Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(3):310-319. doi:10.7150/ijms.38602 This issue Cite
Research Paper
Hydrogen Sulfide Inhibits Homocysteine-Induced Neuronal Senescence by Up-Regulation of SIRT1
Xuan Kang1,2*, Cheng Li2,3*, Xi Xie2,4*, Ke-Bin Zhan2,4 ![]() , San-Qiao Yang1,2, Yi-Yun Tang2, Wei Zou2,5, Ping Zhang2,5, Xiao-Qing Tang1,2
, San-Qiao Yang1,2, Yi-Yun Tang2, Wei Zou2,5, Ping Zhang2,5, Xiao-Qing Tang1,2 ![]()
1. Institute of Neurology, the First Affiliated Hospital, University of South China, Hengyang, 42100, Hunan, P.R. China.
2. Institute of Neuroscience, Hengyang Medical College, University of South China, Hengyang, 42100, Hunan, P.R. China.
3. Department of Emergency, Affiliated Nanhua Hospital, University of South China, Hengyang, 421001, Hunan, P. R. China.
4. Department of Neurology, the Second Affiliated Hospital, University of South China, Hengyang, 421001, Hunan, P.R. China.
5. Department of Neurology, Affiliated Nanhua Hospital, University of South China, Hengyang, 421001, Hunan, P. R. China.
*Xuan Kang, Cheng Li and Xi Xie contributed equally to this work.
Received 2019-7-21; Accepted 2019-12-8; Published 2020-1-17
Abstract
Homocysteine (Hcy) accelerates neuronal senescence and induces age-related neurodegenerative diseases. Silence signal regulating factor 1 (SIRT1) prolongs lifespan and takes neuroprotective effects. We have previously demonstrated that hydrogen sulfide (H2S) prevents Hcy-induced apoptosis of neuronal cells and has neuroprotective effect. In the present work, we aimed to investigate whether H2S protects HT22 cells against Hcy-induced neuronal senescence and whether SIRT1 mediates this role of H2S. We found that Hcy induced cellular senescence in HT22 cells, as determined by β-galactosidase staining, expressions of P16INK4a, P21CIPL, and trypan blue Staining, which are the markers of cellular senescence. However, sodium hydrosulfide (NaHS, the donor of H2S) significantly reversed Hcy-induced cellular senescence. Interestingly, NaHS not only up-regulated the expression of SIRT1 in HT22 cells but also reversed Hcy-downregulated the expression of SIRT1 in HT22 cells. Furthermore, we found that pretreatment with Sirtinol (an inhibitor of SIRT1) markedly reversed the protection of NaHS against Hcy-induced HT22 cells senescence and apoptosis. Our findings illustrated that H2S protects HT22 cells against Hcy-induced senescence by up-regulating SIRT1.
Keywords: cell senescence, homocysteine, hydrogen sulfide, SIRT1
Introduction
Homocysteine (Hcy) is a sulfur-containing nonprotein amino acid derive from methionine metabolism [1]. Hcy promotes neuronal degeneration and thus it contributes to age-related neurodegenerative diseases, such as dementia, Alzheimer's disease (AD), Parkinson's disease(PD), and stroke [2,3]. The emerging evidence suggests that a potentially important contributor to aging and age-related neurodegenerative diseases is cellular senescence [4], a process that imposes permanent proliferative arrest on cells responding to various stressors [5]. Therapeutic strategies that safely interfere with the detrimental effects of cellular senescence are gaining significant attention [4,6]. Interestingly, accumulating evidence showed that cellular senescence is causally implicated in Hcy-generated age-related diseases [7-9]. Thus, finding a new strategy to antagonize Hcy-induced neuronal senescence has important value in the prevention and treatment of age-related neurodegenerative diseases.
Hydrogen sulfide (H2S) is a novel gaseous molecule with an extremely unpleasant odor. In the brain, H2S is an important neuroprotective agent and has therapeutic potential in neurodegenerative diseases of aging, such as AD and PD [10,11]. Recently, H2S was identifiedas a new approach to prolong lifespan [12]. H2S inhibits the production of mitochondrial ROS [13], decreases oxidative stress [14,15], and repairs DNA damage [16] to protect cells from senescence. Previous studies revealedthat H2S protects endothelial cells against nicotinamide- [17] and H2O2- [18] induced cellular senescence. Our previous study indicated that H2S prevents Hcy-induced neurotoxicity [19,20]. In this study, we explored whether ameliorating Hcy-induced neuronal senescence contributes to the protection of H2S against Hcy-induced neurotoxicity.
Silent mating type information regulator 2 homolog 1 (SIRT1) is a NAD+-dependent deacetylase of lysine residue of the target protein. SIRT1 extends lifespan [21] and improves cell tolerance to inhibit environmental stress [22,23]. Recently, it has shown that H2S extends lifespan by activation of SIRT1 [12]. Furthermore, Our previous data demonstrated that H2S up-regulates the expression of SIRT1 in PC12 cells [20]. Thus, we further investigated whether SIRT1 mediates the inhibitory role of H2S in Hcy-induced neuronal senescence.
The present studies demonstrated the ability of H2S to inhibit Hcy-induced senescence in HT-22 cells and identified the mediatory role of SIRT1 in the protective action of H2S from Hcy-induced neuronal senescence. It provides novel strategy to prevent Hcy-induced neurotoxicity via inhibition of senescence-associated neuronal aging. Thus, our research identifies SIRT-1 as a potent therapeutic target and H2S as a potent treatment for Hcy-related neurodegenerative diseases.
Materials and Methods
Materials
Sodium hydrosulfide, homocystein, sirtinol, dimethyl sulfoxide (DMSO), and propidium iodide (PI) were purchased from Sigma (St. Louis, MO, USA). Rh123 was supplied by Dojindo Molecular Technologies, Inc. (Rockvile, MD, USA). Specific monoclonal anti-SIRT1 antibody was obtained from Abcam (Hong Kong, China). Specific monoclonal antibodies of P16INK4a, P21CIPL were purchased from OriGene Biotech Inc. (Burlingame, UK). Annexin V was bought from Nanjing Key GEN Biotech Co., Ltd. (Nanjing, China). DMEM medium, horse serum and fetal bovine serum were supplied by Gibico, BRL (Ground Island, NY, USA).
Cell viability assay
Cell viability was detected by Cell Counting Kit-8 (CCK-8) assay. HT22 Cells of logarithmic phase growth were seeded into a 96-well plate at a density of 1 × 104 cells/well. The cells were treated with 2.5, 5.0, 10.0 mM Hcy, or/and 100, 200, 400 μmol/L NaHS and routinely incubated for 24 hours, 48 hours, and 72 hours. After treatment, viable cells were stained with 5μL of CCK-8 solution (CCK-8, Sigma, USA) and incubated at 37°C for 4 h. Absorbance was measured at 450 nm by a microplate reader (ELX-800, BIO-TEK, USA). The experiment was performed three times.
β-galactosidase (SA-β-gal) staining assay
Senescent cells were detected by the SA-β-gal staining. HT22 Cells were seeded into a 6-well plate at a density of 1 × 104 cells/well. After incubated with 2ml 10% Dulbecco's Modified Eagle Medium (DMEM, Gibco, USA) for 24h, HT22 cells were treated with various concentrations of 2.5, 5.0, 10.0 mM Hcy, or/and 100, 200, 400 μmol/L NaHS for 48 h. The plates were stained daily using Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, Danvers, MA, 9860) following the manufacturer's recommendations. All wells were counted in five randomized fields.
Trypan blue Staining
The cells were grew at concentration of 1 × 104 cells/ml in 24 well-plates to 60-70% confluence by incubation for 24 h, then treated with 2.5mM, 5.0mM, 10mM Hcy or and 100, 200, 400 μmol/L NaHS for 24 h, 48 h, 72 h. Floating and adhering cells were collected, washed once with PBS (pH 7.4), centrifuged, suspended in 10% DMEM and stained with 0.4% trypan blue (sigma, St. Louis, MO, USA) at room temperature. About 10 μl of sample was loaded on hemocytometer chamber and numbers of blue cells and non-blue cells were counted under a light microscope (Mike audi, China). The results were expressed as percentage of the control.
Western blotting
Cells of logarithmic phase growth were plated in 50ml culture bottle. After incubated with DMEM for 24h, the cells were treated with Hcy or/ and NaHS at different concentrations for 48 h. Then the treated cells were resuspended in 100 uM cell lysis buffers and PMSF incubation on ice for 30 min. The supernatant was collected after centrifuged at 12,000 rpm for 10 min at 4˚C. The protein concentrations were detected by BCA method (Solarbio, Beijing, China). Equal amounts of total protein extracts were electrophoresed through 10% or 12% SDS-PAGE gel, then transferred to polyvinylidene difluoride membranes (Solarbio, China). After blocked with TBST (50 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 0.1% Tween-20) containing 5% BSA (Sigma, USA) for 2 h, the membranes were incubated with primary antibodies including monoclonal antibody for P16INK4a, P21CIPL, and SIRT1(dilution, 1:1,000) at 4˚C overnight. After washed with TBST for 5 min 5 times, the membranes were incubated with secondary antibodies including Goat anti-rabbit (Proteintech, USA) (dilution, 1: 5,000) at room temperature for 2 h. After washed for three times with TBST for 3 times, the membranes were visualized with Western Blotting Chemiluminescence Reagent (Solarbio, Beijing, China), followed by apposition of the membranes with autoradiographic films (Kodak, China). The expression of β-actin for each sample was used as a control.
Flow cytometry analysis
HT22 cells of the log phase were grew in 6-well plates and treated with Hcy or NaHS for 48h. Cells were stained 50 μg/ml propidium iodide (PI) (Roche, Mannheim, Germany) and 10 μg/ml RNase A (Sigma, St. Louis, MO, USA). Percentages of cells existing within the various phases (G0/G1, S, G2/M) of the cell cycle were calculated by the mean fluorescence intensity.
Statistical analysis
Experiments were repeated at least three times. Values were expressed as mean ± standard error of the mean (S.E.M.). All data were analyzed by SPSS version 21.0. Data were evaluated for statistical significance with using one-way analysis of variance (ANOVA) followed by LSD post hoc tests. P < 0.05 was considered to indicate a statistically significant difference.
Results
Hcy induced the cellular senescence in HT22 cells
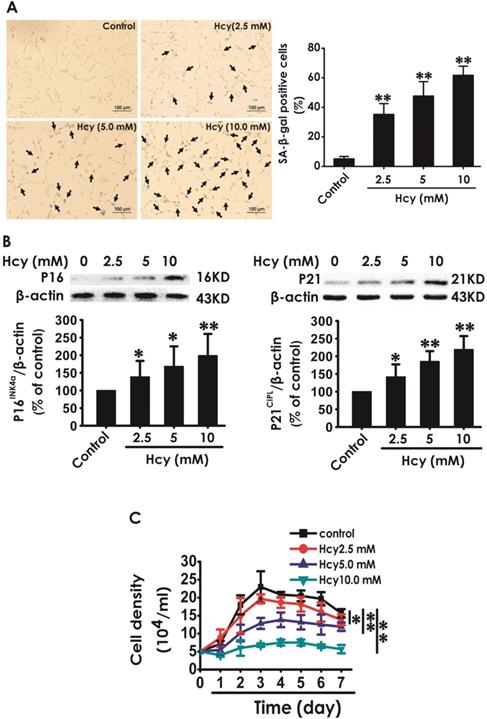
We first explored whether Hcy induces cellular senescence in HT22 cells. After treatment with Hcy (2.5, 5, 10 mM) for 48 h, the percentage of senescence-associated beta-galactosidase (SA-β-Gal)-positive cells was increased (Fig. 1A), the expressions of P16INK4a and P21CIPL were upregulated (Fig. 1B), and the cell density was decreased (Fig. 1C) in HT22 cells, which indicated that Hcy induces the cellular senescence in HT22 cells.
Effect of Hcy on the cellular senescence in HT22 cells. A, HT22 cells were stained with SA-β-gal and the SA-β-gal positive cell was quantitatively analyzed (magnification: × 10; black arrows point the SA-β-gal staining positive cells). B, the expressions of P16INK4a and P21CIPL in HT22 cells were measured by western blotting. C, the cell density was determined by trypan blue analysis and the growth curve for 7 d was drawn. Values are means ± SEM (n = 3). *P<0.05, **P<0.01, vs control group.
H2S prevented Hcy-induced cellular senescence in HT22 cells
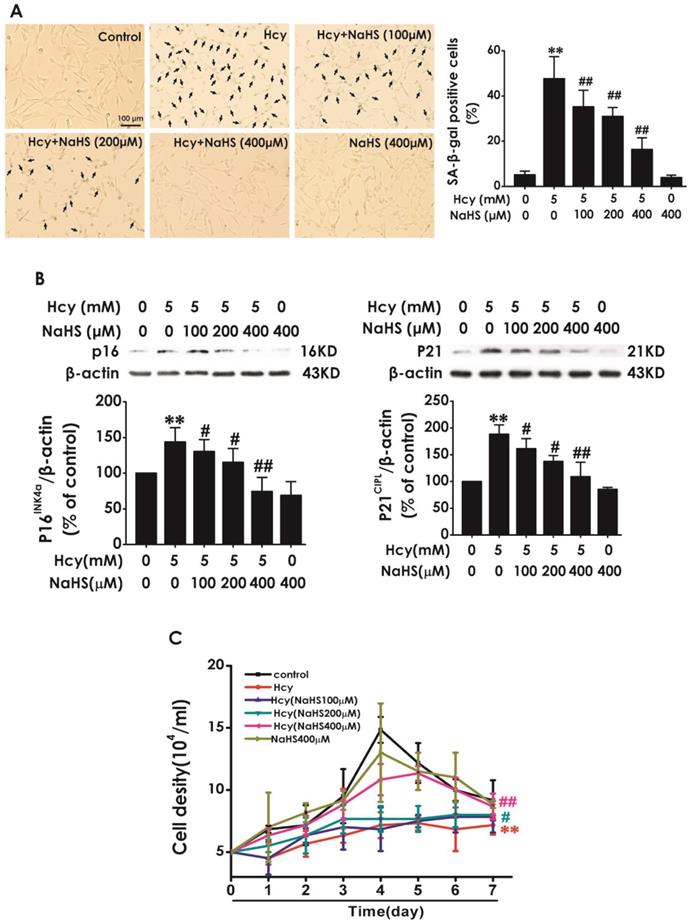
Next, we explored the effect of H2S on Hcy-induced cellular senescence in HT22 cells. HT22 cells were pretreated with NaHS (100, 200, and 400 μM) for 30 min and then cotreated with 5 mM Hcy for 48 h. We found that pretreatment of NaHS (100, 200, or 400 mM) significantly decreased the percentage of SA-β-gal-positive cells (Fig. 2A) and the expressions of P16INK4a and P21CIPL (Fig. 2B), while increased the cell density (Fig. 2C) in Hcy-treated HT22 cells. These results demonstrated that H2S prevented Hcy-induced cellular senescence in HT22 cells.
Effect of H2S on Hcy-induced cellular senescence in HT22 cells. A, HT22 cells were stained with SA-β-gal and the SA-β-gal positive cell was quantitatively analyzed (magnification: × 10; black arrows point the SA-β-gal staining positive cells). B, the expressions of P16INK4a and P21CIPL in HT22 cells were measured by western blotting. C, the cell density was determined by trypan blue analysis and the growth curve for 7 d was drawn. Values are means ± SEM (n = 3). **P<0.01, vs control group; #P<0.05, #P<0.01, vs Hcy-treated group.
NaHS upregulated the expression of SIRT1 in HT22 cells
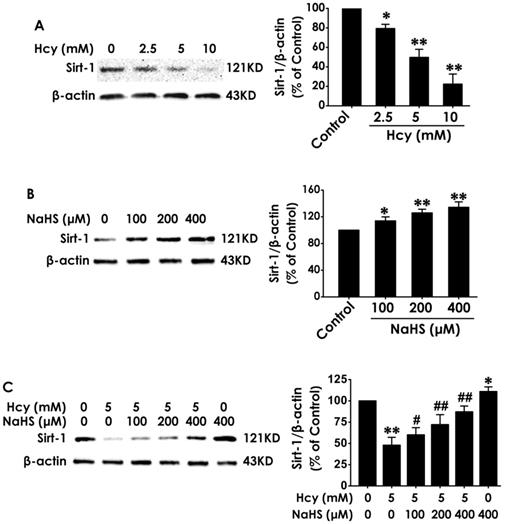
To explore the mediatory role of SIRT1 in the protection of H2S against Hcy-induced cellular senescence in HT22 cells, we first investigated the expression of SIRT1 in different treated HT22 cells. After the expression of SIRT1 in HT22 cells was markedly down-regulated by treatment with Hcy (2.5, 5.0, 10.0 mM) for 48 h (Fig. 3A), while was up-regulated by treatment with NaHS (100, 200, and 400 μM) alone for 48 h (Fig. 3B). Furthermore, preteatment with NaHS (100, 200, and 400 μM) restored the expression of SIRT1 in Hcy-treated HT22 cells (Fig. 3C). These results suggest that NaHS not only upregulated the expression of SIRT1 in HT22 cells but also reversed the down-regulation of SIRT1 in Hcy-treated HT22 cells.
Effects of Hcy and NaHS on the expression of SIRT1 in HT22 cells. The expressions of SIRT1 in HT22 cells treated with 48-h Hcy (2.5, 5.0, 10.0 mM) alone (A), 48-h NaHS (100, 200, 400 μmol/L) alone (B), or 48- h cotreatment with Hcy (5.0) and NaHS (100, 200, 400 μmol/L) (C) were detected by western blotting. Values are means ± SEM (n = 3), *P<0.05, **P<0.01, vs control group; #P<0.05, ##P<0.01, vs Hcy-treated alone group.
Sirtinol reversed the protection of NaHS against Hcy-induced cellular senescence in HT22 cells
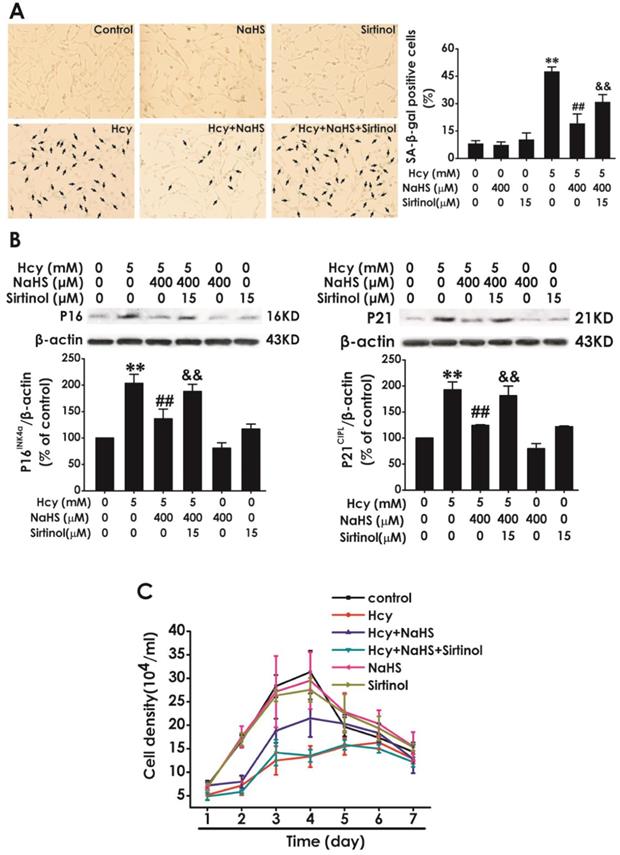
To further confirm whether SIRT1 mediates the protection of NaHS against Hcy-induced cellular senescence in HT22 cells, we explored whether sirtinol, a specific inhibitor of SIRT1, reversed this protective role of NaHS. We found that pretreatment with sirtinol (15 μM, for 30 min) increased the percentage of SA-β-gal-positive cells (Fig. 4A) as well as the expressions of P16INK4a, P21CIPL (Fig. 4B), while decreased the cell density (Fig. 4C) in HT22 cells cotreated with 5 mM Hcy and 400 μM NaHS for 48 h. These findings verified that sirtinol reverses the protection of NaHS against Hcy-induced cellular senescence in HT22 cells.
Effect of Sirtinol on NaHS-attenuated cellular senescence in Hcy-exposed HT22 cells. A, HT22 cells were stained with SA-β-gal and the SA-β-gal positive cell was quantitatively analyzed (magnification: × 10; black arrows point the SA-β-gal staining positive cells). B, the expressions of P16INK4a and P21CIPL in HT22 cells were measured by western blotting. C, the cell density was determined by trypan blue analysis and the growth curve for 7 d was drawn. Values are mean ± SEM (n = 3). **P<0.01, vs control group; ##P<0.01, vs Hcy-treated alone group; &P<0.05 && P<0.01 vs cotreatment with Hcy and NaHS group.
Sirtinol reversed the protective effect of H2S on Hcy-indcued neurotoxicity to HT22 cells
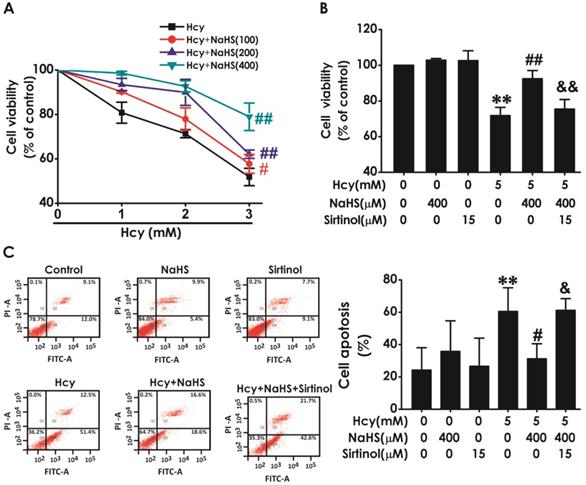
We have previously demonstrated that H2S inhibits Hcy-induced neurotoxicity. Therefore, we next investigated whether blockage of SIRT1 reverses the protective effect of H2S against Hcy-induced neurotoxicity to HT22 cells. As shown in Fig. 5A, pretreatment of NaHS (100, 200, or 400 μM) significantly increased the cell viability of HT22 cells exposed to 5 mM of Hcy for 48 h, which indicated that NaHS inhibites Hcy-induced cytotoxicity to HT22 cells. However, pretreatment with sirtinol (15 μM) markedly decreased the cell viability of HT22 cells cotreated with Hcy (5 mM) and NaHS (400 μM)(Fig. 5B), which indiccated that sirtinol reverses the protection of NaHS against Hcy-induced cytotoxicity to HT22 cells. In addition, pretreatment of NaHS (400 μM, for 30 min) significantly decreased the apoptosis in HT22 cells treated with 5 mM of Hcy for 48 h, while pretreatment with sirtinol (15 μM, for 30 min) increased the apoptosis in HT22 cells cotreated with Hcy and NaHS (Fig. 5C), which indiccated that sirtinol reverses the protection of NaHS against Hcy-induced apoptosis to HT22 cells. These results demonstrated that inhibition of SIRT1 reverses the protection of H2S against Hcy-induced neurotoxicity in HT22 cells.
Effect of Sirtinol on the protection of H2S against Hcy-induced neurotoxicity to HT22 cells. A, the cells were treated with 2.5, 5.0, 10.0 mM Hcy, and 100, 200, 400 μmol/L NaHS and routinely incubated for 48 hours. B-C, after preincubated with Sirtinol (15 mM) for 30 min, HT22 cells were pretreated with NaHS (400 uM) for 30 min, then cotreated with Hcy (5M) for 48h. A-B, the cell viability was determined by CCK-8 assay. C, the apoptosis of HT22 cells was assessed by flow cytometry after PI and Annexin V double staining (The annexin-V2/PI2 population is made up of normal healthy cells, while annexin-V+/PI2 cells exist in early apoptotic stage, and annexin-V+/PI+ cells exist in late apoptotic stage). Values are the mean ± SEM (n = 5). *P<0.05 **P<0.01 vs control group; #P<0.05 ##P<0.01 vs Hcy-treated alone group; &P<0.05 vs cotreated with NaHS and Hcy group.
Discussion
Our previous studies have demonstrated that H2S has a protective effect against Hcy-evoked neurotoxicity [20,24-26]. Considering the cellular senescence is prominent in the neurotoxicity of Hcy [3,27-29], the present work was designed to explore whether the protection of H2S against the neurotoxicity of Hcy is associated with regulating neuronal senescence. The main findings of the present work are the following: (i) H2S suppressed Hcy-induced cellular senescence in HT22; (ii) H2S up-regulated the expression of SIRT1 in Hcy-exposed HT22 cells. (iii) Inhibition of SIRT1 reversed the protective effect of H2S against Hcy-induced senescence and neurotoxicity in HT22 cells. Together, we demonstrated that the protection of H2S against Hcy-induced neurotoxicity involves inhibition of neuronal senescence through upregulating SIRT1 signaling.
Cellular senescence, a process that imposes permanent proliferative arrest on cells in response to various stressors, has emerged as a potentially important contributor to aging and age-related disease, and it represents an attractive target for therapeutic exploitation [4]. Hcy is an independent risk factor for neurological and cardiovascular disease [30,31]. The level of Hcy in bodies increases with age [32]. Therefore, understanding whether Hcy induces neuronal senescence is necessary for a greater understanding of the neurotoxicity of Hcy, which may be part of the mechanisms leading to age-relate neurological diseases [33,34]. SA-β-Gal staining remains as the most widely used biomarker for cellular senescence [35,36]. P21CIPL is a cyclin-dependent kinase inhibitor, with regulating cell cycle progression at G1 and S phase [37]. P16INK4a plays a critical role on controlling cellular senescence [38]. The present work demonstrated that the percentage of SA-β-Gal positive cells and the expressions of age-related markers P16INK4a as well as P21CIPL were significantly increased in Hcy-treated HT22 cells. Furthermore, we found that Hcy led to cell growth arrest. These data concluded that Hcy induces cellular senescence in HT22 cells. Therefore, modulation of neuronal senescence might represent a novel therapeutic strategy to overcome Hcy-induced neurotoxicity.
H2S, a well-known regulator of inflammation [39], ER stress [40] and cytotoxicity [41], is recognized as a new approach to prolong lifespan [12]. H2S has been described to prevent endothelial cell senescence [18,42] and delays aging [43]. Furthermore, we have previously found that H2S inhibits formaldehyde-induced cellular senescence [44] and protects against Hcy-induced neurotoxicity [25]. Therefore, it is imperative to assess the effect of H2S on Hcy-induced cellular senescence. In the present work, we found that H2S increased the growth curve and decreased the percentage of SA-β-Gal positive cells as well as the expressions of P16INK4a and P21CIPL in Hcy-treated HT22 cells. Therefore, we for the first demonstrated that H2S prevents Hcy-induced cellular senescence in HT22 cells. It has been confirmed that H2S prevents the process of senescence in the endothelium [42], kidney [45,46], vascular [47], heart [48] and brain [43] of mice. Furthermore, increasing the content of endogenous H2S by proper diet extends the life span of the aged mice [49,50]. These previous findings offered a reasonable explanation for the results obtained in the present study. Therefore, the regulation of neuronal senescence offers insights into the protection of H2S against the neurotoxicity of Hcy. In our previous work, we used PC12 cells as a cell model to explore the protection of H2S against Hcy-induced endoplasmic reticulum (ER) stress [20]. This previous work focused on the protective role of H2S in Parkinson's disease. Thus, we used PC12 cells to explore the protection of H2S against Hcy-induced ER stress because PC12 cells are dopaminergic neurons and are commonly used as the cell model of PD. In the present study, the purpose is to explore the effect of H2S on Hcy-induced impairment in learning and memory. Learning and memory are closely related to hippocampus [51]. Thus, HT22 cells were used in the present work because they are hippocampal neurons [52]. Hcy may be also induced ER stress in HT22 cells. In our future, we will explore the role of ER stress in Hcy-induced neuronal senescence in HT22 cells.
The present study also investigated the possible underlying mechanism for the protective role of H2S in Hcy-induced senescence. Sirtuin was regarded as the lifespan-extending gene in the past years [53]. SIRT1, an important member of sirtuin family, also takes an important role in delaying cellular senescence and extending longevity [29,54]. Numerous studies have demonstrated that SIRT1 ameliorate neurodegenerative disease, such as AD [55], PD [56], subarachnoid hemorrhage [57], which reveal that SIRT1 delays senescence in the brain. Our previous study demonstrated that SIRT1 mediates the protective effect of H2S on Hcy-induced neurotoxicity [20]. Therefore, we speculated whether SIRT1 mediates the protection of H2S against Hcy-induced senescence in HT22 cells. How might Hcy mediate the blockade of SIRT1? Previous study suggests that Hcy increases ROS generation by activating protein kinase C-β (PKCβ), which in turn improves SIRT1 degradation through a proteasome-dependent mechanism [58]. In line with this finding, our present work demonstrated that Hcy downregulated the expression of SIRT1 in HT22 cells. Notably, we found that H2S not only increased the expressions of SIRT1 in HT22 cells, but also reversed Hcy-reduced the expressions of SIRT1 in HT22 cells. These results implied that the upregulation of SIRT1 contributed to the protective effect of H2S on Hcy-induced senescence. To further confirm whether the SIRT1 mediates the protection of H2S against Hcy-induced senescence, we explored whether the blockage of SIRT1 abolishes this protection of H2S. We utilized Sirtinol as the inhibitor of SIRT1, which get a broaden acceptance [59]. Our results showed that inhibited SIRT1 by Sirtinol abolished the protection of H2S against Hcy-induced increase in the percentage of SA-β-Gal positive cells, up-regulations of P16INK4a and P21CIP1, and the arrest of cell growth. Taken together, these results indicated that SIRT1 mediates H2S-exerted protection against Hcy-induced senescence in HT-22 cells. Simultaneously, the blockage of SIRT1 also eliminated the protection of H2S against Hcy-induced neurotoxicity, which further demonstrated that the involvement of reduced cellular senescence in the H2S-exerted protection against the neurotoxicity of Hcy. The possible molecular mechanism of H2S action on SIRT1 is unknown. A recent study has described that H2S increases intracellular NAD+ levels that are known to fuel SIRT1 activity [60] and that SIRT1 increases phosphorylation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), which may be related to the regulation of anti-apoptosis and stress response [61,62]. Although the precise molecular mechanisms of H2S action on SIRT1 remain to be determined, the most promising candidates are NAD+ or the pathway of PKCβ-ROS. Future works will be required to precisely define whether NAD+ and ERK1/2 implicate in the mechanisms underlying the regulatory role of H2S in the action of SIRT1.
Conclusion
In this study, we demonstrated that treatment with NaHS, a donor of H2S, attenuated Hcy-induced cellular senescence and upregulated SIRT1 expression in Hcy-exposed HT22 cells. Furthermore, we showed the reversing role of inhibited SIRT1 in the protection of NaHS against Hcy-induced cellular senescence and neurotoxicity. These data suggested that H2S inhibits Hcy-induced cellular senescence by upregulation of SIRT1. Our results provide important insights into the molecular mechanism underlying H2S-exerted protection against Hcy-induced neurotoxicity and provide a basis for investigating H2S as a therapeutic approach for Hcy-related neurodegenerative disease.
Abbreviations
Hcy: homocysteine; SIRT1: silence signal regulating factor 1; H2S: hydrogen sulfide; NaHS: sodium hydrosulfide; AD: Alzheimer's disease; PD: Parkinson's disease; DMSO: dimethyl sulfoxide; PI: propidium iodide; CCK-8: cell counting kit-8; DMEM: Dulbecco's Modified Eagle Medium; SA-β-Gal: senescence-associated beta-galactosidase.
Acknowledgements
This study was supported by National Natural Science Foundation of China (81671057, 81771178) and Hunan Provincial Natural Science Foundation of China (2019JJ50546).
Author Contributions
Xiao-Qing Tang and Ke-Bin Zhan conceived and designed the study. Xuan Kang, Cheng Li, Xi Xie performed the experiments. San-Qiao Yang, Yi-Yun Tang, Wei Zou and Ping Zhang conducted the analysis. Xuan Kang and Cheng Li wrote the first version of the manuscript. Xiao-Qing Tang and Xuan Kang have maken necessary modifications to the manuscript. All authors have seen and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Prudova A, Bauman Z, Braun A. et al. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Natl Acad Sci U S A. 2006;103:6489-6494
2. Wei Z, Tiandong W, Yang L. et al. Parkinson's Disease and Homocysteine: A Community-Based Study in a Folate and Vitamin B12 Deficient Population. Parkinsons Dis. 2016;2016:9539836
3. McCully KS. Homocysteine Metabolism, Atherosclerosis, and Diseases of Aging. Compr Physiol. 2015;6:471-505
4. Childs BG, Durik M, Baker DJ. et al. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424-1435
5. Campisi J & d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729-740
6. Childs BG, Gluscevic M, Baker DJ. et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16:718-735
7. Zhang D, Sun X, Liu J. et al. Homocysteine accelerates senescence of endothelial cells via DNA hypomethylation of human telomerase reverse transcriptase. Arterioscler Thromb Vasc Biol. 2015;35:71-78
8. Zhu JH, Chen JZ, Wang XX. et al. Homocysteine accelerates senescence and reduces proliferation of endothelial progenitor cells. J Mol Cell Cardiol. 2006;40:648-652
9. McCully KS. Review: Chemical Pathology of Homocysteine VI. Aging, Cellular Senescence, and Mitochondrial Dysfunction. Ann Clin Lab Sci. 2018;48:677-687
10. Cao X, Cao L, Ding L. et al. A New Hope for a Devastating Disease: Hydrogen Sulfide in Parkinson's Disease. Mol Neurobiol. 2018;55:3789-3799
11. Xuan A, Long D, Li J. et al. Hydrogen sulfide attenuates spatial memory impairment and hippocampal neuroinflammation in beta-amyloid rat model of Alzheimer's disease. J Neuroinflammation. 2012;9:202
12. Qabazard B & Sturzenbaum SR. H2S: A New Approach to Lifespan Enhancement and Healthy Ageing? Handb Exp Pharmacol. 2015;230:269-287
13. Olson KR, Gao Y, Arif F. et al. Metabolism of hydrogen sulfide (H2S) and Production of Reactive Sulfur Species (RSS) by superoxide dismutase. Redox Biol. 2018;15:74-85
14. Yang G, Zhao K, Ju Y. et al. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18:1906-1919
15. Xie ZZ, Shi MM, Xie L. et al. Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid Redox Signal. 2014;21:2531-2542
16. Zhao K, Ju Y, Li S. et al. S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 2014;15:792-800
17. Zheng M, Qiao W, Cui J. et al. Hydrogen sulfide delays nicotinamide-induced premature senescence via upregulation of SIRT1 in human umbilical vein endothelial cells. Mol Cell Biochem. 2014;393:59-67
18. Suo R, Zhao ZZ, Tang ZH. et al. Hydrogen sulfide prevents H(2)O(2)-induced senescence in human umbilical vein endothelial cells through SIRT1 activation. Mol Med Rep. 2013;7:1865-1870
19. Tang XQ, Shen XT, Huang YE. et al. Inhibition of endogenous hydrogen sulfide generation is associated with homocysteine-induced neurotoxicity: role of ERK1/2 activation. J Mol Neurosci. 2011;45:60-67
20. Wang CY, Zou W, Liang XY. et al. Hydrogen sulfide prevents homocysteineinduced endoplasmic reticulum stress in PC12 cells by upregulating SIRT1. Mol Med Rep. 2017;16:3587-3593
21. Giblin W, Skinner ME & Lombard DB. Sirtuins: guardians of mammalian healthspan. Trends in genetics: TIG. 2014;30:271-286
22. Maiese K. New Insights for Oxidative Stress and Diabetes Mellitus. Oxid Med Cell Longev. 2015;2015:875961
23. Fujitsuka N, Asakawa A, Morinaga A. et al. Increased ghrelin signaling prolongs survival in mouse models of human aging through activation of sirtuin1. Mol Psychiatry. 2016;21:1613-1623
24. Tang XQ, Shen XT, Huang YE. et al. Hydrogen sulfide antagonizes homocysteine-induced neurotoxicity in PC12 cells. Neurosci Res. 2010;68:241-249
25. Wei HJ, Xu JH, Li MH. et al. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol Sin. 2014;35:707-715
26. Li M, Zhang P, Wei HJ. et al. Hydrogen Sulfide Ameliorates Homocysteine-Induced Cognitive Dysfunction by Inhibition of Reactive Aldehydes Involving Upregulation of ALDH2. Int J Neuropsychopharmacol. 2017;20:305-315
27. Khuzakhmetova V, Yakovleva O, Dmitrieva S. et al. Prenatal hyperhomocysteinemia induces oxidative stress and accelerates 'aging' of mammalian neuromuscular synapses. Int J Dev Neurosci. 2019;75:1-12
28. Bossenmeyer-Pourie C, Smith AD, Lehmann S. et al. N-homocysteinylation of tau and MAP1 is increased in autopsy specimens of Alzheimer's disease and vascular dementia. J Pathol. 2019;248:291-303
29. Ostrakhovitch EA & Tabibzadeh S. Homocysteine and age-associated disorders. Ageing Res Rev. 2019;49:144-164
30. Rozycka A, Jagodzinski PP, Kozubski W. et al. Homocysteine Level and Mechanisms of Injury in Parkinson's Disease as Related to MTHFR, MTR, and MTHFD1 Genes Polymorphisms and L-Dopa Treatment. Curr Genomics. 2013;14:534-542
31. Bonetti F, Brombo G & Zuliani G. The relationship between hyperhomocysteinemia and neurodegeneration. Neurodegener Dis Manag. 2016;6:133-145
32. Ma F, Wu T, Zhao J. et al. Plasma Homocysteine and Serum Folate and Vitamin B12 Levels in Mild Cognitive Impairment and Alzheimer's Disease: A Case-Control Study. Nutrients. 2017:9
33. Herrmann W & Knapp JP. Hyperhomocysteinemia: a new risk factor for degenerative diseases. Clin Lab. 2002;48:471-481
34. Hainsworth AH, Yeo NE, Weekman EM. et al. Homocysteine, hyperhomocysteinemia and vascular contributions to cognitive impairment and dementia (VCID). Biochim Biophys Acta. 2016;1862:1008-1017
35. Itahana K, Campisi J & Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21-31
36. Geng YQ, Guan JT, Xu XH. et al. Senescence-associated beta-galactosidase activity expression in aging hippocampal neurons. Biochem Biophys Res Commun. 2010;396:866-869
37. Ma WD, Xu SR, Yan YL. et al. Expressions of cyclin B1 and p21cipl in adult acute leukemia and their correlation. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2005;13:751-758
38. Hall BM, Balan V, Gleiberman AS. et al. p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). 2017;9:1867-1884
39. Bhatia M. H2S and Inflammation: An Overview. Handb Exp Pharmacol. 2015;230:165-180
40. George AK, Behera J, Kelly KE. et al. Hydrogen sulfide, endoplasmic reticulum stress and alcohol mediated neurotoxicity. Brain Res Bull. 2017;130:251-256
41. Guo R, Wu K, Chen J. et al. Exogenous hydrogen sulfide protects against doxorubicin-induced inflammation and cytotoxicity by inhibiting p38MAPK/NFkappaB pathway in H9c2 cardiac cells. Cell Physiol Biochem. 2013;32:1668-1680
42. Latorre E, Torregrossa R, Wood ME. et al. Mitochondria-targeted hydrogen sulfide attenuates endothelial senescence by selective induction of splicing factors HNRNPD and SRSF2. Aging (Albany NY). 2018;10:1666-1681
43. Zhan JQ, Zheng LL, Chen HB. et al. Hydrogen Sulfide Reverses Aging-Associated Amygdalar Synaptic Plasticity and Fear Memory Deficits in Rats. Front Neurosci. 2018;12:390
44. Zhu WW, Ning M, Peng YZ. et al. Hydrogen Sulfide Inhibits Formaldehyde-Induced Senescence in HT-22 Cells via Upregulation of Leptin Signaling. Neuromolecular Med. 2019;21:192-203
45. Hou CL, Wang MJ, Sun C. et al. Protective Effects of Hydrogen Sulfide in the Ageing Kidney. Oxid Med Cell Longev. 2016;2016:7570489
46. Lee HJ, Feliers D, Barnes JL. et al. Hydrogen sulfide ameliorates aging-associated changes in the kidney. Geroscience. 2018;40:163-176
47. Das A, Huang GX, Bonkowski MS. et al. Impairment of an Endothelial NAD(+)-H2S Signaling Network Is a Reversible Cause of Vascular Aging. Cell. 2018;173:74-89 e20
48. Ma N, Liu HM, Xia T. et al. Chronic aerobic exercise training alleviates myocardial fibrosis in aged rats through restoring bioavailability of hydrogen sulfide. Can J Physiol Pharmacol. 2018:96 902-908
49. Yoshida S, Yamahara K, Kume S. et al. Role of dietary amino acid balance in diet restriction-mediated lifespan extension, renoprotection, and muscle weakness in aged mice. Aging Cell. 2018;17:e12796
50. Hine C, Zhu Y, Hollenberg AN. et al. Dietary and Endocrine Regulation of Endogenous Hydrogen Sulfide Production: Implications for Longevity. Antioxid Redox Signal. 2018;28:1483-1502
51. Jarrard LE. On the role of the hippocampus in learning and memory in the rat. Behavioral and neural biology. 1993;60:9-26
52. Chao XJ, Chen ZW, Liu AM. et al. Effect of tacrine-3-caffeic acid, a novel multifunctional anti-Alzheimer's dimer, against oxidative-stress-induced cell death in HT22 hippocampal neurons: involvement of Nrf2/HO-1 pathway. CNS neuroscience & therapeutics. 2014;20:840-850
53. Fujita Y & Yamashita T. Sirtuins in Neuroendocrine Regulation and Neurological Diseases. Front Neurosci. 2018;12:778
54. Ota H, Akishita M, Eto M. et al. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571-579
55. Gomes BAQ, Silva JPB, Romeiro CFR. et al. Neuroprotective Mechanisms of Resveratrol in Alzheimer's Disease: Role of SIRT1. Oxid Med Cell Longev. 2018;2018:8152373
56. Singh P, Hanson PS & Morris CM. SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson's disease. BMC Neurosci. 2017;18:46
57. Zhang XS, Wu Q, Wu LY. et al. Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats. Cell Death Dis. 2016;7:e2416
58. Hung CH, Chan SH, Chu PM. et al. Homocysteine facilitates LOX-1 activation and endothelial death through the PKCbeta and SIRT1/HSF1 mechanism: relevance to human hyperhomocysteinaemia. Clinical science. 2015;129:477-487
59. Kim MG, Kim DH, Lee HR. et al. Sirtuin inhibition leads to autophagy and apoptosis in porcine preimplantation blastocysts. Biochem Biophys Res Commun. 2017;488:603-608
60. Arumugam TV & Kennedy BK. H2S to Mitigate Vascular Aging: A SIRT1 Connection. Cell. 2018;173:8-10
61. Vauzour D, Vafeiadou K, Rice-Evans C. et al. Activation of pro-survival Akt and ERK1/2 signalling pathways underlie the anti-apoptotic effects of flavanones in cortical neurons. Journal of neurochemistry. 2007;103:1355-1367
62. Uchida S, Yamagata H, Seki T. et al. Epigenetic mechanisms of major depression: Targeting neuronal plasticity. Psychiatry and clinical neurosciences. 2018;72:212-227
Author contact
![]() Corresponding authors: Xiao-Qing Tang, Institute of Neuroscience, Hengyang Medical College, University of South China, 28 W Chang sheng Road, Hengyang, 421001, Hunan, P. R. China, E-mail address: tangxq-uscedu.cn; tangxq-usccom and Ke-Bin Zhan, Department of Neurology, the Second Affiliated Hospital, University of South China, Hengyang, 421001, Hunan, P.R. China, E-mail address: zhankebincom; Zhankb-uscedu.cn
Corresponding authors: Xiao-Qing Tang, Institute of Neuroscience, Hengyang Medical College, University of South China, 28 W Chang sheng Road, Hengyang, 421001, Hunan, P. R. China, E-mail address: tangxq-uscedu.cn; tangxq-usccom and Ke-Bin Zhan, Department of Neurology, the Second Affiliated Hospital, University of South China, Hengyang, 421001, Hunan, P.R. China, E-mail address: zhankebincom; Zhankb-uscedu.cn