Med. 2015;109:1576-81
124. Sánchez-Díez S, Munoz X, Ojanguren I, Romero-Mesones C, Espejo D, Villar A. et al. YKL-40 and KL-6 Levels in Serum and Sputum of Patients Diagnosed with Hypersensitivity Pneumonitis. J Allergy Clin Immunol Pract. 2022;10:2414-23
125. Wang Z, Wang W, Xiang T, Gong B, Xie J. Serum Uric Acid as a Diagnostic Biomarker for Rheumatoid Arthritis-Associated Interstitial Lung Disease. Inflammation. 2022;45:1800-14
126. Harlow L, Rosas IO, Gochuico BR, Mikuls TR, Dellaripa PF, Oddis CV. et al. Identification of citrullinated hsp90 isoforms as novel autoantigens in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheum. 2013;65:869-79
127. Fu Q, Bai Y, Liu Y, Zhou J, Zheng Y. The serum level and significance of lysyl oxidase-like 2 in patients with rheumatoid arthritis-associated interstitial lung disease. Clin Rheumatol. 2018;37:193-8
128. Pulito-Cueto V, Atienza-Mateo B, Batista-Liz JC, Sebastián Mora-Gil M, Mora-Cuesta VM, Iturbe-Fernández D. et al. Matrix metalloproteinases and their tissue inhibitors as upcoming biomarker signatures of connective tissue diseases-related interstitial lung disease: towards an earlier and accurate diagnosis. Mol Med. 2025;31:70
129. Shi L, Han XL, Guo HX, Wang J, Tang YP, Gao C. et al. Increases in tumor markers are associated with primary Sjögren's syndrome-associated interstitial lung disease. Ther Adv Chronic Dis. 2020;11:2040622320944802
130. Shi L, Fu Q, Chen N, Liu R, Zheng Y. Angiopoietin-like protein 2 as a novel marker for patients with primary Sjogren's syndrome-related interstitial lung disease. Clinical and Experimental Medicine. 2020;20:393-9
131. Dong J, Wang Z, Xu Y, Liang S. Exploration common biomarkers and pathogenesis of primary Sjögren's syndrome and interstitial lung disease by machine learning and weighted gene co-expression networks. PLoS One. 2025;20:e0333070
132. Matsuda S, Kotani T, Kuwabara H, Suzuka T, Kiboshi T, Fukui K. et al. CCL2 produced by CD68+/CD163+ macrophages as a promising clinical biomarker of microscopic polyangiitis-interstitial lung disease. Rheumatology. 2021;60:4643-53
133. Conticini E, d'Alessandro M, Bergantini L, Castillo D, Cameli P, Frediani B. et al. KL-6 in ANCA-Associated Vasculitis Patients with and without ILD: A Machine Learning Approach. Biology (Basel). 2022 11
Author contact
![]() Corresponding author: Feng Li, MD. PhD, Department of Pulmonary and Critical Care Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University School of Medicine, No. 241, West Huaihai Road, Xuhui, Shanghai, 200030, P.R. China; Telephone:0086-21-22200000*3408; Email: lifeng741com.
Corresponding author: Feng Li, MD. PhD, Department of Pulmonary and Critical Care Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University School of Medicine, No. 241, West Huaihai Road, Xuhui, Shanghai, 200030, P.R. China; Telephone:0086-21-22200000*3408; Email: lifeng741com.