Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Experimental Procedure
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2026; 23(5):1595-1604. doi:10.7150/ijms.128068 This issue Cite
Research Paper
Effects of Ranolazine on Vascular Adrenergic Receptors in Rabbit Aorta
Adrian Jorda1,2, Maria Dolores Mauricio1, Solanye Guerra-Ojeda1, Jose M. Vila1, Soraya L. Valles1, Martin Aldasoro1 ![]()
1. School of Medicina, University of Valencia, Spain.
2. School of Nursing and Podiatry, University of Valencia, Spain.
Received 2025-11-7; Accepted 2026-2-10; Published 2026-3-17
Abstract

Background: Different mechanisms of action have been proposed for Ranolazine (Rn), mainly the inhibition of the late sodium current and antagonism of α₁-adrenergic receptors. In the present study, we evaluated the possible involvement of other adrenergic receptors, specifically α₂, β₂, and β₃, as mediators of the vascular effects of Rn.
Methods: Segments of rabbit aorta were mounted in an organ bath. Electrical field stimulation (EFS; 2, 4, and 8 Hz) induced frequency-dependent contractions that were abolished by tetrodotoxin, prazosin, or guanethidine (10⁻⁶ M), confirming the neural origin of the vascular responses. The effects of Rn on vascular responses to adrenergic stimulation were evaluated by incubating the preparations with increasing concentrations of the drug (10⁻⁷-10⁻⁴ M) for 20 minutes prior to neural stimulation (4 Hz). The involvement of α₁-, α₂-, β₂-, or β₃-adrenergic receptors was assessed using specific antagonists (10⁻⁶ M): prazosin (α₁), yohimbine (α₂), butaxamine (β₂), and SR59230A (β₃). Subsequently, the sequence of electrical field stimulations was performed in the presence of Rn. Expression levels of α₁-, α₂-, β₂-, and β₃-adrenergic receptors were determined by Western blot analysis.
Results: Rn decreases the contractile effect induced by adrenergic nerve stimulation in the rabbit aorta. In the presence of prazosin or yohimbine, the vasoconstrictor response was significantly reduced. However, incubation with butaxamine or SR59230A significantly increased the contractile response to adrenergic nerve stimulation. The protein expression of α1 and α2 receptors significantly decreased compared to the control when incubated with Rn. In contrast, the expression of β2 and β3 receptors increased only at 10⁻⁷ M, a concentration lower than that reached with therapeutic doses of Rn.
Conclusion: Rn inhibits the vasoconstrictor response to adrenergic nerve stimulation through an antagonistic effect on α1 and α2 receptors and enhancing the vasodilatory responses mediated by β2 and β3 adrenergic receptors.
Keywords: Ranolazine, adrenergic α1, α2, β2, β3 receptors, adrenergic receptor expression, vasoconstriction, vasodilatation.
Introduction
Ranolazine [(+)-N-(2,6-dimethylphenyl)-4(2-hydroxy-3-(2-methoxyphenoxy)-propyl)-1-piperazine acetamide] (Rn) is a piperazine-derived drug approved by the FDA in 2006, whose clinical indications are mainly focused on the treatment of chronic coronary ischemia refractory to other therapies [1, 2]. However, in recent years, the medical indications for Rn have expanded, including its use in the treatment of cardiac arrhythmias such as atrial fibrillation, and in coronary endothelial dysfunction, by increasing the release of various vasodilator factors such as nitric oxide and prostanoids. Moreover, it has also been shown to reduce oxidative stress by inhibiting fatty acid oxidation and promoting greater glucose oxidation [3].
Different metabolic effects have also been described, including a reduction in blood glucose and glycated hemoglobin (hemoglobin A1C, HbA1c) levels [4, 5], as well as improvement in insulin secretion and β-cell survival in diabetic mice [6]. Moreover, evidence suggests that Rn may improve various cognitive processes in type II diabetes [7, 8]. In our laboratory, we have demonstrated that Rn enhances tissular insulin sensitivity both in the vascular wall and in cells of the central nervous system [9, 10].
Other effects of Rn target cells in the central nervous system by reducing neuronal excitability, thus acting as an anticonvulsant agent [11, 12]. In addition, it has been proposed as a potential treatment for neuropathic pain [13]. There is also evidence of a protective effect against the development of different types of dementia, particularly those related to oxidative and inflammatory mechanisms [14, 9, 15].
Although the mechanism of action of Rn is not exactly known, it has been shown that, at therapeutic concentrations, it selectively inhibits the late inward Na⁺ current (I_NaL), thus reducing the intracellular concentration of Na⁺, which would inhibit the activity of the Na⁺-Ca²⁺ exchanger and the subsequent entry of Ca²⁺ into the cells (Ca²⁺_i). In this way, intracellular ionic homeostasis would be preserved, thereby reducing the tension of both ventricular and vascular muscle fibers [16, 17]. Rn also acts on other cellular ion channels such as TASK-1 potassium channels, whose inhibition would contribute to its antiarrhythmic effects [18].
Other mechanisms of action have also been described for Rn, among which the antagonistic effect it exerts on α₁-adrenergic receptors stands out. In fact, in our laboratory we have demonstrated an antagonism with α₁ adrenergic receptors in the saphenous vein used in coronary bypass surgery [19]. Nevertheless, it is possible that Rn also interacts with other adrenergic receptors, particularly those of the beta type. The aim of this study was to evaluate the possible interactions between Rn and the β₂-, β₃-, α₁-, and α₂-adrenergic receptors.
Materials and Methods
Animal Model
The investigation was carried out in accordance with the ethical standards in animal experimentation established by EU Directive 2010/63 and Spanish Royal Decree (RD) 1201/2005. The experiments were carried out using tissue samples according to procedure 2017/VSC/PEA/00049 type 2, authorized by the Bioethics Committee of the University of Valencia, Spain. Forty-two male New Zealand white rabbits, weighing 3.2-3.8 kg, were used in this study and housed in a 12:12 h light/dark cycle at a constant room temperature of 22 ºC and 60% humidity. Animals were euthanized following heparinization and anesthesia (sodium thiopental 60 mg/kg i.v.).
Preparation of Vascular Rings
Organ bath experiments were carried out as previously described [20]. Abdominal aorta was isolated and cut into 4-mm rings for isometric recording of tension. Two stainless steel L-shaped pins were introduced through the lumen of the vascular rings. One pin was fixed to the wall of the organ bath, and the other one was connected to a force-displacement transducer (FT03; Grass Instruments, West Warwick, RI, USA). Variations in isometric force were registered on a Macintosh computer (Apple Corp., Cupertino, CA, USA) using the Chart, version 7, and a MacLab/8e data acquisition system (AD Instruments). Individual rings were suspended in a 5 mL bath with a modified Krebs-Henseleit solution containing (mM) NaCl, 115; CaCl2, 2.5; KCl, 4.6; MgCl2.6H2O, 1.2; NaHCO3, 25; glucose, 11.1; and disodium EDTA, 0.01, with 95% O2 and 5% CO2 to obtain a pH 7.3-7.4, and temperature was held at 37 ºC. The optimal resting tension for vascular rings was 3.5 g, and aortic preparations were allowed to equilibrate for 3 h. The contractile capacity of vascular smooth muscle was evaluated by the maximum response to KCl (60 mM). The endothelium was considered functional if relaxation to acetylcholine (10-6 M), in aortic rings precontracted with noradrenaline, was ≥ 70%. Vascular rings with dysfunctional endothelium in the control conditions were excluded.
Experimental Procedure
Periarterial Adrenergic Nerve Stimulation
To obtain adrenergic nerve stimuli, electrical field stimulation (EFS) was applied through two platinum electrodes placed on both sides of the aortic segments with a 5-mm separation between the two electrodes. The electrodes were connected to a multichannel stimulator (Grass S88). The correspondence between frequency and vasomotor response was studied in a determinate range of frequencies, specifically 2 and 4 Hz, with the application of 25 V stimuli (supramaximal voltage) of 0.25 ms duration for each pulse for 30 s of total duration of stimulation. The evaluation of the neurogenic nature of the contractile response to EFS was carried out by incubating the vascular segments for 15 min with tetrodotoxin (TTX) (10-6 M), a blocker of voltage dependent Na+ channels and, therefore, an inhibitor of the nerve conduction of the neural fibers present in the vascular wall; guanethidine (10-6 M), a blocker of the release of noradrenaline, neurotransmitter of the adrenergic nervous axons; therefore, it also inhibits adrenergic neurotransmission; or with an α1 adrenergic postsynaptic receptor antagonist, prazosin (10-6 M). Different stimulation rounds (2 and 4 Hz) were provoked, with 5 min intervals between each stimulus of increasing frequency. These series of stimuli were applied again 10 min after adding TTX (10-6 M), guanethidine (10-6 M), or prazosin (10-6 M), to the organ bath. As a control group, in another series of vascular segments, electrical field stimuli were performed without the presence of adrenergic nervous system blockers.
Interaction between Rn with the nervous adrenergic system
The possible interaction of Rn with the sympathetic-adrenergic nervous system was assessed by incubating the vascular rings with Rn (10-7-10-4 M) for fifteen minutes prior to the application of adrenergic nervous stimuli at frequency of 4 Hz.
Involvement of α₁-adrenergic receptors in the vascular responses to Rn
The involvement of α₁-adrenergic receptors in the responses of the abdominal aorta to Rn after stimulation of the adrenergic nervous system was evaluated by incubating the vascular segments with Prazosin (10⁻⁶ M) for fifteen minutes prior to the application of EFS (4 Hz) and Rn (10⁻⁷-10⁻⁴ M).
Involvement of α₂-adrenergic receptors in the vascular responses to Rn
The involvement of α₂-adrenergic receptors in the responses of the abdominal aorta to Rn after stimulation of the adrenergic nervous system was evaluated by incubating the vascular segments with Yohimbine (10⁻⁶ M) for fifteen minutes prior to the application of EFS (4 Hz) and Rn (10⁻⁷-10⁻⁴ M).
Involvement of β₂-adrenergic receptors in the vascular responses to Rn
The involvement of β₂-adrenergic receptors in the responses of the abdominal aorta to Rn after stimulation of the adrenergic nervous system was evaluated by incubating the vascular segments with Butaxamine (10⁻⁶ M) for fifteen minutes prior to the application of EFS (4 Hz) and Rn (10⁻⁷-10⁻⁴ M).
Involvement of β₃-adrenergic receptors in the vascular responses to Rn
The involvement of β₃-adrenergic receptors in the responses of the abdominal aorta to Rn after stimulation of the adrenergic nervous system was evaluated by incubating the vascular segments with SR59230A (10⁻⁶ M) for fifteen minutes prior to the application of EFS (4 Hz) and Rn (10⁻⁷-10⁻⁴ M).
Drugs
The drugs used were Potassium Chloride (KCl, Merck, Darmstadt, Germany), Insulin, Acetylcholine, Noradrenaline Hydrochloride, Prazosin, Tetrodotoxin, Guanethidine, Yohimbine, Butaxamine, SR59230A and Ranolazine [(+)-N-(2,6-dimetilfenil)-4(2-hidroxi- 3-(2-metoxifenoxi)-propil)-1-piperazina acetamida], (3,4-dihydro-4-(2-pyrimidinylmethyl)-7-[4-(trifluoromethoxy)phenyl]-1,4benzoxazepin-5(2H)-one) (Sigma-Aldrich, Madrid, Spain). Concentrated drug solutions were obtained with bi-distilled water, except for prazosin, which were dissolved in ethanol.
Western-blot analysis
Protein extracts from vascular rings of abdominal aorta were mixed with an equal volume of SDS buffer (0.125 M Tris-HCl, pH 6.8, 2% SDS, 0.5% (v/v) 2-mercaptoethanol, 1% bromophenol blue, and 19% glycerol) and then heated for 5 minutes. Protein concentration was measured using a modified Lowry method. Proteins were separated via SDS-PAGE and transferred to nitrocellulose membranes using standard techniques. Membranes were blocked with 5% dried milk in TBS containing 0.05% Tween-20, then incubated with the appropriate antibodies according to the manufacturer's instructions. The blots were washed three times for 15 minutes each with washing buffer (phosphate-buffered saline, 0.2% Tween-20), followed by a 1-hour incubation with a secondary horseradish peroxidase-linked anti-rabbit or anti-mouse IgG antibody (Cell Signaling Technologies, Barcelona, Spain). Afterward, the blots were washed three times and developed using the enhanced chemiluminescence (ECL) method as per the manufacturer's instructions (Pharmacia Biotech, San Francisco, CA, USA). Autoradiographic signals were quantified with a Bio-Rad scanning densitometer. The following antibodies were used: Anti-α1 Adrenoreceptor (ab137123), Anti-α2 Adrenoreceptor (ab85570), Anti-β2 Adrenoreceptor (ab176490), Anti-β3 Adrenoreceptor (ab94506) and anti-tubulin (ab6046) (Abcam biotechnology).
Statistical analysis
Data are expressed as means ± SEM; n indicates the number of rabbits. At least eight artery rings were obtained from each case. The results were evaluated statistically by means of paired or unpaired Student's t test or one-way analysis of variance. The probability value of < 0.05 was significant.
Results
Effects of Electrical Field Stimulation (EFS)
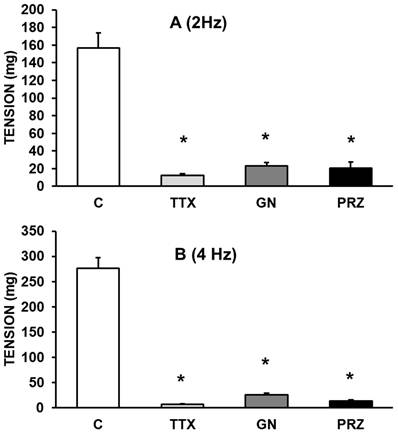
EFS (at 2 and 4 Hz) elicited frequency-dependent contractions of rabbit aortic rings at resting tension. The vasoconstriction induced by EFS was blocked after incubating the aortic segments with TTX (10-6 M), guanethidine (10-6 M), or prazosin (10-6 M). Taken together, these results indicate that the vasoconstrictor effect induced by EFS is mediated by adrenergic fibers that release norepinephrine from nerve axons following its binding to α1-adrenergic receptors (Figure 1, A and B).
Effects of EFS at 2 Hz (A) and 4 Hz (B) on rabbit aorta in the absence (control, n = 8) and in the presence of tetrodotoxin (TTX) (10-6 M, n = 7), guanethidine (GN) (10-6 M, n = 6), or prazosin (PRZ) (10-4 M, n = 6). Values are means ± SEM shown by vertical bars. *p < 0.05 vs. control.
Effects of Ranolazine on the responses of the aortic segments to sympathetic nerve stimulation
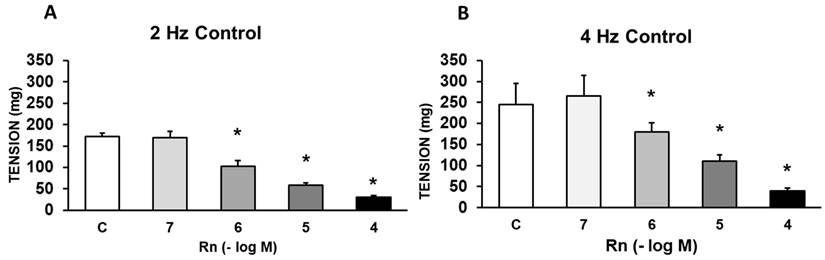
Rn (10-6 - 10-4 M) caused significant concentration-dependent decrease in the vasoconstrictor response induced by adrenergic nerve stimulation (4 Hz). Lower concentrations of Rn (10-7) do not cause any vascular effect (Figure 2).
Effects of EFS at 2 Hz (A) and 4 Hz (B) on rabbit aorta in the absence (control, n = 6) and in the presence of Rn (10-7-10-4 M, n = 7). Values are means ± SEM shown by vertical bars. *p < 0.05 vs. control.
Interactions between Ranolazine and α1 adrenergic receptors
In the presence of Prazosin (10⁻⁶ M), an α₁-adrenergic receptor antagonist, the contractile response induced by EFS was reduced by 48-56%. Rn (10⁻⁷ - 10⁻⁴ M) inhibited the contractile responses to EFS by a percentage similar to that observed in experiments without Prazosin (Figure 3A). Furthermore, Rn, at all concentrations tested, decreased the protein expression of α₁-adrenergic receptors (Figure 3B).
(A) Contractile effects of EFS at 4 Hz on rabbit aorta in the absence (control, n = 7) and in the presence of Rn (10-7-10-4 M) and without PRZ (□) (n = 7) or previously incubated with PRZ (■) (10-6 M, n = 6). Values are means ± SEM shown by vertical bars. *p < 0.05 vs. control with PRZ. (B) Protein expression of α1 adrenergic receptors by Western-blot. A representative immunoblot is shown in each panel. Data are the mean ± SD of four independent experiments. *p < 0.05 vs. control.
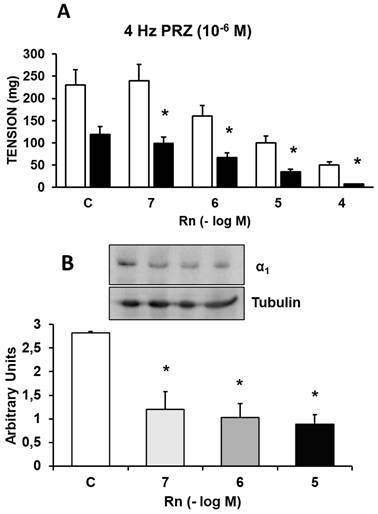
Together, these results indicate that Rn continues to inhibit the contractile response to EFS, which was previously partially blocked by Prazosin (10⁻⁶ M). Given that Prazosin at a concentration of 10⁻⁴ M completely inhibits the contractile response to EFS, it is reasonable to assume that the relaxation induced by Rn under these conditions is due to α₁-adrenergic antagonism, consistent with the reduced expression of α₁ receptors in the presence of Rn.
Interactions between Ranolazine and α2 adrenergic receptors
Incubation with Yohimbine (10⁻⁶ M), an α₂-adrenergic receptor antagonist, decreased the contractile response induced by EFS by 23-31%. Rn (10⁻⁷ - 10⁻⁴ M) continued to inhibit the contractile responses to EFS by a percentage similar to that observed in experiments without Yohimbine (Figure 4A). Additionally, Rn, at all concentrations tested, decreased the protein expression of α₂-adrenergic receptors (Figure 4B).
(A) Contractile effects of EFS at 4 Hz on rabbit aorta in the absence (control, n = 8) and in the presence of Rn (10-7-10-4 M) and without Yohimbine (□) (n = 8) or previously incubated with Yohimbine (■) (10-6 M, n = 7). Values are means ± SEM shown by vertical bars. *p < 0.05 vs. control with Yohimbine. (B) Protein expression of α2 adrenergic receptors by Western-blot. A representative immunoblot is shown in each panel. Data are the mean ± SD of four independent experiments. *p < 0.05 vs. control.
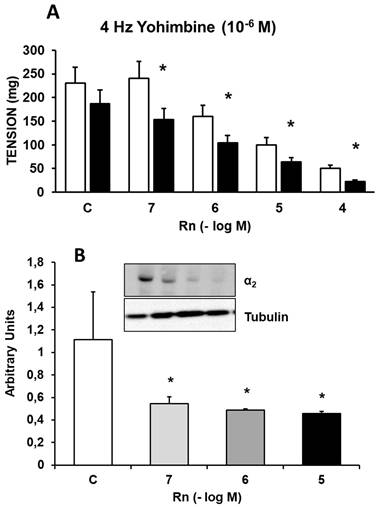
Together, these results indicate that Rn continues to inhibit the contractile response to EFS, which was previously partially blocked by Yohimbine (10⁻⁶ M). Overall, these findings suggest that Rn also acts as an antagonist of α₂-adrenergic receptors.
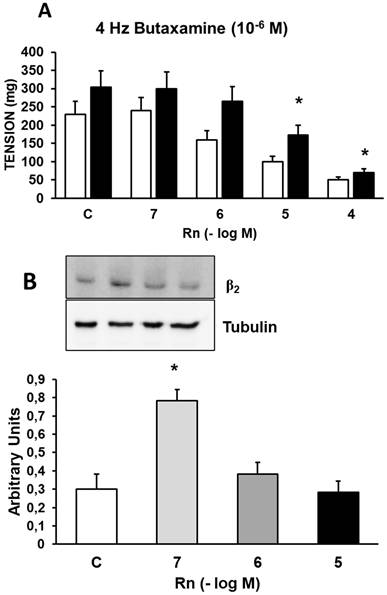
Interactions between Ranolazine and β2 adrenergic receptors
Incubation with Butaxamine (10⁻⁶ M), a β₂-adrenergic receptor antagonist, increased the contractile response induced by EFS by 21% (Figure 5A) compared to control values (Figure 2A). Rn (10⁻⁷ and 10⁻⁶ M) did not induce changes in the contractile response to EFS. On the other hand, Rn (10⁻⁵ and 10⁻⁴ M) reduced the contractile responses to EFS (Figure 5A), but to a lesser extent than in aortic segments not incubated with Butaxamine (Figure 2A). Additionally, Rn induced a very significant increase in β₂ receptor expression only at the concentration of 10⁻⁷ M (Figure 5B). These data suggest that β₂ receptors may be involved in the relaxing responses elicited by Rn.
(A) Contractile effects of EFS at 4 Hz on rabbit aorta in the absence (control, n = 7) and in the presence of Rn (10-7-10-4 M) and without Butaxamine (□) (n = 8) or previously incubated with Butaxamine (■) (10-6 M, n = 8). Values are means ± SEM shown by vertical bars. *p < 0.05 vs. control with Butaxamine. (B) Protein expression of β2 adrenergic receptors by Western-blot. A representative immunoblot is shown in each panel. Data are the mean ± SD of five independent experiments. *p < 0.05 vs. control. # < 0,05 vs. control sin Butaxamine.
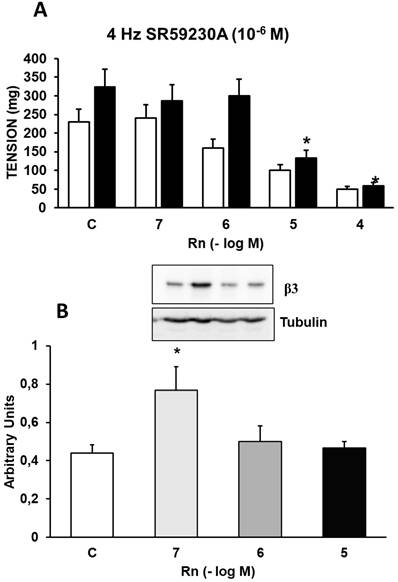
Interactions between Ranolazine and β3 adrenergic receptors
Incubation with SR59230A (10⁻⁶ M), a β₃-adrenergic receptor antagonist, increased the contractile response induced by EFS by 29% (Figure 6A) compared to control values (Figure 2A). Rn (10⁻⁷ and 10⁻⁶ M) did not induce changes in the contractile response to EFS. On the other hand, Rn (10⁻⁵ and 10⁻⁴ M) reduced the contractile responses similarly to aortic segments not incubated with SR59230A (Figure 6A). Additionally, Rn induced a very significant increase in β₃ receptor expression only at the concentration of 10⁻⁷ M (Figure 6B). These data suggest that β₃ receptors may be involved in the relaxing responses elicited by Rn.
(A) Contractile effects of EFS at 4 Hz on rabbit aorta in the absence (control, n = 8) and in the presence of Rn (10-7-10-4 M) and without SR59230A (□) (n = 8) or previously incubated with SBR59230A (■) (10-6 M, n = 9). Values are means ± SEM shown by vertical bars. *p < 0.05 vs. control with SR59230A. (B) Protein expression of β3 adrenergic receptors by Western-blot. A representative immunoblot is shown in each panel. Data are the mean ± SD of five independent experiments. *p < 0.05 vs. control. # < 0,05 vs. control sin SR59230A.
Discussion
In our study, we analyzed the effects of sympathetic nervous stimulation on the rabbit aorta by using electrical field stimuli (EFS) capable of causing the release of specific neurotransmitters from the sympathetic nervous system. The response was abolished in the presence of tetrodotoxin, guanethidine, or prazosin.
Rn (10⁻⁶-10⁻⁴ M) inhibited the contractile response to adrenergic nerve stimulation at all stimulation frequencies used in a concentration-dependent manner. In the presence of prazosin, an α₁-adrenergic receptor antagonist, the contractile response to EFS was reduced, while the relaxation induced by Rn began at lower concentrations of Rn (10⁻⁷-10⁻⁴ M). Similarly, Rn decreased the protein expression of α₁-adrenergic receptors.
Yohimbine, an α₂-adrenergic receptor antagonist, like prazosin, reduced the vasoconstrictor response induced by EFS and shifted the vascular relaxation induced by Rn such that this relaxation started at lower concentrations of Rn (10⁻⁷-10⁻⁴ M). Rn decreased the protein expression of α2-adrenergic receptors.
Incubation with butaxamine, a β₂-adrenergic receptor antagonist, increased the contractile response to EFS and decreased the relaxation induced by Rn at concentrations of 10⁻⁷ and 10⁻⁶ M. Rn significantly increased the protein expression of β₂-adrenergic receptors only at the concentration of 10⁻⁷ M.
The β₃-adrenergic receptor inhibitor SR59230A potentiated the vasoconstriction induced by EFS and decreased the relaxation induced by Rn at concentrations of 10⁻⁷ and 10⁻⁶ M. Rn significantly increased the protein expression of β₃-adrenergic receptors only at the concentration of 10⁻⁷ M.
Vascular responses to adrenergic nerve stimulation depend on the interaction of noradrenaline with adrenergic receptors located in the vascular wall. In general, the response is vasoconstrictive in both animal [10] and human [20, 21] vessels. This vasoconstrictive response is typically mediated by the binding of noradrenaline to α1-adrenergic receptors located on the membrane of vascular smooth muscle fibers [20]. In some cases, α2-adrenergic receptors [22] also participate, facilitating this vasoconstrictive response. However, other adrenergic receptors such as β2 [23] or β3 [24] produce a vasodilatory effect when NA binds to them. β2 receptors are normally present in vascular smooth muscle, whereas β3 receptors are located in vascular endothelial cells. Therefore, the final response to adrenergic nerve stimulation results from the sum of the partial responses generated by each of the receptors. In our study, we observed that Rn inhibits the contractile response to adrenergic nerve stimulation, with its effect beginning at a concentration of 10⁻⁶ M. In the presence of prazosin, an α1-adrenergic receptor antagonist, Rn continues to block the contractile response, but this inhibition occurs at lower Rn concentrations (10⁻⁷ M). This finding, together with the fact that Rn inhibits α1-adrenergic receptor expression at all concentrations tested, suggests that Rn exerts a clear antagonistic effect on these receptors. These results are consistent with previous studies that proposed an antagonistic effect of Rn on α1-adrenergic receptors [25, 19].
As previously mentioned, α2-adrenergic receptors also generate a vasoconstrictive response when NA binds to them. In our study, we observed that in the presence of yohimbine, an α2-adrenergic receptor antagonist, Rn continued to inhibit the contractile response starting at a concentration of 10⁻⁷ M, like what was observed with prazosin incubation. Likewise, Rn inhibited the protein expression of α2-adrenergic receptors at all concentrations tested. Therefore, these results suggest that Rn may also exert an antagonistic effect on α2-adrenergic receptors. This antagonistic effect of Rn on α2-adrenergic receptors has not been previously described. Regarding β2 receptors, our results show that in the presence of butaxamine, an inhibitor of these receptors, the vasoconstrictor response to adrenergic stimulation was greater than the control, and the vasodilatory effect of Rn was abolished at both 10⁻⁷ and 10⁻⁶ M concentrations. Rn increased the expression of β2-adrenergic receptors, but only at the 10⁻⁷ M concentration. Therefore, Rn appears to exert a β2-agonist effect at the lowest concentration tested. Interactions between Rn and β2-adrenergic receptors have been previously reported [26]. It has also been reported that ranolazine may exert a weak antagonistic effect on β2-adrenergic receptors [27], whereas other studies have found no evidence of interactions between Rn and β2 receptors [28]. Furthermore, it has been suggested that Rn may act as a weak agonist of β2 and β3 receptors [29]. β3-adrenergic receptors induce endothelium-dependent vasodilation. Their activation stimulates endothelial nitric oxide synthase (eNOS) via the PI3K/Akt pathway, increasing the production of nitric oxide (NO) [24, 30]. Likewise, β3 receptors promote the release of endothelium-derived hyperpolarizing factor (EDHF) [31], generating a protective pathway against endothelial dysfunction. β3 receptors act as a counterregulatory mechanism opposing α1-adrenergic receptor-mediated vasoconstriction under conditions of high sympathetic activity, thereby limiting excessive increases in peripheral vascular resistance. This function is particularly relevant in pathological conditions such as hypertension, heart failure, and metabolic syndrome [32], and in facilitating endothelial function in patients with hyperglycemia [33]. On the other hand, activation of β3 receptors can induce the controlled production of reactive oxygen species (ROS), which act as second messengers for eNOS activation. This redox-functional coupling distinguishes β3 receptors from other β subtypes, particularly because of their endothelial signaling profile [34]. In addition, β3 receptors play a role in protecting against oxidative stress and endothelial inflammation, which is important for preventing vascular dysfunction and the development of cardiovascular diseases [35]. Activation of β3-adrenergic receptors has been associated with the facilitation of neurocognitive processes [36]. SR59230A, a β3 antagonist, blocks responses mediated by these receptors, including lipolysis, thermogenesis, and endothelium-dependent vasodilation induced by β3 agonists or catecholamines [37, 38]. This effect is associated with reduced activation of the PI3K/Akt/eNOS pathway, leading to decreased activating phosphorylation of eNOS (Ser¹¹⁷⁷) and reduced NO bioavailability [24, 32]. Blockade of β3 receptors with SR59230A abolishes their counterregulatory role against α1-adrenergic vasoconstriction. Therefore, an enhanced vasoconstrictor response to different adrenergic agents is observed, accompanied by an increase in basal vascular tone. This effect has been described in resistance vessels and may be key in situations of sympathetic hyperactivity [39]. Several studies suggest that SR59230A promotes a specific type of endothelial dysfunction characterized by reduced NO availability and an altered redox balance, such that blockade of β3 signaling leads to a predominance of deleterious ROS and functional uncoupling of eNOS [34].
In our study, Rn at a concentration of 10⁻⁷ M increases the expression of β2 and β3 receptors. However, at higher concentrations (10⁻⁶ and 10⁻⁵ M), Rn does not induce any change in the expression of these receptors. There may be several explanations for the mechanisms underlying this response. One possibility is that Rn, at low doses, activates the protein kinase A (PKA) signaling pathway [40], which could lead to an increase in β3 receptor expression. In contrast, at higher doses, it could activate the protein kinase C (PKC) signaling pathway, which may inhibit the expression of β3 receptors [41, 42]. It has been shown that β₂-adrenergic receptors undergo desensitization following sustained activation, which involves PKA- and GRK-dependent phosphorylation, recruitment of β-arrestins, and subsequent receptor internalization, ultimately reducing their expression on the cell surface [29, 43]. Another possibility is that at low doses, Rn could bind to β3 receptors, whereas at higher doses it might interact with other adrenergic receptors, such as α1, which could inhibit the expression of β2 or β3 receptors [25]. Furthermore, it has been reported that ranolazine can exert an antagonistic effect on β2-adrenergic receptors at doses corresponding to therapeutic concentrations of Rn (10⁻⁶-10⁻⁵ M). There are no studies on a potential antagonistic effect of Rn at these concentrations on β3 receptors, but this could be a possible mechanism [27]. This may be due to unequal selectivity of ranolazine for different adrenergic receptors [44].
These interactions with adrenergic receptors could explain various effects attributed to Rn. Among these, the cardiovascular effects in the treatment of coronary ischemia [2] and different cardiac arrhythmias [45, 46], such as atrial fibrillation, are particularly noteworthy. Rn improves coronary endothelial dysfunction [47], reduces levels of asymmetric dimethylarginine and C-reactive protein, and increases endothelial release of vasodilator mediators such as nitric oxide [48]. Furthermore, it improves both systolic and diastolic heart failure [49], prevents oxidative stress [50], and reduces hypertrophic cardiomyopathy [51]. Similarly, the interactions of Rn with adrenergic receptors could be involved in the drug's metabolic effects. In patients with type 2 diabetes, Rn decreases blood glucose and glycated hemoglobin (HbA1c) levels [52, 53]. It has also been observed that Rn may reduce glucagon release through the opening of sodium channels, thereby improving pre-prandial and postprandial glucose levels [54]. It has also been demonstrated that Rn improves cognitive processes in these patients [7]. In addition, Rn exhibits anticonvulsant properties in epileptic seizures [55], has shown efficacy in the treatment of neuropathic pain [13], and exerts neuroprotective effects in different types of dementia [15]. However, the mechanisms underlying these effects have not yet been fully elucidated. It is possible that different adrenergic receptors are involved in these processes. In recent years, evidence has emerged suggesting that Rn may reduce tissue insulin resistance by increasing sensitivity to the hormone. In a previous study conducted in our laboratory, we demonstrated that Rn improves vascular sensitivity to insulin. In this effect, α1-adrenergic antagonism induced by Rn plays an important role [10]. It would be interesting to further investigate the potential involvement of α- or β-adrenergic receptors in the mechanisms facilitating tissue insulin sensitivity or in other effects attributed to Rn.
Conclusions
According to the data obtained in our study, Rn inhibits vasoconstrictor responses to adrenergic nerve stimulation in the rabbit aorta. This inhibition is concentration-dependent, and it may be due to an antagonistic effect of Rn on α1- and α2-adrenergic receptors, as well as an enhancing effect on β2- and β3-adrenergic receptors. Furthermore, Rn, at all concentrations tested, decreases the expression of the vasoconstrictor α1- and α2-receptors and increases the expression of the vasodilatory β2- and β3-receptors, but only at the 10⁻⁷ M concentration, which corresponds to a dose lower than that used in clinical practice. This finding may represent a point of clinical interest, as it reflects a potential therapeutic effect.
Abbreviations
Rn: ranolazine; EFS: electrical field stimulation; PRZ: prazosin; TTX: tetrodotoxin; GN: Guanethidine.
Acknowledgements
Funding
Generalitat Valenciana (Spain) (CIAICO/2023/143).
Ethics approval and consent to participate
The present study was carried out in accordance with the ethical standards in animal experimentation established by EU Directive 2010/63 and Spanish Royal Decree (RD) 1201/2005. The experiments were developed using shared tissue samples from procedure 2017/VSC/PEA/00049 type 2, authorized by the Bioethics Committee of the University of Valencia, Spain.
Authorship contribution statement
Adrian Jorda: Software, Formal analysis, Investigation. Maria Doroles Mauricio: Investigation, Formal analysis. Sol Guerra-Ojeda: Methodology, Investigation, Formal analysis. Jose M. Vila: Software, Formal analysis. Soraya L. Valles: Writing - review & editing. Martin Aldasoro: Writing - review & editing. Conceptualization.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Siddiqui MA, Keam SJ. Ranolazine: a review of its use in chronic stable angina pectoris. Drugs. 2006;66(5):693-710 doi: 10.2165/00003495-200666050-00010
2. Sossalla S, Maier LS. Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol Ther. 2012Mar;133(3):311-23 doi: 10.1016/j.pharmthera.2011.11.003
3. McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996Jan1;93(1):135-42 doi: 10.1161/01.cir.93.1.135
4. Morrow DA, Scirica BM, Chaitman BR, McGuire DK, Murphy SA, Karwatowska-Prokopczuk E, McCabe CH, Braunwald E; MERLIN-TIMI 36 Investigators. Evaluation of the glycometabolic effects of ranolazine in patients with and without diabetes mellitus in the MERLIN-TIMI 36 randomized controlled trial. Circulation. 2009Apr21;119(15):2032-9 doi: 10.1161/CIRCULATIONAHA.107.763912
5. Gilbert BW, Sherard M, Little L, Branstetter J, Meister A, Huffman J. Antihyperglycemic and Metabolic Effects of Ranolazine in Patients with Diabetes Mellitus. Am J Cardiol. 2018Feb15;121(4):509-512 doi: 10.1016/j.amjcard.2017.11.021
6. Ning Y, Zhen W, Fu Z, Jiang J, Liu D, Belardinelli L, Dhalla AK. Ranolazine increases β-cell survival and improves glucose homeostasis in low-dose streptozotocin-induced diabetes in mice. J Pharmacol Exp Ther. 2011Apr;337(1):50-8 doi: 10.1124/jpet.110.176396
7. Cassano V, Leo A, Tallarico M, Nesci V, Cimellaro A, Fiorentino TV, Citraro R, Hribal ML, De Sarro G, Perticone F, Sesti G, Russo E, Sciacqua A. Metabolic and Cognitive Effects of Ranolazine in Type 2 Diabetes Mellitus: Data from an in vivo Model. Nutrients. 2020Jan31;12(2):382 doi: 10.3390/nu12020382
8. Cassano V, Tallarico M, Armentaro G, De Sarro C, Iannone M, Leo A, Citraro R, Russo E, De Sarro G, Hribal ML, Sciacqua A. Ranolazine Attenuates Brain Inflammation in a Rat Model of Type 2 Diabetes. Int J Mol Sci. 2022Dec18;23(24):16160 doi: 10.3390/ijms232416160
9. Jordá A, Aldasoro M, Campo-Palacio I, Vila JM, Aldasoro C, Campos-Campos J, Colmena C, Singh SK, Obrador E, Valles SL. Facilitation of Insulin Effects by Ranolazine in Astrocytes in Primary Culture. Int J Mol Sci. 2022Oct9;23(19):11969 doi: 10.3390/ijms231911969
10. Guerra-Ojeda S, Jorda A, Aldasoro C, Vila JM, Valles SL, Arias-Mutis OJ, Aldasoro M. Improvement of Vascular Insulin Sensitivity by Ranolazine. Int J Mol Sci. 2023Aug31;24(17):13532 doi: 10.3390/ijms241713532
11. Peters CH, Sokolov S, Rajamani S, Ruben PC. Effects of the antianginal drug, ranolazine, on the brain sodium channel Na(V)1.2 and its modulation by extracellular protons. Br J Pharmacol. 2013Jun;169(3):704-16 doi: 10.1111/bph.12150
12. Park YY, Johnston D, Gray R. Slowly inactivating component of Na+ current in peri-somatic region of hippocampal CA1 pyramidal neurons. J Neurophysiol. 2013Mar;109(5):1378-90 doi: 10.1152/jn.00435.2012
13. Gould HJ 3rd, Garrett C, Donahue RR, Paul D, Diamond I, Taylor BK. Ranolazine attenuates behavioral signs of neuropathic pain. Behav Pharmacol. 2009Dec;20(8):755-8 doi: 10.1097/FBP.0b013e3283323c90
14. Aldasoro M, Guerra-Ojeda S, Aguirre-Rueda D, Mauricio MD, Vila JM, Marchio P, Iradi A, Aldasoro C, Jorda A, Obrador E, Valles SL. Effects of Ranolazine on Astrocytes and Neurons in Primary Culture. PLoS One. 2016Mar7;11(3):e0150619 doi: 10.1371/journal.pone.0150619
15. Samir SM, Hassan HM, Elmowafy R, ElNashar EM, Alghamdi MA, AlSheikh MH, Al-Zahrani NS, Alasiri FM, Elhadidy MG. Neuroprotective effect of ranolazine improves behavioral discrepancies in a rat model of scopolamine-induced dementia. Front Neurosci. 2024Jan12;17:1267675 doi: 10.3389/fnins.2023.1267675
16. Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004Aug24;110(8):904-10 doi: 10.1161/01.CIR.0000139333.83620.5D
17. Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006 Jul;92 Suppl 4(Suppl 4):iv6-iv14. doi: 10.1136/hrt. 2005 078790
18. Ratte A, Wiedmann F, Kraft M, Katus HA, Schmidt C. Antiarrhythmic Properties of Ranolazine: Inhibition of Atrial Fibrillation Associated TASK-1 Potassium Channels. Front Pharmacol. 2019Nov26;10:1367 doi: 10.3389/fphar.2019.01367
19. Marchio P, Guerra-Ojeda S, Aldasoro M, Valles SL, Martín-Gonzalez I, Martínez-León JB, Mauricio MD, Vila JM. Relaxant and antiadrenergic effects of ranolazine in human saphenous vein. Eur J Cardiothorac Surg. 2020Aug1;58(2):277-285 doi: 10.1093/ejcts/ezaa034
20. Aldasoro M, Martínez C, Vila JM, Flor B, Lluch S. Endothelium-dependent component in the contractile responses of human omental arteries to adrenergic stimulation. Eur J Pharmacol. 1993Nov30;250(1):103-7 doi: 10.1016/0014-2999(93)90626-s
21. Aldasoro M, Martínez C, Vila JM, Medina P, Lluch S. Influence of endothelial nitric oxide on adrenergic contractile responses of human cerebral arteries. J Cereb Blood Flow Metab. 1996Jul;16(4):623-8 doi: 10.1097/00004647-199607000-00012
22. Borbujo J, García-Villalón AL, Valle J, Gómez B, Diéquez G. Postjunctional alpha-1 and alpha-2 adrenoceptors in human skin arteries. An in vitro study. J Pharmacol Exp Ther. 1989Apr;249(1):284-7
23. Gaballa MA, Peppel K, Lefkowitz RJ, Aguirre M, Dolber PC, Pennock GD, Koch WJ, Goldman S. Enhanced vasorelaxation by overexpression of beta 2-adrenergic receptors in large arteries. J Mol Cell Cardiol. 1998May;30(5):1037-45 doi: 10.1006/jmcc.1998.0668
24. Dessy C, Moniotte S, Ghisdal P, Havaux X, Noirhomme P, & Balligand J. L. (2004). Endothelial β3-adrenoceptors mediate nitric oxide-dependent vasorelaxation. Journal of Clinical Investigation, 114(3), 371-379. https://doi.org/10.1172/JCI200420613.
25. Virsolvy A, Farah C, Pertuit N, Kong L, Lacampagne A, Reboul C, Aimond F, Richard S. Antagonism of Nav channels and α1-adrenergic receptors contributes to vascular smooth muscle effects of ranolazine. Sci Rep. 2015Dec10;5:17969 doi: 10.1038/srep17969
26. Flenner F, Friedrich FW, Ungeheuer N, Christ T, Geertz B, Reischmann S, Wagner S, Stathopoulou K, Söhren KD, Weinberger F, Schwedhelm E, Cuello F, Maier LS, Eschenhagen T, Carrier L. Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovasc Res. 2016Jan1;109(1):90-102 doi: 10.1093/cvr/cvv247
27. Létienne R, Vié B, Puech A, Vieu S, Le Grand B, John GW. Evidence that ranolazine behaves as a weak beta1- and beta2-adrenoceptor antagonist in the rat [correction of cat] cardiovascular system. Naunyn Schmiedebergs Arch Pharmacol. 2001Apr;363(4):464-71 doi: 10.1007/s002100000378
28. Allely MC, Brown CM, Kenny BA, Kilpatrick AT, Martin A, Spedding M. Modulation of alpha 1-adrenoceptors in rat left ventricle by ischaemia and acyl carnitines: protection by ranolazine. J Cardiovasc Pharmacol. 1993Jun;21(6):869-73 doi: 10.1097/00005344-199306000-00004
29. Allen TJ, Chapman RA. Effects of ranolazine on L-type calcium channel currents in guinea-pig single ventricular myocytes. Br J Pharmacol. 1996May;118(2):249-54 doi: 10.1111/j.1476-5381.1996.tb15395.x
30. Moniotte S, Balligand J. L, & Gauthier, C. (2007). β3-adrenoceptor in the cardiovascular system: from ligand binding to therapeutic implications. Journal of Molecular and Cellular Cardiology, 42(4), 754-765. https://doi.org/10.1016/j. yjmcc. 2007 01.010
31. Dessy C, Moniotte S, Ghisdal P, Havaux X, Noirhomme P, Balligand JL. Endothelial beta3-adrenoceptors mediate vasorelaxation of human coronary microarteries through nitric oxide and endothelium-dependent hyperpolarization. Circulation. 2004Aug24;110(8):948-54 doi: 10.1161/01.CIR.0000139331.85766.AF
32. Rozec B, & Gauthier C. (2006). β3-adrenoceptors in the cardiovascular system: putative roles in human pathologies. Pharmacology & Therapeutics, 111(3), 652-673. https://doi.org/10.1016/j. pharmthera. 2005 12.002
33. Karimi Galougahi K, Liu CC, Garcia A, Gentile C, Fry NA, Hamilton EJ, Hawkins CL, Figtree GA. β3 Adrenergic Stimulation Restores Nitric Oxide/Redox Balance and Enhances Endothelial Function in Hyperglycemia. J Am Heart Assoc. 2016Feb19;5(2):e002824 doi: 10.1161/JAHA.115.002824
34. Balligand J. L. (2016). Cardiac β3-adrenergic receptors: from bench to bedside. Cardiovascular Research, 109(3), 323-334. https://doi.org/10.1093/cvr/cvw006.
35. Zhang M, Xu Y, Chen J, Qin C, Liu J, Guo D, Wang R, Hu J, Zou Q, Yang J, Wang Z, Niu X. Beta3-Adrenergic Receptor Activation Alleviates Cardiac Dysfunction in Cardiac Hypertrophy by Regulating Oxidative Stress. Oxid Med Cell Longev. 2021 Oct 4;2021:3417242. doi: 10.1155/2021/3417242. Retraction in: Oxid Med Cell Longev. 2025Oct16;2025:9832368 doi: 10.1155/omcl/9832368
36. Hutchinson DS, Summers RJ, Gibbs ME. Beta2- and beta3-adrenoceptors activate glucose uptake in chick astrocytes by distinct mechanisms: a mechanism for memory enhancement? J Neurochem. 2007Nov;103(3):997-1008 doi: 10.1111/j.1471-4159.2007.04789.x. Epub 2007 Aug 6. PMID: 17680985
37. Nisoli E, Tonello C, Landi M, & Carruba M. O. (1996). SR59230A antagonizes β3-adrenoceptor-mediated responses. British Journal of Pharmacology, 119(5), 981-986. https://doi.org/10.1111/j.1476-5381. 1996 tb15760.x
38. Arch J. R. S. (2002). β3-Adrenoceptor agonists and antagonists. British Journal of Pharmacology, 136(6), 781-788. https://doi.org/10.1038/sj.bjp.0704774.
39. Rozec B, Quang T. T, Noireaud, J, & Gauthier, C. (2009). Mixed β3-adrenergic receptor antagonist/β1-β2 partial agonist properties of SR59230A. British Journal of Pharmacology, 157(7), 1376-1384. https://doi.org/10.1111/j.1476-5381. 2009 00330.x
40. Robidoux J, Kumar N, Daniel KW, Moukdar F, Cyr M, Medvedev AV, Collins S. Maximal beta3-adrenergic regulation of lipolysis involves Src and epidermal growth factor receptor-dependent ERK1/2 activation. J Biol Chem. 2006Dec8;281(49):37794-802 doi: 10.1074/jbc.M605572200. Epub 2006 Oct 10. PMID: 17032647
41. Kathöfer S, Röckl K, Zhang W, Thomas D, Katus H, Kiehn J, Kreye V, Schoels W, Karle C. Human beta(3)-adrenoreceptors couple to KvLQT1/MinK potassium channels in Xenopus oocytes via protein kinase C phosphorylation of the KvLQT1 protein. Naunyn Schmiedebergs Arch Pharmacol. 2003Aug;368(2):119-26 doi: 10.1007/s00210-003-0772-x. Epub 2003 Jul 19. PMID: 12879210
42. Gao ZG, Gao RR, Meyer CK, Jacobson KA. A2B adenosine receptor-triggered intracellular calcium mobilization: Cell type-dependent involvement of Gi, Gq, Gs proteins and protein kinase C. Purinergic Signal. 2025Jun;21(3):499-513 doi: 10.1007/s11302-025-10070-1. Epub 2025 Feb 11. PMID: 39934472; PMCID: PMC12222587
43. Fan X, Gu X, Zhao R, Zheng Q, Li L, Yang W, Ding L, Xue F, Fan J, Gong Y, Wang Y. Cardiac β2-Adrenergic Receptor Phosphorylation at Ser355/356 Regulates Receptor Internalization and Functional Resensitization. PLoS One. 2016Aug19;11(8):e0161373 doi: 10.1371/journal.pone.0161373. PMID: 27541735; PMCID: PMC4991819.)
44. Hale SL, Shryock JC, Belardinelli L, Sweeney M, Kloner RA. Late sodium current inhibition as a new cardioprotective approach. J Mol Cell Cardiol. 2008Jun;44(6):954-967 doi: 10.1016/j.yjmcc.2008.03.019. Epub 2008 Apr 8. PMID: 18462746
45. Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011Aug;8(8):1281-90 doi: 10.1016/j.hrthm.2011.03.045
46. Murai K, Vasigh A, Alexy T, Tóth K, Czopf L. The Role of Ranolazine in the Treatment of Ventricular Tachycardia and Atrial Fibrillation: A Narrative Review of the Clinical Evidence. Biomedicines. 2024Jul26;12(8):1669 doi: 10.3390/biomedicines12081669
47. Deshmukh SH, Patel SR, Pinassi E, Mindrescu C, Hermance EV, Infantino MN, Coppola JT, Staniloae CS. Ranolazine improves endothelial function in patients with stable coronary artery disease. Coron Artery Dis. 2009Aug;20(5):343-7 doi: 10.1097/MCA.0b013e32832a198b
48. Nusca A, Bernardini F, Mangiacapra F, Maddaloni E, Melfi R, Ricottini E, Piccirillo F, Manfrini S, Ussia GP, Grigioni F. Ranolazine Improves Glycemic Variability and Endothelial Function in Patients with Diabetes and Chronic Coronary Syndromes: Results from an Experimental Study. J Diabetes Res. 2021Dec31;2021:4952447 doi: 10.1155/2021/4952447
49. Banerjee K, Ghosh RK, Kamatam S, Banerjee A, Gupta A. Role of Ranolazine in cardiovascular disease and diabetes: Exploring beyond angina. Int J Cardiol. 2017Jan15;227:556-564 doi: 10.1016/j.ijcard.2016.10.102
50. Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res. 2011Oct;64(4):381-92 doi: 10.1016/j.phrs.2011.06.018
51. Mosqueira D, Mannhardt I, Bhagwan JR, Lis-Slimak K, Katili P, Scott E, Hassan M, Prondzynski M, Harmer SC, Tinker A, Smith JGW, Carrier L, Williams PM, Gaffney D, Eschenhagen T, Hansen A, Denning C. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J. 2018Nov14;39(43):3879-3892 doi: 10.1093/eurheartj/ehy249
52. Fu Z, Zhao L, Chai W, Dong Z, Cao W, Liu Z. Ranolazine recruits muscle microvasculature and enhances insulin action in rats. J Physiol. 2013Oct15;591(20):5235-49 doi: 10.1113/jphysiol.2013.257246
53. Lisi D, Andrews E, Parry C, Hill C, Ombengi D, Ling H. The Effect of Ranolazine on Glycemic Control: a Narrative Review to Define the Target Population. Cardiovasc Drugs Ther. 2019Dec;33(6):755-761 doi: 10.1007/s10557-019-06917-6
54. Bell DSH, Goncalves E. Diabetogenic effects of cardioprotective drugs. Diabetes Obes Metab. 2021Apr;23(4):877-885 doi: 10.1111/dom.14295
55. Kahlig KM, Hirakawa R, Liu L, George AL Jr, Belardinelli L, Rajamani S. Ranolazine reduces neuronal excitability by interacting with inactivated states of brain sodium channels. Mol Pharmacol. 2014Jan;85(1):162-74 doi: 10.1124/mol.113.088492
Author contact
![]() Corresponding author: Dr. Martin Aldasoro. Department of Physiology. Faculty of Medicine. University of Valencia. email: martin.aldasoroes.
Corresponding author: Dr. Martin Aldasoro. Department of Physiology. Faculty of Medicine. University of Valencia. email: martin.aldasoroes.