Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(7):698-704. doi:10.7150/ijms.19800 This issue Cite
Research Paper
Autophagy was involved in the protective effect of metformin on hyperglycemia-induced cardiomyocyte apoptosis and Connexin43 downregulation in H9c2 cells
Guang-Yu Wang1, Ya-Guang Bi1, Xiang-Dong Liu1, Yu Zhao1, Jun-Feng Han2, Meng Wei1, Qing-Yong Zhang1 ![]()
1. Affiliation: Department of Cardiology, Shanghai Sixth People's Hospital, Shanghai Jiao Tong University, Shanghai, China;
2. Affiliation: Department of Endocrinology and Metabolism, Shanghai Sixth People's Hospital, Shanghai Jiao Tong University, Shanghai, China.
Received 2017-2-24; Accepted 2017-4-23; Published 2017-6-23
Abstract
Background: Increased cardiomyocyte apoptosis under high glucose condition contributes to diabetic cardiomyopathy. Degradation of cardiac Connexin43 (Cx43) has been associated with cardiac dysfunction in diabetic heart. Clinical and experimental studies suggested that metformin (Met) exhibits cardioprotective properties against diabetes.
Aim: The aim of this study was to investigate the effect and underlying signaling mechanisms of metformin on apoptosis and Cx43 expression in H9c2 cells presenting with hyperglycemia conditions.
Methods: In the present study, H9c2 cardiac cells were incubated with 5.5 mM glucose, 33.3 mM glucose, 33.3 mM glucose with metformin at two dose (100 μM, 1 mM) for 96 hours, and 1 mM metformin with chloroquine (50 μM) in 33.3 mM glucose medium. Cell viability was determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) cell survival assay. Cytotoxicity was determined by the release of lactate dehydrogenase (LDH). The expression of Cx43, autophagic maker protein (LAMP-1, Beclin-1, p62 and LC3) and apoptosis maker protein (Bcl-2 and Bax) were determined by western blot.
Results: The results showed that high glucose increased apoptosis and decreased Cx43 expression. Interestingly, metformin attenuated hyperglycemia-increased apoptosis and restored Cx43 expression. Moreover, this treatment caused autophagy as well, which indicated by up-regulation of autophagy-related proteins LAMP-1, Beclin-1, p62 and reduction in the ratio of LC3-II/LC3-I. In addition, administration autophagy inhibitor chloroquine (CQ) did not block the effect of metformin on Cx43 expression while increasing Cx43 content, together with an increased apoptosis.
Conclusion: Administration metformin can protect the H9c2 cells against hyperglycemia-induced apoptosis and Cx43 down-regulation, in part, mediated through the induction of autophagy pathway.
Keywords: autophagy, connexin43, hyperglycemia, metformin, apoptosis.
Introduction
Diabetes caused serious complications in cardiovascular system including diabetic cardiomyopathy and arrhythmias, yet the underlying molecular mechanism of how these occurs remained unclear. An increasing number of studies have demonstrated that cardiomyocyte apoptosis induced by hyperglycemia was observed in diabetic patients and animals [1, 2].
Cardiac connexin43 (Cx43) is the major connexin in ventricular cardiomyocytes, which formed communication channels for proper electric and metabolic coupling between cardiomyocytes [3]. Expectedly, alteration of Cx43 expression has been associated with a variety of pathological conditions such as myocardial ischemia [4], heart failure [5], hypertrophy [6], diabetes [7], and arrhythmias [8]. Moreover, previous studies demonstrated that changed Cx43 expression could compromise gap junction intercellular communication in diabetic heart, providing an arrhythmogenic substrate for various arrhythmias [7]. It has been demonstrated that decreased Cx43 expression in the diabetic heart induced a decrease in the conductivity. Several studies have implicated that autophagy process was involved in the regulation of Cx43 turnover in the heart [5, 9].
Autophagy is a conserved process for bulk degradation and recycling of cytoplasmic proteins and organelles in lysosomes, providing free fatty acids and amino acids for maintain energy production and protein synthesis. In order to accomplish these work orderly, autophagy related proteins including microtubule associated protein light chain 3 (LC3), LAMP-1, Beclin-1 and p62 (also known as sequestosome-1) were all indispensable for cytoplasm-to-lysosome delivery [10-13]. In addition to rest conditions, activation of autophagy has also been implicated in a variety of pathological state such as ischaemia-reperfusion [14], starvation [15], heart failure [5], hypertensive [6], and diabetic heart [2] , suggesting that autophagy may play a vital role in heart diseases. Interestingly, cardiac dysfunction was improved when administrate metformin in diabetic heart by activation of autophagy [2].
Metformin (Met), the most commonly anti-diabetic drug, improved many clinical parameters and reduced all-cause mortality and cardiovascular disease events compared to lifestyle changes alone in Chinese type 2 diabetes mellitus patients [16]. Furthermore, a population-based dynamic cohort and in vitro studies showed that metformin treatment decreased the risk of atrial fibrillation in patients with type 2 DM, probably via attenuation of atrial cell tachycardia-induced myolysis and oxidative stress [17]. Experimental investigations demonstrated that metformin serves as a therapeutic strategy for diabetic cardiomyopathy through autophagy pathway [2, 18]. To our knowledge, the relationship of metformin, autophagy, apoptosis and Cx43 under hyperglycemia condition remains to be established. Thus, this study aims to evaluate the effect of metformin on autophagy, Cx43 and apoptosis in H9c2 cells.
Materials and methods
Materials
Metformin and chloroquine were purchased from Sigma (St. Louis, MO). The H9c2 cells were obtained from the Cell Bank of Chinese Academy of Science (Shanghai, China). LDH activity assay kit and MTT were purchased from Beyotime (Shanghai, China). Antibodies against Cx43, LAMP-1, Beclin-1, p62, LC3, Bax and Bcl-2 were obtained from Cell Signaling Technology (CST, Danver, MA, USA).
Cell culture and treatment
The cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) at 37°C in a humidified incubator consisting of 5% CO2 and 95% air. Confluent cells (60-70% confluence) were employed to the experiments. The H9c2 cells were exposed to five conditions: medium containing 5.5 mM glucose (Con group), 33.3 mM glucose (HG group), 33.3 mM glucose with 0.1 mM metformin (Met 0.1 group), 33.3 mM glucose with 1 mM metformin (Met 1 group), 33.3 mM glucose with 1 mM metformin in the presence of chloroquine (50 μM, CQ group) for 96 hours.
Cell viability assay
Cell viability was measured by 3-(4,5-dimethylthiazol 2-yl)-2,5-(diphenyltetrazolium bromide) (MTT) experiments. The H9c2 cells were seeded in 96-well plates at 2.0 x 104 cells/well. At the end of incubation period, MTT solution (final concentration of 0.5 mg/ml) was added to each well and incubated for 4 h at 37 °C. After the medium was removed, DMSO was added to dissolve the blue-colored formazan product. Absorbance was measured with a microplate reader at 490nm. Cell survival rates were expressed as the percentage of the absorbance of treated group to con group.
Lactate Dehydrogenase (LDH) release
Cell death was assessed by the amount of LDH, which was used to assess the damage of cells. According to the LDH activity assay kit manufacturer's instructions, cell medium was collected and mixed with LDH reaction buffer for 30 min at room temperature. The absorbance was read at 450nm when the reaction stopped. Cell death rates were expressed as the percentage of the absorbance of treated group to con group.
Western blotting
H9c2 cells were harvested and lysed with RIPA buffer (50 mM Tris-HCl, PH 7.4, 150 mM NaCl, 0.1% SDS, 1% sodium deoxycholate) supplemented with protease and phosphatase inhibitor cocktails (Roche, Germany). The supernatant fractions were collected and protein concentration was determined using bicinchoninic acid (BCA) kit (Beyotime, shanghai, China). Lysate protein was separated by 10%-12% SDS-polyacrylamide gel and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were incubated with primary antibodies at 4°C overnight and then incubated for 2 h with goat anti-rabbit secondary antibody conjugated to horseradish peroxidase at room temperature. After reaction with electrochemiluminescence (ECL) regent (Millipore, Billerica, MA, USA), the bands were captured using the image reader LAS-4500 mini system and the density of bans was quantified by Gel-Pro32 Analyzer software.
Statistical analysis
The data were expressed as mean ± standard deviation. One-way analysis of variance test was used to identify significant difference between HG group and metformin groups. Student's t-test was performed to compare the difference between con group and HG group. A probability value of P<0.05 was used as the criterion for statistical significance.
Results
Effect of metformin on H9c2 cells cell viability
Although H9c2 cells of all the four groups (Con group, HG group, Met 0.1 group and Met 1 group) remained viable for 96 hours, the viability of H9c2 cells in Met group was significantly increased than HG group. Compared with Met 1 group, cell survival was attenuated in CQ group (figure 1A).
Effect of metformin on cell survival in H9c2 cells under hyperglycemia condition. H9c2 cells were incubated in high glucose medium containing metformin in the absence or presence of chloroquine. MTT cell survival assay was performed with MTT assay kit. LDH serves as a maker of cell death was performed with LDH assay kit. Representative images of three different samples, and each experiment was repeated at least three times. Results are expressed as mean ± SD. n=3. *P<0.05 vs. Con, #P<0.05 vs. HG, **P<0.05 vs. Met1.
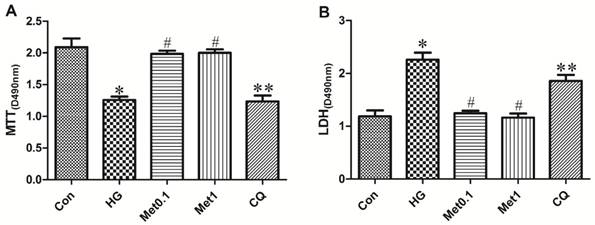
The cell death was indicated by LDH activity in the medium (Figure 2). The LDH activity in HG group was significantly higher than Con group. The LDH activity in Met group significantly decreased than HG group while treatment with chloroquine in CQ group significantly increased cell death than Met 1 group (figure 1B).
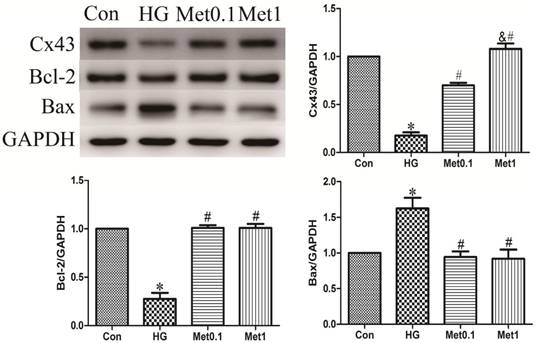
Metformin prevented Cx43 down-regulation and apoptosis under high glucose condition. Western blotting was performed with H9c2 cells lysates treated with metformin as mentioned in text for the expression of Cx43 and apoptosis marker protein. The target protein density was normalized to the control cells. Representative images of three different samples, and each experiment was repeated at least three times. Results are expressed as mean ± SD. n=3.*P<0.05 vs. Con, #P<0.05 vs. HG.
Effect of metformin on apoptosis protein expression
The levels of Bcl-2 and Bax protein were used to assess apoptosis situation. The results showed that Bcl-2 expression decreased in HG group, which was prevented by metformin administration in a concentration-independent manner. Compared with Con group, the Bax expression in HG group was increased. However, metformin reduced the expression of Bax in a concentration-independent manner (figure 2).
Effect of metformin on Cx43 expression
Cx43 expression was analysis by western blot in all groups. Cx43 expression in HG group was significantly decreased than Con group, which was prevented by metformin treatment in a concentration-dependent manner (figure 2).
Effect of metformin on autophagy activity
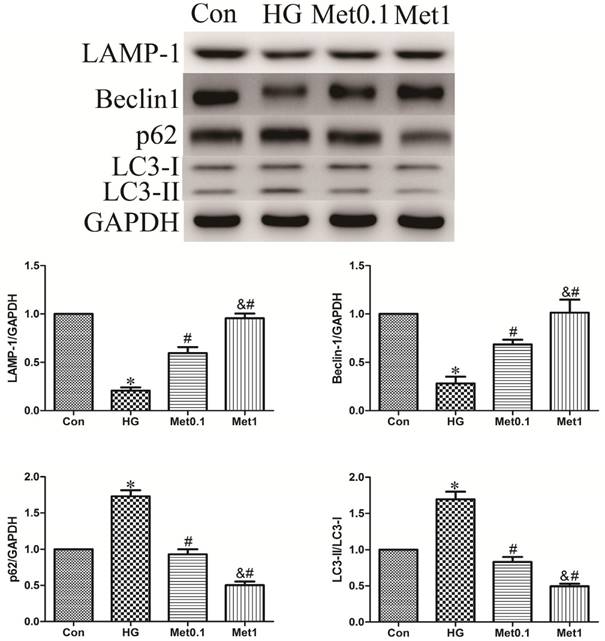
To assess the activity of autophagy in H9c2 cells incubation with hyperglycemia, we measured autophagic marker including LAMP-1, Beclin-1, p62 and LC3. As a result, the expression of LAMP-1 and Beclin-1 were decreased, while the level of p62 and the ratio of LC3-II/LC3-I were increased in HG group versus con group. Remarkably, the effect of hyperglycemia on autophagy activity was abrogated by metformin administration indicated by increased LAMP-1 and Beclin-1 expression, and decreased p62 content and the ratio of LC3-II/LC3-I. The efficiency of metformin was in a concentration-dependent manner (figure 3).
Effect of metformin on autophagy protein in H9c2 cells under high glucose condition. Western blotting was performed with H9c2 cells lysates treated with metformin as mentioned in text for the expression of autophagic marker protein. The target protein density was normalized to the control cells. Representative images of three different samples, and each experiment was repeated at least three times. Results are expressed as mean ± SD. n=3.*P<0.05 vs. Con, #P<0.05 vs. HG, & P<0.05 vs. Met0.1
Effect of chloroquine on cell viability and apoptosis
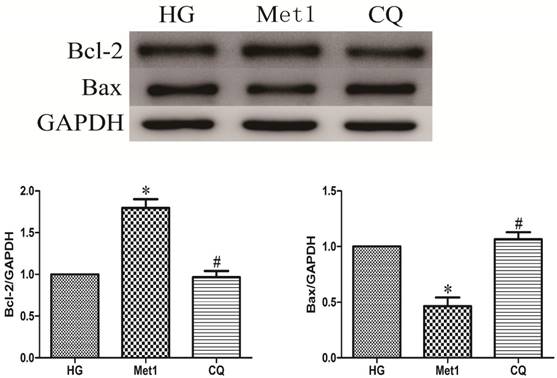
Above indicators (MTT, LDH, Bcl-2, Bax) were also used to determine effect of chloroquine on cell viability and apoptosis under metformin conditions. Compared with Met 1 group, the cell viability significantly decreased in CQ group (figure 1). As expected, the similar results were also observed in CQ group compared with Met group, indicated by decreased Bcl-2 and increased Bax levels (figure 4).
Chloroquine treatment increased apoptosis. Western blotting was performed with H9c2 cells lysates treated with chloroquine as mentioned in text for the expression of apoptosis marker proteins. The target protein density was normalized to the control cells. Representative images of three different samples, and each experiment was repeated at least three times. Results are expressed as mean ±SD. n=3. *P<0.05 vs. Con, #P<0.05 vs. HG.
Effect of chloroquine on Cx43 expression
As mentioned above, metformin increased autophagic flux in H9c2 cells under hyperglycemia condition, along with up-regulation of Cx43 expression. Compared with Met 1 group, the expression of Cx43 was further increased in CQ group, which indicated autophagy pathway was involved in Cx43 degradation (figure 5).
Effect of chloroquine on Cx43 expression and autophagy activity in H9c2 cells under high glucose condition. Western blotting was performed with H9c2 cells lysates treated with chloroquine as mentioned in text for the expression of Cx43 and autophagic marker protein. The target protein density was normalized to the control cells. Representative images of three different samples, and each experiment was repeated at least three times. Results are expressed as mean ± SD. n=3.*P<0.05 vs. Con, #P<0.05 vs. HG.
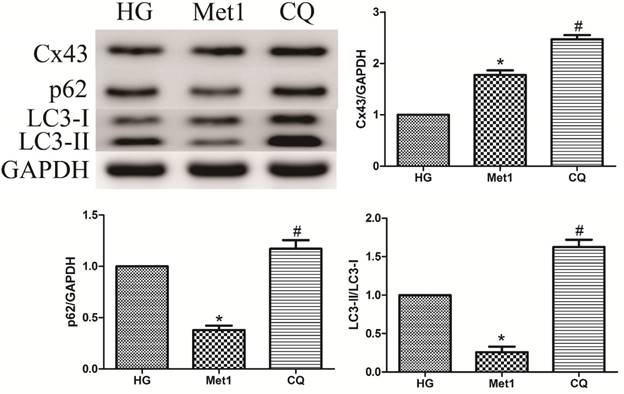
Effect of Chloroquine on autophagy activation
Compared with Met 1 group, the p62 amount and the ratio of LC3-II/LC3-I were increased in CQ group, suggesting that metformin increased autophagic turnover or flux in H9c2 cells (figure 5). However, LAMP-1 and Beclin-1 expression was unaffected by chloroquine treatment (Data not shown).
Discussion
In the present study, we found that metformin inhibited the apoptosis and Cx43 down-regulation caused by hyperglycemia, which was associated with activation of cardiac autophagy. However, the protective effect of metformin was abolished by chloroquine treatment. Intriguingly, administration autophagy inhibitor chloroquine did not block the effect of metformin on Cx43 expression while further increased Cx43 content. These results suggested that autophagy was inhibited in the pathogenesis of diabetes and metformin performed cardiac protection, at least in part, by increasing autophagy activities.
Previous studies have been demonstrated the interaction between cardiomyocytes apoptosis and autophagy pathway under hyperglycemia conditions [2, 18]. Autophagy was a self-digestion process, which provided energy and amino acids for cell growth by degrading the aggregated proteins or damaged organelles. He et al. have demonstrated that autophagy was suppressed in diabetic rats, however, administration of metformin enhanced autophagic activity and protected cell against hyperglycemia insults [18]. A recent study suggested that metformin attenuated the up-regulation of ubiquitinated protein via activating autophagy and then confers cardiac protection roles in diabetic hearts [2]. However, this opinion was not supported by several studies. Although autophagic flux was inhibited both in vivo and in vitro under hyperglycemia conditions, the reduction of autophagy appeared to be an adaptive response which limited hyperglycemia-induced injury [10, 19]. Thus, autophagy has a dual role in cell survival depending on cell type and environment. In our study, we found that metformin enhanced autophagy activity and reduced cell apoptosis, but these effects were absent in CQ group. These observations suggest that induction of autophagy by metformin confers a protective effect against hyperglycemia-induced cardiomyocytes apoptosis.
Another key finding from this study was that autophagy plays an essential role in mediating Cx43 expression. Several studies have demonstrated that autophagy pathway was implicated in the regulation of Cx43 turnover in various heart diseases [20]. Martins et al. demonstrated that both in vivo and in vitro ischemia-induced cardiac Cx43 degradation resulted in gap junction intercellular communication impairment, which can be restored by autophagy inhibition [14]. It was also reported that Cx43 expression decreased in aged spontaneously hypertensive rat hearts and elevated after supplement with aliskiren by decreasing autophagy [6]. In line with previous studies, our results showed that metformin lead to Cx43 up-regulation in H9c2 cells through activating autophagy in hyperglycemia medium. The levels of LAMP-1 and Beclin-1 were increased, but p62 content and the ratio of L3-II/LC3-I were decreased in this context.
Interestingly, the change of the ratio of LC3-II/LC3-I was not supported by previous studies. Previous studies suggested that the increase of LC3-II/LC3-I ratio indicated that metformin enhanced autophagic flux [2, 18]. However, guidelines for monitoring autophagy suggested that LC3-II/LC3-I ratio would decrease if the degradation process of LC3-II by lysosomal is rapid. Furthermore, guidelines point out that LC3 changes may be particularly rapid, while clearance of substrates of autophagy may need a longer time [13]. Taken the results from our study into consideration, we have considerable reason to believe that metformin enhanced autophagy activity in our study. In order to verify this view, we used chloroquine to inhibit autophagy pathway. It was well known that chloroquine inhibited lysosome fusion with autophagosomes and elevated lysosomal PH, thereby preventing the final digestion step and inhibiting lysosomal activity [21]. As expected, the ratio of LC3-II/LC3-I and p62 content increased in CQ group. Intriguingly, the amount of Cx43 increased when treated with metformin, which was further enhanced induced by chloroquine. On the basis of our data, we presume that metformin-mediated activation of autophagy may have a dual role in Cx43 turnover. The process of autophagy provided amino acids and free fatty acids to maintain energy homeostasis and protein synthesis by degradation of damage proteins and organelles. Thus, these in vitro data indicated that the metformin-mediated autophagy contributed to both Cx43 synthesis and degradation, but the net effect of metformin was promoting Cx43 synthesis. Further investigations are required to explore the detailed mechanisms.
It was consistent with recent study that metformin have cardioprotective effects in multiple cardiovascular diseases, which was supported by elevated cell viability and Cx43 expression in our study. Since myocardial cell rarely proliferate, the loss of myocardial cell would lead to cardiac dysfunction. Administration of metformin protected cardiomyocytes from death under hyperglycemia condition, which could improve cardiac function. Additionally, the remodeling of cardiac Cx43 was associated with various heart diseases, particularly arrhythmia. Diabetic patients showed a higher incidence of cardiac arrhythmias, including atrial fibrillation (AF) [22, 23]. Recent studies indicated that the remodeling of Cx43 can facilitate development of AF in old guinea pig hearts and patients [24, 25]. Chang et al. demonstrated that metformin treatment decreased the risk of AF in diabetic patients and inhibited the generation of ROS and myofibril degradation [17]. To our knowledge, there were rare reports on the effect of metformin on cardiac Cx43 expression in H9c2 cells incubation with hyperglycemia medium. More important, we found that metformin induced Cx43 up-regualtion in H9c2 cells via increasing autophagy activity, which may account for the decrease of AF incidence induced by metformin. Future investigations are needed to verify this hypothesis on animal models.
In summary, our findings demonstrated that a reduction in cardiac autophagy and the subsequent decrease of cell viability and Cx43 expression were implicated in cardiac dysfunction in diabetic heart. Metformin prevented apoptosis and Cx43 down-regulation by enhancing autophagy activity, which may represent a novel mechanism for the cardioprotective effects of metformin.
Acknowledgements
The present study was supported by Shanghai Committee of Science and Technology, China (Grant NO.:13ZR1431500)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Frustaci A, Kajstura J, Chimenti C. et al. Myocardial cell death in human diabetes. Circ Res. 2000;87:1123-32
2. Xie Z, Lau K, Eby B. et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770-8
3. Vozzi C, Dupont E, Coppen SR. et al. Chamber-related differences in connexin expression in the human heart. J Mol Cell Cardiol. 1999;31:991-1003
4. Rutledge CA, Ng FS, Sulkin MS. et al. c-Src kinase inhibition reduces arrhythmia inducibility and connexin43 dysregulation after myocardial infarction. J Am Coll Cardiol. 2014;63:928-34
5. Hesketh GG, Shah MH, Halperin VL. et al. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ Res. 2010;106:1153-63
6. Zhang W, Zhao G, Hu X. et al. Aliskiren-attenuated myocardium apoptosis via regulation of autophagy and connexin-43 in aged spontaneously hypertensive rats. J Cell Mol Med. 2014;18:1247-56
7. Lin H, Ogawa K, Imanaga I. et al. Remodeling of connexin 43 in the diabetic rat heart. Mol Biol Cell. 2006;290:69-78
8. Greener ID, Sasano T, Wan X. et al. Connexin43 gene transfer reduces ventricular tachycardia susceptibility after myocardial infarction. J Am Coll Cardiol. 2012;60:1103-10
9. Bejarano E, Girao H, Yuste A. et al. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell. 2012;23:2156-69
10. Xu X, Kobayashi S, Chen K. et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077-92
11. Pankiv S, Clausen TH, Lamark T. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45
12. Kabeya Y, Mizushima N, Ueno T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-8
13. Klionsky DJ, Abdelmohsen K, Abe A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
14. Martins-Marques T, Catarino S, Zuzarte M. et al. Ischaemia-induced autophagy leads to degradation of gap junction protein connexin43 in cardiomyocytes. Biochem J. 2015;467:231-45
15. Lichtenstein A, Minogue PJ, Beyer EC. et al. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci. 2011;124:910-20
16. Fung CS, Wan EY, Wong CK. et al. Effect of metformin monotherapy on cardiovascular diseases and mortality: a retrospective cohort study on Chinese type 2 diabetes mellitus patients. Cardiovasc Diabetol. 2015;14:137-51
17. Chang SH, Wu LS, Chiou MJ. et al. Association of metformin with lower atrial fibrillation risk among patients with type 2 diabetes mellitus: a population-based dynamic cohort and in vitro studies. Cardiovasc Diabetol. 2014;13:123-31
18. He C, Zhu H, Li H. et al. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270-81
19. Kobayashi S, Xu X, Chen K. et al. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy. 2012;8:577-92
20. Martins-Marques T, Catarino S, Marques C. et al. To beat or not to beat: degradation of Cx43 imposes the heart rhythm. Biochem Soc Trans. 2015;43:476-81
21. Kanamori H, Takemura G, Goto K. et al. Resveratrol reverses remodeling in hearts with large, old myocardial infarctions through enhanced autophagy-activating AMP kinase pathway. Am J Pathol. 2013;182:701-13
22. January CT, Wann LS, Alpert JS. et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130:2071-104
23. Huxley RR, Filion KB, Konety S. et al. Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol. 2011;108:56-62
24. Shinohara D, Matsushita S, Yamamoto T. et al. Reduction of c-kit positive cardiac stem cells in patients with atrial fibrillation. J Cardiol. 2017;69:712-8
25. Nagibin V, Egan Benova T, Viczenczova C. et al. Ageing related down-regulation of myocardial connexin-43 and up-regulation of MMP-2 may predict propensity to atrial fibrillation in experimental animals. Physiol Res. 2016;65:91-100
Author contact
![]() Corresponding author: Qingyong Zhang, department of cardiology, Shanghai Sixth People's Hospital, Shanghai Jiao Tong University, Shanghai, China, No. 600 Yishan Road, Shanghai 200233, China. Fax: (86-21)-64369181. E-mail: zhangqingyong6thcom
Corresponding author: Qingyong Zhang, department of cardiology, Shanghai Sixth People's Hospital, Shanghai Jiao Tong University, Shanghai, China, No. 600 Yishan Road, Shanghai 200233, China. Fax: (86-21)-64369181. E-mail: zhangqingyong6thcom