Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(1):132-139. doi:10.7150/ijms.105295 This issue Cite
Review
Research Progress of Regulatory Cell Death in Coronary Microembolization
Chen Chang1#, Wan-Zhong Huang2#, Ru-Ping Cai3, Li-Rong Mo2 ![]() , Qiang Wu4
, Qiang Wu4 ![]() , Qiang Su2
, Qiang Su2 ![]()
1. Department of Cardiology, The First Affiliated Hospital of Xi'an Medical University, Xi'an 710077, Shanxi, People's Republic of China.
2. Department of Cardiology, Jiangbin Hospital of Guangxi Zhuang Autonomous Region, Nanning 530021, Guangxi, People's Republic of China.
3. Department of Cardiology, The First Affiliated Hospital of Shandong First Medical University, Taian 271016, Shandong, People's Republic of China.
4. Senior Department of Cardiology, The Sixth Medical Center, Chinese PLA General Hospital, Beijing 100048, People's Republic of China.
#Contributed equally.
Received 2024-10-16; Accepted 2024-11-14; Published 2025-1-1
Abstract

Coronary microembolization (CME) is defined as atherosclerotic plaque erosion, spontaneous rupture, or rupture of the plaque while undergoing interventional therapy resulting in the formation of tiny emboli that obstruct the coronary microcirculatory system. For percutaneous coronary intervention, CME is a major complication, with a periprocedural incidence of up to 25%. Recent studies have demonstrated that regulatory cell death (RCD) exerts a profound influence on CME through its modulation of inflammatory responses, oxidative stress, cell death, and angiogenesis. RCD, including apoptosis, autophagy, and pyroptosis, is a unique class of genetically highly regulated death patterns pervasive in instances of coronary microembolization. The aim of this review is to summarize the currently known molecular mechanisms underlying CME. Further investigations of the RCD mechanisms may unravel new avenues for the prevention and treatment of CME.
Keywords: coronary microembolization, regulatory cell death, apoptosis, autophagy, pyroptosis
Introduction
According to a 2020 report from the World Health Organization, about 17.9 million people died from cardiovascular disease in 2019, accounting for about 32% of global mortality [1]. Numerous studies have shown that cardiovascular diseases, particularly acute myocardial infarction, are the leading cause of disability and death [2-5]. Currently, primary percutaneous coronary intervention is the treatment of choice for AMI [6]. The prevalence of coronary microembolization (CME) in primary percutaneous coronary intervention is about 25%, which substantially burdens healthcare resources [7]. This is attributed to the rupture of capillaries and bleeding caused by myocardial ischemia-reperfusion following interventional therapy, which promotes the occurrence of CME [8]. Currently, there are no effective measures to prevent myocardial ischemia-reperfusion injury. The CME refers to the formation of microemboli that block the coronary microcirculatory system as a result of erosion of atherosclerotic plaque, spontaneous rupture, or rupture of the plaque while undergoing interventional therapy [9, 10]. These microemboli have a complex composition, mainly consisting of platelet aggregates, fibrin, hyaluronic acid, and substances from atherosclerotic plaques, including cholesterol [10]. A previous report based on a pathological examination of the hearts of 44 patients who experienced sudden death due to coronary heart disease indicated that 89% of the affected vessel calibers from microcirculatory embolism were within 120 μm [11]. Of this 89%, 46% were in the range of 40 to 80 μm, while 39% were less than 40 μm [11]. Plaque rupture or erosion also leads to the release of soluble pro-thrombotic, vasoconstrictive and pro-inflammatory factors [8]. CME induces vasoconstriction and inflammation, which may lead to myocardial contractile dysfunction and myocardial microinfarction, as well as the development of arrhythmias [12]. In clinical practice, CME is considered one of the main factors contributing to the no-reflow or slow-flow phenomenon after percutaneous coronary intervention [13]. No-reflow or slow-flow is a common complication during percutaneous coronary intervention, characterized by incomplete restoration of blood flow despite successful opening of the coronary vessels, leading to persistent myocardial ischemia symptoms [13]. The commonly used clinical treatments (thrombolytic therapy, inhibition of platelet aggregation, and vasodilatation) cannot improve the clinical outcome of CME patients [9]. Interestingly, mechanical ischaemic conditioning approaches, involving brief cycles of ischaemia-reperfusion in the heart or a tissue remote from the heart, reduce myocardial infarct size and coronary microvascular damage [8]. Although percutaneous coronary intervention with manual thrombus aspiration demonstrated better ST-segment resolution and less distal embolization on angiography compared to primary percutaneous coronary intervention alone, clinical outcome (cardiovascular death, re-infarction, cardiogenic shock, or NYHA class IV heart failure) did not show significant improvement [10]. Cardiomyocytes in adult mammals are non-renewable cells. Therefore, the reversal of myocardial damage is crucial for restoring cellular function and preventing cardiomyocyte death [14].
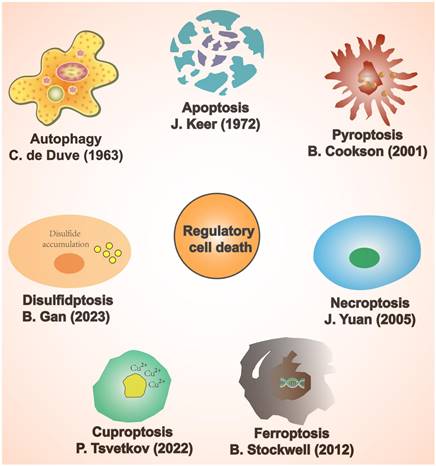
Cell death patterns include accidental cell death and regulatory cell death (RCD) [15]. Accidental cell death is a non-regulated cell death, usually caused by a sudden external injury or stimulus (strong chemicals, radioactive radiation, and physical damage) that exceeds the normal range of cellular response [16]. RCD is characterised by a precise molecular mechanism and it is regulated by specific signal transduction pathways. Furthermore, RCD can undergo pharmacological intervention and is regulated by interfering with gene expression and gene-mediated signaling pathways [17, 18]. The known forms of RCD include apoptosis, autophagy, pyroptosis, ferroptosis, cuproptosis, disulfidptosis, and necroptosis (Figure 1) [19-25]. RCD is closely related to cardiovascular diseases [26]. In addition, numerous studies have indicated that RCD plays a significant regulatory role in coronary microembolization (CME) by mediating various signaling pathways involved in its development [27-29]. Therefore, precision-targeted therapies can be obtained by modulating the expression of RCD-associated signature genes or their mediated signaling pathways. Although cuproptosis, disulfidptosis, and necroptosis exert a significant influence on the pathogenesis of human disease, they remain understudied in the context of CME. Thus, the aim of this review is to summarize the currently known molecular mechanisms related to RCD (apoptosis, autophagy, pyroptosis, and ferroptosis) in the context of CME (Figure 2).
Classification of regulatory cell death.
The mechanism of regulatory cell death in coronary microembolization. AKT: protein kinase B; BIM: Bcl-2-like protein 11; BMF: Bcl-2 modifying factor; GPX4: glutathione peroxidase 4; GSK-3β: glycogen synthase kinase 3; PI3K: phosphoinositide 3-kinase; POP1: pyrin only protein 1; SIRT1: sirtuin1.
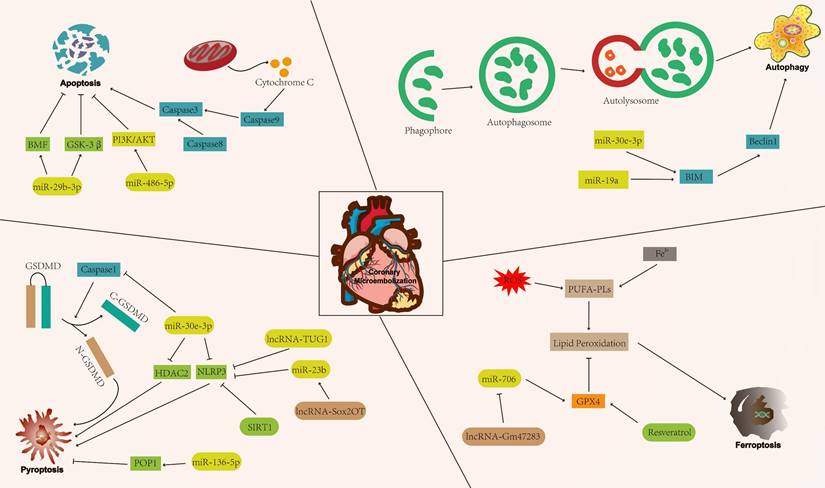
RCD in CME
Apoptosis and CME
Apoptosis is widespread in organisms and is a physiological phenomenon mediated by specific genes [30]. Apoptosis is characterized by several key features, including chromatin condensation, cellular shrinkage, DNA fragmentation, formation of apoptotic body, and membrane blebbing [31]. Previous study found the presence of apoptosis in the CME model [32]. Apoptosis-inducing pathways can be classified into three main categories: mitochondrial, endoplasmic reticulum (ER), and death receptor pathways [33].
The mitochondrial pathway represents a crucial endogenous apoptotic pathway, whereby the activation of the mitochondria-mediated endogenous apoptotic pathway results in a notable reduction in mitochondrial membrane potential, thereby leading to a considerable enhancement in mitochondrial membrane permeability [34]. Liu et al. [35] showed that the expression levels of ectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), cytochrome c and caspase-9 are significantly increased in a CME model established in Bama miniature pigs. This suggests that CME promotes cardiomyocyte apoptosis and exacerbates CME-induced myocardial injury, possibly through the LOX-1-dependent mitochondrial pathway. A study revealed that rosuvastatin inhibits apoptosis mediated by the mitochondrial pathway and CME-induced cardiac dysfunction in rat CME models by up-regulating B-cell lymphoma-2 (BCL-2) expression and reducing caspase-3, cytochrome c, and BCL-2-associated X protein levels [27]. Furthermore, puerarin and resveratrol exhibit comparable efficacy in inhibiting apoptosis and mitigating CME-induced cardiotoxicity, partly due to the increased expression of phosphatidylinositol 3-kinase and protein kinase B in the phosphorylated form [36, 37]. MiR-29b-3p expression is significantly reduced in rat CME model [38]. Further study showed that miR-29b-3p overexpression mediates neovascularisation, inhibits apoptosis mediated by the mitochondrial pathway, and reduces the area of myocardial microinfarction in rat CME [38]. Qin et al. [38] demonstrated that miR-29b-3p overexpression mitigates CME-induced myocardial injury, possibly due to the suppression of glycogen synthase kinase 3 and BCL-2 modifying factor (BMF) expression. Moreover, miR-486-5p can mediate the activation of the phosphatidylinositol 3-kinase/protein kinase B axis, thereby attenuating CME-induced cardiomyocyte apoptosis [39]. The term "death receptor pathway" is employed to delineate the process by which a cell binds a specific death receptor (Fas or tumour necrosis factor (TNF) receptor) to its ligand (Fas ligand or TNF-α), forming a death signaling complex [40]. This complex then initiates a series of intracellular signaling events that ultimately result in apoptosis. Fas-associated death domain (FADD) is an adapter molecule that bridges the interaction between receptor-interacting protein 1 and aspartate-specific caspase-8 [41]. The caspase-8-mediated death receptor pathway also played an important role in the CME model established in Bama minipigs [35]. Notably, TNF-α has been identified as an important causative factor for myocardial contractile dysfunction in CME [7]. Leukocyte count and TNF-α contents were increased in the CME posterior myocardium [42]. Pretreatment with antibodies to TNF-α appears to prevent contractile dysfunction after CME, whereas in the absence of CME, intracoronary injection of exogenous TNF-α induces contractile dysfunction [42]. In conclusion, TNF-α is considered to be an important cause of progressive myocardial contractile dysfunction after CME [42]. Zhou et al. [43] observed that TNF-α can trigger apoptosis mediated by receptor-interacting protein 1 (RIP1)/FADD/caspase-8 in astrocytes. Furthermore, Su et al. [44] indicated that the level of TNF-α is markedly increased in the CME model constructed using Bama miniature pigs. The disruption of the RIP1-FADD complex has been shown to exacerbate myocardial damage [45]. However, the effect of TNF-α triggering the RIP1/FADD/caspase-8 signaling pathway on the cardiac in the CME model requires further experimental support. In addition, apoptosis is associated with ER stress, a cellular stress response to the accumulation of unfolded or misfolded proteins in the endoplasmic reticulum lumen [46]. The ER-mediated apoptotic pathway has been demonstrated to be an important mechanism of hypoxic injury in cardiomyocytes [47]. AMP-activated protein kinase (AMPK) is a key regulatory enzyme involved in energy homeostasis during hypoxia [47]. Hypoxia induces activation of the ER-mediated apoptotic pathway in cardiomyocytes, and endogenous activation of AMPK partially reverses these effects [47]. In the CME model, targeting the JNK/p38 MAPK pathway was observed to activate the ER stress pathway and induce cardiomyocyte apoptosis, which may be associated with hyperphosphorylation of JNK and p38 [48]. In conclusion, apoptosis mediates multiple signaling pathways involved in the process of CME.
Autophagy and CME
Autophagy is a biological process whereby an organism eliminates aberrant proteins or cellular components through the activation of specific genes and their associated signaling pathways, and mainly includes macroautophagy, microautophagy, and chaperone-mediated autophagy [49]. Notably, the three forms of autophagy eliminate aberrant cellular components and macromolecules, including proteins, through lysosomes [50]. Microautophagy represents a process whereby cytoplasmic carriers are directly phagocytosed through lysosomal membrane invaginations, without the formation of autophagosomes [51]. Chaperone-mediated autophagy is the selective degradation of proteins with KFERQ sequences in the cytoplasm via the lysosomal pathway and does not require autophagosome formation [51]. A recent study has demonstrated that the activation of chaperone-mediated autophagy provides protection for cardiomyocytes against hypoxic cell death [52]. Although microautophagy and chaperone-mediated autophagy have been shown to have important roles in a variety of diseases, their impact in the CME has yet to be extensively studied. Currently, macroautophagy (later referred to as “autophagy” if not otherwise stated) is considered to be the main autophagic branch regulating physiological and pathological mechanisms in the cardiovascular system [53].
Beclin 1, microtubule-associated protein II light chain 3 (LC3-II), and sequestosome 1 are widely employed as indicators to assess autophagy status. Specifically, autophagy activation increases the expression levels of Beclin 1 and LC3-II, while decreasing the levels of sequestosome 1 protein [54-56]. Notably, miR-30e-3p expression is elevated under autophagy activation [57]. Besides, miR-30e-3p levels are negatively correlated with sequestosome 1 levels in the rat CME model [57]. These findings suggest that targeting miR-30e-3p is a promising approach for CME treatment. Moreover, miR-30e-3p directly targets the 3'-UTR of BCL-2-like protein 11 (BIM), decreasing BIM expression, thus activating autophagy and preserving the functional integrity of human-induced pluripotent stem cell-derived cardiomyocytes while mitigating CME-induced cardiac impairment [58]. Similarly, miRNA-19a regulates autophagic flux and maintains cardiomyocyte integrity by inhibiting the expression of the pro-apoptotic protein BIM [59]. Reduced expression of early growth response factor 1 in the rat model of CME further inhibits BIM expression and up-regulates the level of beclin 1, modulating autophagic flux, thus alleviating CME-induced cardiac impairment [60]. Lysosome-associated membrane protein 2a (LAMP2a) protein levels were used as both a primary indicator and driver of CMA function [52]. Increased levels of LAMP2a protein were observed in hypoxia-treated cardiomyocytes and in the serum of patients with heart failure [52]. In fact, increased levels of LAMP2a protein were thought to be a stress response in cardiomyocytes [52]. Furthermore, Ghosh et al. showed a significant enhancement of both macroautophagy and chaperone-mediated autophagy activity by increasing LAMP2a protein levels [52]. However, the overall effect of the above mechanisms on CME-induced myocardium requires extensive experimental validation. Furthermore, autophagy exerts a dual influence on the regulation of the organism [61]. Moderate autophagy is beneficial to the stability of the intracellular environment, while excessive autophagy may lead to cell death, possibly due to basal autophagy in normal cellular activities and induced autophagy under various adverse stimuli [61, 62]. Overall, the effects of moderate activation or inhibition of autophagy on the organism should be explored in depth due to the dual effects of autophagy and the complexity of the disease.
Pyroptosis and CME
The inflammasome is a multi-protein complex essential for regulating the innate immune inflammatory response. NOD-like receptor thermal protein domain associated protein 3 (NLRP3), a member of the NOD-like receptor family, has been extensively studied [63]. Pyroptosis is mainly induced by the inflammasome and mediated by gasdermin family proteins [64, 65]. Pyroptosis can be divided into classical (caspase-1 mediated) and non-classical (caspase-4, caspase-5, and caspase-11 mediated) pathways [64]. In the classical pathway, the inflammasome induces the activation of caspase-1, which specifically cleaves the N-terminal structural domain of gasdermin-D and induces its oligomerization, leading to the disruption of the cell membrane [66]. This causes the release of its contents and inflammatory factors (IL-1β, IL-18), ultimately triggering pyroptosis [66]. In concrete terms, the NLRP3 inflammasome activates caspase-1, cleaving pro-IL-1β and pro-IL-18 into their active forms, IL-1β and IL-18 [67]. This triggers pyroptosis and exacerbates the inflammatory response, leading to cardio-depressive effects and cardiac remodeling [68]. The non-classical pathway does not require the involvement of inflammatory vesicles but directly activates gasdermin-D via caspase-4/caspase-5/caspase-11, leading to cell membrane rupture and pyroptosis [69].
Recently, NLRP3 inflammasome was proposed as a new biomarker of cardiovascular diseases and predictor of hospitalization and death for myocardial injurie [70]. Pyrin domain-containing 1 inhibits excessive NLRP3 inflammasome activity and thereby ameliorates auto-inflammatory disease [71]. Pyrin domain-containing 1 regulates the innate immune response by inhibiting nuclear factor-kappa B (NF-κB) transcription factor activity and pro-caspase-1 activation [72]. Cai et al. [29] demonstrated that miR-136-5p overexpression can increase the level of pyrin domain-containing 1, which inhibits pyroptosis and alleviates CME-induced myocardial injury. Furthermore, miR-30e-3p overexpression reduces the expression of caspase-1 and NLRP3 in the CME rat model [73]. Further research showed that miR-30e-3p alleviates CME-induced pyroptosis and inflammatory responses by targeting and inhibiting the expression of histone deacetylase (HDAC) 2, partly due to the reduction of HDAC2 levels, which attenuates the inhibition of mothers against decapentaplegic homolog 7 expression [73]. Additionally, miR-142-3p overexpression can target the ataxin 1/HDAC3 axis, promoting the deacetylation modification of histone H3 and inhibiting CME-induced myocardial pyroptosis in rats [74]. Zhou et al. [75] found that overexpression of lncRNA-taurine up-regulated gene 1 can target the miR-186-5p/x-linked inhibitor of apoptosis protein axis in rat CME models, thus inhibiting NLRP3-mediated pyroptosis and exerting a cardioprotective effect. MiR-200a-3p can also alleviate cardiac dysfunction caused by CME by inhibiting NLRP3-mediated pyroptosis [76]. Besides microRNAs, lncRNAs participate in the development of CME-related pyroptosis. LncRNA-Sox2OT can act as a molecular sponge for miR-23b. Also, lncRNA-Sox2OT silencing promotes the binding of miR-23b to the 3'UTR of TLR4 mRNA, thereby inhibiting its downstream NF-κB-mediated signaling pathways, thus alleviating CME-induced cardiomyocyte pyroptosis [77]. Besides microRNAs, lncRNAs participate in the development of CME-related pyroptosis. LncRNA-Sox2OT can act as a molecular sponge for miR-23b. Also, lncRNA-Sox2OT silencing promotes the binding of miR-23b to the 3'UTR of TLR4 mRNA, thereby inhibiting its downstream NF-κB-mediated signaling pathways, thus alleviating CME-induced cardiomyocyte pyroptosis [78, 79]. Liu et al. [80] showed that nicorandil can reduce the expression of thioredoxin-interacting protein and inhibit NLRP3-mediated pyroptosis, thus maintaining the function of rat cardiomyocytes. Additionally, Li et al. [81] showed that colchicine can promote the expression of silent information regulator 1 and inhibit NLRP3-mediated cardiomyocyte pyroptosis in rat CME models. Therefore, targeting pyroptosis and related signaling pathways is a potential strategy for CME treatment.
Ferroptosis and CME
Ferroptosis was first identified by Dixon et al. [22] in 2012. Ferroptosis is an iron-dependent cell death that is morphologically and genetically distinct from other RCDs [22]. Iron is an essential trace element that mediates various biological processes and maintains the normal life activities of the organism [82]. However, excessive accumulation of intracellular Fe2+ can contribute to the generation of lipid reactive oxygen radicals and the accumulation of lipid peroxides, which induces ferroptosis [83, 84].
Ferroptosis status is widely detected by measuring intracellular Fe2+ concentration, malondialdehyde levels, and the ratio of reduced glutathione/oxidised glutathione [85]. Liu et al. [86] reported reduced levels of glutathione peroxidase (GPX) 4 and elevated levels of prostaglandin endoperoxide synthase 2, malondialdehyde, and Fe2+ by constructing a rat model of CME, suggesting that CME induces the ferroptosis. Further pretreatment of the CME model using desferrioxamine (an inhibitor of ferroptosis) and atorvastatin increased GPX4 expression levels, decreased peroxisomal synthase 2 levels, decreased malondialdehyde and Fe2+ levels within the prostaglandins, reduced inflammatory response in the lesion area and significantly improved cardiac function of the rats. Gao et al. [87] indicated that miR-706 is a molecular sponge of lncRNA-Gm47283. Furthermore, they showed that knockdown of lncRNA-Gm47283 in the rat myocardial infarction model up-regulates miR-706 levels while decreasing the expression of prostaglandin endoperoxide synthase 2, arachidonic acid 15-lipoxygenase, and GPX4 [87]. This suggests that lncRNA-Gm47283 knockdown can inhibit ferroptosis and protect cardiac function by targeting miR-706. In addition, resveratrol pretreatment can increase the expression of lysine acetyltransferase 5 and GPX4 in rat myocardial infarction model, suggesting that resveratrol can inhibit cardiomyocyte ferroptosis and alleviate cardiac dysfunction caused by myocardial infarction [88]. However, further studies should assess whether lncRNA-Gm47283 and resveratrol exert the same effect of antagonising ferroptosis in the CME model. Ischemia-reperfusion injury is considered a significant cause of CME. Research has confirmed that galangin suppressed ferroptosis through nuclearfactor erythroidderived 2-like 2/ GPX4 signaling pathway activation [89]. This suggests that the aforementioned effects may be present in CME. Epidemiological results show that severity of heart disease is related to degree of environmental contamination [90]. Di(2-ethylhexyl) phthalate, an environmental pollutant, causes lipid peroxidation and elevated Fe2+ levels in cardiomyocytes [90]. Further study showed that di(2-ethylhexyl) phthalate induced the onset of ferroptosis in cardiomyocytes by upregulating heme-oxygense-1 [90]. Obviously, ferroptosis is closely related to the integrity of cardiac function and requires in-depth study in the context of CME.
Summary and Future Perspectives
In conclusion, RCD is triggered by specific signals that elicit distinct death patterns associated with CME progression. However, the related research has mainly focused on apoptosis, autophagy, pyroptosis and ferroptosis, ignoring other RCD forms, such as cuproptosis, disulfidptosis, and necroptosis. The mechanisms of RCD are multifactorial and complex. Different forms of RCD are associated with distinct characteristic genes and signaling pathways. Furthermore, certain molecular crosstalk occurs between various forms of RCD, which further limits the research. Therefore, a more comprehensive understanding of RCD and CME may facilitate the clinical translation of existing findings. Nonetheless, further studies should comprehensively assess the potential regulatory mechanisms of RCD in CME to provide a definitive reference for the treatment of cardiovascular diseases, including CME.
Abbreviations
AKT: protein kinase B; BIM: BCL-2-like protein 11; BMF: BCL-2 modifying factor; CME: coronary microembolization; Caspase: cysteine aspartate protease; GPX4: glutathione peroxidase 4; GSK-3β: glycogen synthase kinase 3; HDAC: histone deacetylase; LC3-II: microtubule-associated protein II light chain 3; LOX-1: lipoprotein receptor-1; NLRP3: NOD-like receptor thermal protein domain associated protein 3; NF-κB: nuclear factor-kappa B; PI3K: phosphoinositide 3-kinase; POP1: pyrin only protein 1; RCD: regulatory cell death; SIRT1: sirtuin1; TNF: tumour necrosis factor.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant number 82260072]; Guangxi Natural Science Foundation [grant number 2020GXNSFFA297002].
Author contributions
Chen Chang and Wan-Zhong Huang searched the literature and drafted the manuscript; Qiang Su, Li-Rong Mo and Qiang Wu conceived and designed the review; Chen Chang and Ru-Ping Cai constructed the figures; Qiang Su, Li-Rong Mo and Qiang Wu made critical revisions of the review. All the authors contributed to the article and approved the final version for submission.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kumar N, Mohammadnezhad M, Narayan R. 'Some of my patients only come to renew their prescriptions. They are not interested in any additional advice or support'. Physicians' perceptions on their roles in cardiovascular diseases risk reduction and management in Fiji. Prim Health Care Res Dev. 2023;24:e11
2. Chang C, Cai R, Wu Q. et al. Uncovering the Genetic Link between Acute Myocardial Infarction and Ulcerative Colitis Co-Morbidity through a Systems Biology Approach. CVIA. 2023;8(1):e978
3. Cai R, Chang C, Zhong X. et al. Lowering of Blood Lipid Levels with a Combination of Pitavastatin and Ezetimibe in Patients with Coronary Heart Disease: A Meta-Analysis. CVIA. 2023;7(1):e985
4. Li D H, Wu Q, Lan J S. et al. Plasma metabolites and risk of myocardial infarction: a bidirectional Mendelian randomization study. J Geriatr Cardiol. 2024;21(2):219-231
5. Wu Q, He C, Huang W. et al. Gastroesophageal reflux disease influences blood pressure components, lipid profile and cardiovascular diseases: Evidence from a Mendelian randomization study. J Transl Int Med. 2024;12(5):510-525
6. Jain M, Dev R, Doddapattar P. et al. Integrin α9 regulates smooth muscle cell phenotype switching and vascular remodeling. JCI Insight. 2021;6(10):e147134
7. Heusch G, Kleinbongard P, Böse D. et al. Coronary microembolization: from bedside to bench and back to bedside. Circulation. 2009;120(18):1822-1836
8. Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17(12):773-789
9. Su Q, Lv X, Sun Y. et al. Role of TLR4/MyD88/NF-κB signaling pathway in coronary microembolization-induced myocardial injury prevented and treated with nicorandil. Biomed Pharmacother. 2018;106:776-784
10. Kleinbongard P, Heusch G. A fresh look at coronary microembolization. Nat Rev Cardiol. 2022;19(4):265-280
11. Schwartz R S, Burke A, Farb A. et al. Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death: relation to epicardial plaque histopathology. J Am Coll Cardiol. 2009;54(23):2167-2173
12. Yuan Y, Li B, Peng W. et al. Protective effect of glycyrrhizin on coronary microembolization-induced myocardial dysfunction in rats. Pharmacol Res Perspect. 2021;9(1):e00714
13. Kleinbongard P, Böse D, Baars T. et al. Vasoconstrictor potential of coronary aspirate from patients undergoing stenting of saphenous vein aortocoronary bypass grafts and its pharmacological attenuation. Circ Res. 2011;108(3):344-352
14. Qiao P, Zhang B, Liu X. et al. Effects of Escin on Oxidative Stress and Apoptosis of H9c2 Cells Induced by H(2)O(2). Dis Markers. 2022;2022:7765353
15. Xue Y, Jiang X, Wang J. et al. Effect of regulatory cell death on the occurrence and development of head and neck squamous cell carcinoma. Biomark Res. 2023;11(1):2
16. Fernandez Rico C, Konate K, Josse E. et al. Therapeutic Peptides to Treat Myocardial Ischemia-Reperfusion Injury. Front Cardiovasc Med. 2022;9:792885
17. Xie D, Wang Q, Wu G. Research progress in inducing immunogenic cell death of tumor cells. Front Immunol. 2022;13:1017400
18. Chang C, Cai R P, Su Y M. et al. Mesenchymal Stem Cell-Derived Exosomal Noncoding RNAs as Alternative Treatments for Myocardial Ischemia-Reperfusion Injury: Current Status and Future Perspectives. J Cardiovasc Transl Res. 2023;16(5):1085-1098
19. Kerr J F, Wyllie A H, Currie A R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-257
20. Pandarathodiyil A K, Vijayan S P, Ramanathan A. et al. Autophagy: The "Pac-Man" within Us-Ally or Adversary? J Contemp Dent Pract. 2021;22(10):1079-1081
21. Cookson B T, Brennan M A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113-114
22. Dixon S J, Lemberg K M, Lamprecht M R. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072
23. Tsvetkov P, Coy S, Petrova B. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254-1261
24. Liu X, Nie L, Zhang Y. et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25(3):404-414
25. Degterev A, Huang Z, Boyce M. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112-119
26. Heusch G. Myocardial ischemia/reperfusion: Translational pathophysiology of ischemic heart disease. Med. 2024;5(1):10-31
27. Cao Y, Chen Z, Jia J. et al. Rosuvastatin Alleviates Coronary Microembolization-Induced Cardiac Injury by Suppressing Nox2-Induced ROS Overproduction and Myocardial Apoptosis. Cardiovasc Toxicol. 2022;22(4):341-351
28. Mao Q, Liang X, Wu Y. et al. Resveratrol Attenuates Cardiomyocyte Apoptosis in Rats Induced by Coronary Microembolization Through SIRT1-Mediated Deacetylation of p53. J Cardiovasc Pharmacol Ther. 2019;24(6):551-558
29. Cai R, Xu Y, Ren Y. et al. MicroRNA-136-5p protects cardiomyocytes from coronary microembolization through the inhibition of pyroptosis. Apoptosis. 2022;27(3-4):206-221
30. Zambonatto R F, Teixeira R N, Poma S O. et al. Features of Neutrophils From Atopic and Non-Atopic Elite Endurance Runners. Front Immunol. 2021;12:670763
31. Wu L, Xu G, Li N. et al. Curcumin Analog, HO-3867, Induces Both Apoptosis and Ferroptosis via Multiple Mechanisms in NSCLC Cells with Wild-Type p53. Evid Based Complement Alternat Med. 2023;2023:8378581
32. Heusch G, Schulz R, Baumgart D. et al. Coronary microembolization. Prog Cardiovasc Dis. 2001;44(3):217-230
33. Huang Y P, Hsia T C, Yeh C A. et al. PW06 Triggered Fas-FADD to Induce Apoptotic Cell Death In Human Pancreatic Carcinoma MIA PaCa-2 Cells through the Activation of the Caspase-Mediated Pathway. Oxid Med Cell Longev. 2023;2023:3479688
34. Li Y, Guo F, Guan Y. et al. Novel Anthraquinone Compounds Inhibit Colon Cancer Cell Proliferation via the Reactive Oxygen Species/JNK Pathway. Molecules. 2020;25(7):1672
35. Liu T, Zhou Y, Wang J Y. et al. Coronary Microembolization Induces Cardiomyocyte Apoptosis in Swine by Activating the LOX-1-Dependent Mitochondrial Pathway and Caspase-8-Dependent Pathway. J Cardiovasc Pharmacol Ther. 2016;21(2):209-218
36. Chen Z Q, Zhou Y, Huang J W. et al. Puerarin pretreatment attenuates cardiomyocyte apoptosis induced by coronary microembolization in rats by activating the PI3K/Akt/GSK-3β signaling pathway. Korean J Physiol Pharmacol. 2021;25(2):147-157
37. Li T, Chen Z, Zhou Y. et al. Resveratrol Pretreatment Inhibits Myocardial Apoptosis in Rats Following Coronary Microembolization via Inducing the PI3K/Akt/GSK-3β Signaling Cascade. Drug Des Devel Ther. 2021;15:3821-3834
38. Qin Z, Wang X, Zhou Y. et al. Upregulation of miR-29b-3p alleviates coronary microembolization-induced myocardial injury via regulating BMF and GSK-3β. Apoptosis. 2023;28(1-2):210-221
39. Zhu H H, Wang X T, Sun Y H. et al. MicroRNA-486-5p targeting PTEN Protects Against Coronary Microembolization-Induced Cardiomyocyte Apoptosis in Rats by activating the PI3K/AKT pathway. Eur J Pharmacol. 2019;855:244-251
40. Green D R. The Death Receptor Pathway of Apoptosis. Cold Spring Harb Perspect Biol. 2022;14(2):a041053
41. Park Y H, Han C W, Jeong M S. et al. DED Interaction of FADD and Caspase-8 in the Induction of Apoptotic Cell Death. J Microbiol Biotechnol. 2022;32(8):1034-1040
42. Thielmann M, Dörge H, Martin C. et al. Myocardial dysfunction with coronary microembolization: signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha, and sphingosine. Circ Res. 2002;90(7):807-813
43. Zhou H, Zhou M, Hu Y. et al. TNF-α Triggers RIP1/FADD/Caspase-8-Mediated Apoptosis of Astrocytes and RIP3/MLKL-Mediated Necroptosis of Neurons Induced by Angiostrongylus cantonensis Infection. Cell Mol Neurobiol. 2022;42(6):1841-1857
44. Su Q, Li L, Zhou Y. et al. Induction of myocardial PDCD4 in coronary microembolization-related cardiac dysfunction: evidence from a large-animal study. Cell Physiol Biochem. 2014;34(2):533-542
45. Dhingra R, Lin J, Kirshenbaum L A. Disruption of RIP1-FADD Complexes by MicroRNA-103/107 Provokes Necrotic Cardiac Cell Death. Circ Res. 2015;117(4):314-316
46. Liu Q, Duan S B, Wang L. et al. Apelin-13 alleviates contrast-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Ren Fail. 2023;45(1):2179852
47. Terai K, Hiramoto Y, Masaki M. et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25(21):9554-9575
48. Liu T, Zhou Y, Liu Y C. et al. Coronary Microembolization Induces Cardiomyocyte Apoptosis Through the LOX-1-Dependent Endoplasmic Reticulum Stress Pathway Involving JNK/P38 MAPK. Can J Cardiol. 2015;31(10):1272-1281
49. Cheng A, Zhang H, Chen B. et al. Modulation of autophagy as a therapeutic strategy for Toxoplasma gondii infection. Front Cell Infect Microbiol. 2022;12:902428
50. Wu C, Yao W, Kai W. et al. Mitochondrial Fusion Machinery Specifically Involved in Energy Deprivation-Induced Autophagy. Front Cell Dev Biol. 2020;8:221
51. Carinci M, Palumbo L, Pellielo G. et al. The Multifaceted Roles of Autophagy in Infectious, Obstructive, and Malignant Airway Diseases. Biomedicines. 2022;10(8):1944
52. Ghosh R, Gillaspie J J, Campbell K S. et al. Chaperone-mediated autophagy protects cardiomyocytes against hypoxic-cell death. Am J Physiol Cell Physiol. 2022;323(5):C1555-c1575
53. Gatica D, Chiong M, Lavandero S. et al. The role of autophagy in cardiovascular pathology. Cardiovasc Res. 2022;118(4):934-950
54. Wang X, Li Y, Lu J. et al. Engineering Nanoplatform for Combined Cancer Therapeutics via Complementary Autophagy Inhibition. Int J Mol Sci. 2022;23(2):657
55. Zhang G, He C, Wu Q. et al. Impaired Autophagy Induced by oxLDL/β2GPI/anti-β2GPI Complex through PI3K/AKT/mTOR and eNOS Signaling Pathways Contributes to Endothelial Cell Dysfunction. Oxid Med Cell Longev. 2021;2021:6662225
56. Ye X, Chen L. Protective role of autophagy in triptolide-induced apoptosis of TM3 Leydig cells. J Transl Int Med. 2023;11(3):265-274
57. Wang X T, Wu X D, Lu Y X. et al. Potential Involvement of MiR-30e-3p in Myocardial Injury Induced by Coronary Microembolization via Autophagy Activation. Cell Physiol Biochem. 2017;44(5):1995-2004
58. Mo B, Wu X, Wang X. et al. miR-30e-5p Mitigates Hypoxia-Induced Apoptosis in Human Stem Cell-Derived Cardiomyocytes by Suppressing Bim. Int J Biol Sci. 2019;15(5):1042-1051
59. Gao Y H, Qian J Y, Chen Z W. et al. Suppression of Bim by microRNA-19a may protect cardiomyocytes against hypoxia-induced cell death via autophagy activation. Toxicol Lett. 2016;257:72-83
60. Wang X T, Wu X D, Lu Y X. et al. Egr-1 is involved in coronary microembolization-induced myocardial injury via Bim/Beclin-1 pathway-mediated autophagy inhibition and apoptosis activation. Aging (Albany NY). 2018;10(11):3136-3147
61. Zhou J, Wang Z, He Y. et al. Qiliqiangxin reduced cardiomyocytes apotosis and improved heart function in infarcted heart through Pink1/Parkin -mediated mitochondrial autophagy. BMC Complement Med Ther. 2020;20(1):203
62. Eum K H, Lee M. Targeting the autophagy pathway using ectopic expression of Beclin 1 in combination with rapamycin in drug-resistant v-Ha-ras-transformed NIH 3T3 cells. Mol Cells. 2011;31(3):231-238
63. Hseu Y C, Tseng Y F, Pandey S. et al. Coenzyme Q(0) Inhibits NLRP3 Inflammasome Activation through Mitophagy Induction in LPS/ATP-Stimulated Macrophages. Oxid Med Cell Longev. 2022;2022:4266214
64. Zhang K J, Wu Q, Jiang S M. et al. Pyroptosis: A New Frontier in Kidney Diseases. Oxid Med Cell Longev. 2021;2021:6686617
65. Zuo Q, He L, Ma S. et al. Canagliflozin Alleviates Atherosclerosis Progression through Inflammation, Oxidative Stress, and Autophagy in Western Diet-fed ApoE-/- Mice. CVIA. 2024;9(1):e981
66. Schnappauf O, Chae J J, Kastner D L. et al. The Pyrin Inflammasome in Health and Disease. Front Immunol. 2019;10:1745
67. Cote B, Elbarbry F, Bui F. et al. Mechanistic Basis for the Role of Phytochemicals in Inflammation-Associated Chronic Diseases. Molecules. 2022;27(3):781
68. Miteva K, Pappritz K, Sosnowski M. et al. Mesenchymal stromal cells inhibit NLRP3 inflammasome activation in a model of Coxsackievirus B3-induced inflammatory cardiomyopathy. Sci Rep. 2018;8(1):2820
69. Song R, Wu Y, He S. et al. A pilot study on pyroptosis related genes in peripheral blood mononuclear cells of non-small cell lung cancer patients. BMC Pulm Med. 2023;23(1):174
70. Quagliariello V, De Laurentiis M, Cocco S. et al. NLRP3 as Putative Marker of Ipilimumab-Induced Cardiotoxicity in the Presence of Hyperglycemia in Estrogen-Responsive and Triple-Negative Breast Cancer Cells. Int J Mol Sci. 2020;21(20):7802
71. de Almeida L, Khare S, Misharin A V. et al. The PYRIN Domain-only Protein POP1 Inhibits Inflammasome Assembly and Ameliorates Inflammatory Disease. Immunity. 2015;43(2):264-276
72. Crist A M, Hinkle K M, Wang X. et al. Transcriptomic analysis to identify genes associated with selective hippocampal vulnerability in Alzheimer's disease. Nat Commun. 2021;12(1):2311
73. Dai R, Ren Y, Lv X. et al. MicroRNA-30e-3p reduces coronary microembolism-induced cardiomyocyte pyroptosis and inflammation by sequestering HDAC2 from the SMAD7 promoter. Am J Physiol Cell Physiol. 2023;324(2):C222-c235
74. Xu Y, Lv X, Cai R. et al. Possible implication of miR-142-3p in coronary microembolization induced myocardial injury via ATXN1L/HDAC3/NOL3 axis. J Mol Med (Berl). 2022;100(5):763-780
75. Zhou Y, Li T, Chen Z. et al. Overexpression of lncRNA TUG1 Alleviates NLRP3 Inflammasome-Mediated Cardiomyocyte Pyroptosis Through Targeting the miR-186-5p/XIAP Axis in Coronary Microembolization-Induced Myocardial Damage. Front Immunol. 2021;12:637598
76. Chen Z Q, Zhou Y, Chen F. et al. miR-200a-3p Attenuates Coronary Microembolization-Induced Myocardial Injury in Rats by Inhibiting TXNIP/NLRP3-Mediated Cardiomyocyte Pyroptosis. Front Cardiovasc Med. 2021;8:693257
77. Xuan L, Fu D, Zhen D. et al. Long non-coding RNA Sox2OT promotes coronary microembolization-induced myocardial injury by mediating pyroptosis. ESC Heart Fail. 2022;9(3):1689-1702
78. Luo C J, Li T, Li H L. et al. Resveratrol pretreatment alleviates NLRP3 inflammasome-mediated cardiomyocyte pyroptosis by targeting TLR4/MyD88/NF-κB signaling cascade in coronary microembolization-induced myocardial damage. Korean J Physiol Pharmacol. 2023;27(2):143-155
79. Li H L, Li T, Chen Z Q. et al. Tanshinone IIA reduces pyroptosis in rats with coronary microembolization by inhibiting the TLR4/MyD88/NF-κB/NLRP3 pathway. Korean J Physiol Pharmacol. 2022;26(5):335-345
80. Liu Y, Shu J, Liu T. et al. Nicorandil protects against coronary microembolization-induced myocardial injury by suppressing cardiomyocyte pyroptosis via the AMPK/TXNIP/NLRP3 signaling pathway. Eur J Pharmacol. 2022;936:175365
81. Li H, Yang H, Qin Z. et al. Colchicine ameliorates myocardial injury induced by coronary microembolization through suppressing pyroptosis via the AMPK/SIRT1/NLRP3 signaling pathway. BMC Cardiovasc Disord. 2024;24(1):23
82. Xiao Z, Zhao H. Ferroptosis-Related APOE, BCL3 and ALOX5AP Gene Polymorphisms are Associated with the Risk of Thyroid Cancer. Pharmgenomics Pers Med. 2022;15:157-165
83. Luan Y, Huang E, Huang J. et al. Serum myoglobin modulates kidney injury via inducing ferroptosis after exertional heatstroke. J Transl Int Med. 2023;11(2):178-188
84. Liu Y, Liu Y, Ye S. et al. A new ferroptosis-related signature model including messenger RNAs and long non-coding RNAs predicts the prognosis of gastric cancer patients. J Transl Int Med. 2023;11(2):145-155
85. Alarcón-Veleiro C, Mato-Basalo R, Lucio-Gallego S. et al. Study of Ferroptosis Transmission by Small Extracellular Vesicles in Epithelial Ovarian Cancer Cells. Antioxidants (Basel). 2023;12(1):183
86. Liu T, Shu J, Liu Y. et al. Atorvastatin attenuates ferroptosis-dependent myocardial injury and inflammation following coronary microembolization via the Hif1a/Ptgs2 pathway. Front Pharmacol. 2022;13:1057583
87. Gao F, Zhao Y, Zhang B. et al. Suppression of lncRNA Gm47283 attenuates myocardial infarction via miR-706/Ptgs2/ferroptosis axis. Bioengineered. 2022;13(4):10786-10802
88. Liu J, Zhang M, Qin C. et al. Resveratrol Attenuate Myocardial Injury by Inhibiting Ferroptosis Via Inducing KAT5/GPX4 in Myocardial Infarction. Front Pharmacol. 2022;13:906073
89. Yang T, Liu H, Yang C. et al. Galangin Attenuates Myocardial Ischemic Reperfusion-Induced Ferroptosis by Targeting Nrf2/Gpx4 Signaling Pathway. Drug Des Devel Ther. 2023;17:2495-2511
90. Wang J X, Zhao Y, Chen M S. et al. Heme-oxygenase-1 as a target for phthalate-induced cardiomyocytes ferroptosis. Environ Pollut. 2023;317:120717
Author contact
![]() Corresponding authors: Qiang Su, Department of Cardiology, Jiangbin Hospital of Guangxi Zhuang Autonomous Region, Nanning, Guangxi, China, Email: suqiang1983edu.cn; Qiang Wu, Senior Department of Cardiology, The Sixth Medical Center, Chinese PLA General Hospital, Beijing 100048, People's Republic of China, Email: wuqiangcom; Li-Rong Mo, Department of Cardiology, Jiangbin Hospital of Guangxi Zhuang Autonomous Region, Nanning 530021, Guangxi, People's Republic of China, Email: 292318992com.
Corresponding authors: Qiang Su, Department of Cardiology, Jiangbin Hospital of Guangxi Zhuang Autonomous Region, Nanning, Guangxi, China, Email: suqiang1983edu.cn; Qiang Wu, Senior Department of Cardiology, The Sixth Medical Center, Chinese PLA General Hospital, Beijing 100048, People's Republic of China, Email: wuqiangcom; Li-Rong Mo, Department of Cardiology, Jiangbin Hospital of Guangxi Zhuang Autonomous Region, Nanning 530021, Guangxi, People's Republic of China, Email: 292318992com.