Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(14):2807-2823. doi:10.7150/ijms.97169 This issue Cite
Research Paper
Comprehensive Analysis of the Prognostic Implications and Biological Function of HDACs in Liver Hepatocellular Carcinoma
Zhaolei Cui1#, Chaoqiang Zheng1#, Yiqing You1, Shijie He1, Shan Jiang2, Yan Chen1, Yingying Lin1 ![]() , Zhenzhou Xiao1
, Zhenzhou Xiao1 ![]()
1. Laboratory of Biochemistry and Molecular Biology Research, Department of Laboratory Medicine, Clinical Oncology School of Fujian Medical University, Fujian Cancer Hospital, Fuzhou, 350014, China.
2. Department of Gynecology, Clinical Oncology School of Fujian Medical University, Fujian Cancer Hospital, Fuzhou, 350014, China.
#Co-first authors
Received 2024-4-10; Accepted 2024-9-6; Published 2024-10-28
Abstract

Background: The prognostic significance and biological functions of the histone deacetylases (HDACs) gene family in liver hepatocellular carcinoma (LIHC) have not been fully investigated.
Methods: Using Kaplan-Meier and Cox regression analysis, this study determined if HDAC genes were relevant for prognosis in LIHC. A regression model utilizing HDAC genes and the least absolute shrinkage and selection operator (LASSO) was created to foretell LIHC risk. A selective inhibitor of endogenous HDACs, CKD-581, was studied in vitro and in vivo to determine its effects on the development, invasion, migration, and proliferation of LIHC cell lines.
Results: Six HDACs were identified as correlating with the prognosis of LIHC. Overall survival (OS) was found to be shorter in individuals with higher risk scores when compared to those with lower risk scores, according to survival study. Natural killer cell infiltration was higher in individuals with lower risk ratings, which was mainly explained by the type II interferon (IFN) response. Limiting the activity of endogenous HDACs caused LIHC cell death by preventing their migration, invasion, and proliferation. In vivo studies confirmed that blocking HDAC expression inhibited tumor growth in mice. Further mechanistic studies showed that inhibition of HDACs expression elevates the protein levels of P21 and P27, and reduces those of cyclins A2, B1, D1 and E1.
Conclusions: The risk score prognostic model based on HDAC genes could provide a valuable prognostic biomarker for LIHC. CKD-581 prohibits LIHC progression via inhibiting the cell cycle signaling pathway. CKD-581 holds promise as a therapeutic agent for the clinical management of LIHC.
Keywords: histone deacetylases (HDACs), CKD-581, liver hepatocellular carcinoma, prognostic model, cyclin
Introduction
According to the most current cancer statistics from 2022, LIHC is the third most frequent disease worldwide and the sixth most common cancer overall. To add insult to injury, LIHC ranks second in male death rates and has a two- to thrice greater incidence in males than females in most locations [1]. Because tumors grow so rapidly and are so hard to detect in their early stages, most patients with LIHC do not receive a diagnosis until the disease has progressed or metastasized. This highlights the tremendous clinical and societal importance of developing accurate techniques of prognosis assessment and targeted treatment approaches [2].
Critical to gene regulation and cellular metabolism is the histone deacetylase (HDAC) family of enzymes[3]. Within this family, there are four distinct classes that make up the 18 isoforms found in mammals: proteins belonging to class I Rpd3-like (HDAC1, HDAC2, HDAC3, and HDAC8), proteins belonging to class II Hda1-like (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10), proteins belonging to class III Sir2-like (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7), and proteins belonging to class IV (HDAC11) [4]. HDACs deacetylate lysine residues on histone tails, which modulates gene expression. Given that epigenetic modifications are crucial in tumorigenesis and tumor progression, these enzymes can significantly influence cancer development [5]. Previous research established that HDACs play a role in hematological malignancies [6], and more recent investigations have shown that glioma [7], renal clear cell carcinoma [8], breast cancer [9], and gastric cancer [10] all exhibit differential expression of HDACs. In addition, studies show that HDAC gene knockdown causes cell cycle arrest and apoptosis in cancers, demonstrating the significant correlation between HDAC expression, tumor cell proliferation, and tumor progression [11].
Studies on the HDAC gene family in LIHC are limited, most have focused on the functional role between only one or two HDAC genes and LIHC [12-16], and research on the prognostic feature of HDACs in LIHC is scarce. Yet to find out what role HDACs play in LIHC, we need to perform a thorough and methodical investigation of the whole HDAC gene family. Alteminostat (CKD-581; molecular weight, 492.61; molecular formula, C27H36N6O3) is an effective, specific inhibitor of HDACs, promoting the acetylation of histone H3 and microtubulin. Recent clinical study results showed that CKD-581 was safe, well-tolerated, and effective against cancer when used alone in treating trefractory lymphoma or multiple myeloma [17]. In this study, our objective was to analyze the expression, mutations, function, and immune infiltration of the HDAC family of genes using online databases, assessing their potential oncogenic and prognostic values for LIHC. Concurrently, we corroborated our hypotheses by examining clinical samples. Utilizing the specific HDACs inhibitor CKD-581, we explored the biological functions and potential molecular mechanisms by which HDAC inhibition affects LIHC at the cellular level, and confirmed our results with in vivo experiments using CKD-581 in mice.
Materials and methods
Tissue samples
Approval No. SQ2015-049-01 was granted by the Clinical Research Ethics Committee of Fujian Cancer Hospital, and all subjects voluntarily gave their informed consent. We used three sets of LIHC tissue and neighboring non-tumor tissue samples that were taken during surgical excisions at Fujian Cancer Hospital between September 2022 and September 2023. All samples were shown to have LIHC by postoperative pathology. We treated one half of the samples in 4% paraformaldehyde and immersed the other half in paraffin for long-term preservation; the other half was kept in liquid nitrogen. Additionally, we acquired LIHC tissue cDNA microarrays (cDNA-HLivH090Su01) from Shanghai Xinchao Biotechnology Co. Ltd, which included data from 64 patients treated from April 2006 to May 2013, with follow-ups ranging from 2 to 9 years.
Data collection and data processing
Expression, clinical, and single nucleotide polymorphism data were extracted from 374 LIHC cases and 50 controls in the Cancer Genome Atlas (TCGA) database. Additionally, clinical and ICGC expression data for LIHC patients were obtained. Furthermore, we obtained expression data for 33 common cancers and copy number variation (CNV) information for LIHC from the Xena database at UC Santa Cruz (UCSC). We next proceeded to analyze 18 HDAC genes in additional detail.
Construction and validation of a prognostic model
Using information from the TCGA database, we created a model to forecast OS. The optimal genes for this prognostic model were selected based on a regression coefficient (coef) value in conjunction with genes that are both differentially expressed and associated to prognosis. The coef was determined by LASSO regression analysis, with replacement 1,000 times to compress the coefs of less impactful HDACs to 0, deeming them insignificant. The larger the absolute value of a coef, the greater its impact on the prognostic outcome.
The following is the formula for determining the risk score of each sample:
risk score = coef1 × gene1 expression + coef2 × gene2 expression + ... + coefn × genen expression, where coef represents the corresponding HDAC coefficient, and gene expression denotes the respective HDAC expression level. Based on their median risk score, each patient was categorized as either high-risk or low-risk. To examine the OS variation among the groups, we used Kaplan-Meier survival curves. For patients undergoing TCGA and ICGC, we used receiver operating characteristic (ROC) curves to assess the accuracy of the prognosis for 1-, 2-, and 3-year OS. To categorize the risks, we performed principal component analysis. It was afterwards determined that the ICGC dataset had adequately verified the predictive model.
Independent prognostic analysis and clinical correlation
In order to verify that the risk score is independent, we used both multivariate and univariate Cox regression analysis. We examined many clinical datasets using stratified analysis to see if risk ratings were associated with clinical features.
Immune infiltration, tumor microenvironment, gene set enrichment, and drug sensitivity analysis
Immune cell scores and functions were quantified for each LIHC patient using a single-sample gene set enrichment analysis (ssGSEA), and these results were then compared across high-risk and low-risk categories [18]. We calculated risk scores, immunological scores, and stromal scores for each category. We then performed enrichment analysis on the TCGA dataset to identify high-risk and low-risk groups. We used the Cell Miner database to retrieve information about gene expression and medication sensitivity. We then used P-values and correlation coefficients to investigate the link between gene expression and drug sensitivity.
Reagents and animals
The Shanghai, China-based provider MCE supplied the CKD-581 medication. The C57BL/6J mice utilized in this research were procured from Shanghai Slake Experimental Animal Co., Ltd. The mice ranged in weight from 20 to 25 g, were eight weeks old, and were male. Procell Life Science&Technology Co.Ltd of Wuhan, China, was contacted in order to obtain Thepa1-6, Huh-7, and Bel7402. The study's mice were housed in a climate-controlled chamber that maintained a constant temperature, relative humidity, and light intensity. They had an endless supply of food and water. The study's method was green-lit by Fujian Medical University's Animal Ethics Center (ethics number: IACUC FJMU 2022-0723), and the mice were well-cared-for throughout.
Cell proliferation, migration, and invasion assays
The proliferation of cells was measured using a Cell Counting Kit-8 from APExBIO in Houston, TX, USA. To summarize, with a density of 2 × 103 cells per well, Bel7402, Huh-7, and Hepa1-6 cells were seeded into 96-well plates. We treated the cells with CKD-581 at doses of 1 µM and 10 µM after assigning five replicate wells to each group. Thermo Fisher Scientific's enzyme calibration was used to quantify the absorbance at 450 nm at 2, 4, 8, and 72 hours, respectively.
The cell migration assay was carried out by placing 2 × 104 cells into one chamber of a 24-well plate (BIOLIF, Guangzhou, China) that did not have a membrane coating. The lower chamber contained 600 µL of complete medium, while this compartment had 200 µL of serum-free media. The cells were cultured at 37°C for 24 hours after being exposed to either 1 µM or 10 µM concentrations of CKD-581 in both chambers. Subsequently, cells were incubated with 4% paraformaldehyde on the membrane surface for 20 minutes, followed by the staining with 0.1% crystal violet. Afterwards, an inverted microscope (Olympus, Tokyo, Japan) was used to ascertain the cell count.
The cell invasion assay was conducted by coating the bottom of the upper chamber with 80 µL of matrix gel (Corning Inc., Corning, NY, USA). The subsequent steps were identical to those used in the migration assay.
Plate cloning assay
A total of 1 × 103 cells per well of six-well plates containing complete medium were used to culture the logarithmic growth phase cells. Subsequently, CKD-581 was introduced to the cells at concentrations of 1 µM and 10 µM. Over the course of three weeks, the medium was swapped out every three days. Upon becoming visible, the clones were rinsed three times with phosphate-buffered saline (PBS, pH 7.4). After that, they were incubated in 4% paraformaldehyde for 30 minutes, dyed crystal violet, and then rinsed. The clones were subsequently photographed in order to count them.
Apoptosis assay
In order to get 1 × 106 cells/mL, the cells were first collected, rinsed twice with 4°C precooled PBS, spun in a centrifuge to collect liquid above the cells, and then resuspended in 1×Binding Buffer. After that, each cell tube was supplemented with 5 µL of Annexin V-PE and 5 µL of 7-AAD. After a gentle mixing, the tubes were incubated in the dark at 25°C for 15 minutes. The reaction was stopped by adding 400 μL of 1× Binding Buffer, and it was tested on a CytoFlex machine (Beckman Coulter Life Sciences, USA) within an hour.
Real-time quantitative PCR (RT-qPCR) analysis
The cells or tissues that were exposed to CKD-581 (1 µM, 10 µM) for 24 hours had their total RNA harvested, and a reverse transcription kit from Roche (Basel, Switzerland) was used to generate complementary DNA. The PCR primers listed in Table 1 were acquired from Sunya in Fuzhou, China. An RT-qPCR assay using a SYBR Green kit from Roche was run on an ABI 7500 System (ABI, Vernon, CA, USA). To find the relative gene expression, the formula (2-ΔΔct) was utilized, and to find ΔCt, the Ct value of each target gene was subtracted from the Ct value of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Primer sequences.
| Name | Sequence (5'→3') | mer | OD/Tube | nmol/Tube | MW | GC% | Tm(°C) |
|---|---|---|---|---|---|---|---|
| HDAC1(M)-F | TGCGTTCTATTCGCCCAGATA | 21 | 1 | 5.3 | 6372.1 | 47.6 | 58.0 |
| HDAC1(M)-R | CCTCCCGTGGACAACTGAC | 19 | 1 | 5.8 | 5733.7 | 63.2 | 61.9 |
| HDAC4(M)-F | CACTGCATTTCCAGCGATCC | 20 | 1 | 5.7 | 6012.9 | 55.0 | 59.9 |
| HDAC4(M)-R | AAGACGGGGTGGTTGTAGGA | 20 | 1 | 4.7 | 6302.1 | 55.0 | 59.9 |
| HDAC5(M)-F | AGCACCGAGGTAAAGCTGAG | 20 | 1 | 4.7 | 6200.1 | 55.0 | 59.9 |
| HDAC5(M)-R | GAACTCTGGTCCAAAGAAGCG | 21 | 1 | 4.7 | 6464.2 | 52.4 | 60.0 |
| HDAC11(M)-F | CTGTGCCTATGCAGACATCAC | 21 | 1 | 5.2 | 6366.1 | 52.4 | 60.0 |
| HDAC11(M)-R | GTGGCGGTTGTAAACATCCAT | 21 | 1 | 4.9 | 6461.2 | 47.6 | 58.0 |
| SIRT6(M)-F | CTCCAGCGTGGTTTTCCACA | 20 | 1 | 5.7 | 6043.9 | 55.0 | 59.9 |
| SIRT6(M)-R | GCCCATGCGTTCTAGCTGA | 19 | 1 | 5.8 | 5779.8 | 57.9 | 59.7 |
| SIRT7(M)-F | GCACTTGGTTGTCTACACGG | 20 | 1 | 5.5 | 6124.0 | 55.0 | 59.9 |
| SIRT7(M)-R | TGTCCATACTCCATTAGGACCC | 22 | 1 | 5.1 | 6630.3 | 50.0 | 60.1 |
| GAPDH(M)-F | AATGGATTTGGACGCATTGGT | 21 | 1 | 4.8 | 6516.3 | 42.9 | 56.1 |
| GAPDH(M)-R | TTTGCACTGGTACGTGTTGAT | 21 | 1 | 5.2 | 6458.2 | 42.9 | 56.1 |
| P21(H)-F | CGATGGAACTTCGACTTTGTCA | 22 | 1 | 4.8 | 6725.4 | 45.5 | 58.2 |
| P21(H)-R | GCACAAGGGTACAAGACAGTG | 21 | 1 | 4.4 | 6513.3 | 52.4 | 60.0 |
| P27(H)-F | AACGTGCGAGTGTCTAACGG | 20 | 1 | 5.0 | 6182.0 | 55.0 | 59.9 |
| P27(H)-R | CCCTCTAGGGGTTTGTGATTCT | 22 | 1 | 5.2 | 6723.3 | 50.0 | 60.1 |
| Cyclin A2(H)-F | GGATGGTAGTTTTGAGTCACCAC | 23 | 1 | 4.5 | 7094.6 | 47.8 | 60.2 |
| Cyclin A2(H)-R | CACGAGGATAGCTCTCATACTGT | 23 | 1 | 4.6 | 7023.6 | 47.8 | 60.2 |
| Cyclin B1(H)-F | TTGGGGACATTGGTAACAAAGTC | 23 | 1 | 4.2 | 7127.7 | 43.5 | 58.4 |
| Cyclin B1(H)-R | ATAGGCTCAGGCGAAAGTTTTT | 22 | 1 | 4.6 | 6789.4 | 40.9 | 56.3 |
| Cyclin D1(H)-F | CAATGACCCCGCACGATTTC | 20 | 1 | 5.5 | 6021.9 | 55.0 | 59.9 |
| Cyclin D1(H)-R | CATGGAGGGCGGATTGGAA | 19 | 1 | 5.0 | 5957.9 | 57.9 | 59.7 |
| Cyclin E1(H)-F | GCCAGCCTTGGGACAATAATG | 21 | 1 | 4.8 | 6455.2 | 52.4 | 60.0 |
| Cyclin E1(H)-R | CTTGCACGTTGAGTTTGGGT | 20 | 1 | 5.5 | 6170.0 | 50.0 | 57.8 |
| P21(M)-F | CGAGAACGGTGGAACTTTGAC | 21 | 1 | 4.7 | 6495.3 | 52.4 | 60.0 |
| P21(M)-R | CCAGGGCTCAGGTAGACCTT | 20 | 1 | 5.3 | 6118.0 | 60.0 | 61.9 |
| P27(M)-F | TCAAACGTGAGAGTGTCTAACG | 22 | 1 | 4.5 | 6783.4 | 45.5 | 58.2 |
| P27(M)-R | CCGGGCCGAAGAGATTTCTG | 20 | 1 | 5.2 | 6158.0 | 60.0 | 61.9 |
| Cyclin A2(M)-F | GCCTTCACCATTCATGTGGAT | 21 | 1 | 5.3 | 6372.1 | 47.6 | 58.0 |
| Cyclin A2(M)-R | TTGCTCCGGGTAAAGAGACAG | 21 | 1 | 4.7 | 6495.3 | 52.4 | 60.0 |
| Cyclin B1(M)-F | TCGTTCACCAGCGATCTGTC | 20 | 1 | 5.7 | 6043.9 | 55.0 | 59.9 |
| Cyclin B1(M)-R | CGAAGCCCTGCCAATACCATA | 21 | 1 | 5.0 | 6344.2 | 52.4 | 60.0 |
| Cyclin D1(M)-F | GCGTACCCTGACACCAATCTC | 21 | 1 | 5.3 | 6311.1 | 57.1 | 61.9 |
| Cyclin D1(M)-R | ACTTGAAGTAAGATACGGAGGGC | 23 | 1 | 4.1 | 7161.7 | 47.8 | 60.2 |
| Cyclin E1(M)-F | GAAAAGCGAGGATAGCAGTCAG | 22 | 1 | 4.1 | 6866.5 | 50.0 | 60.1 |
| Cyclin E1(M)-R | CCCAATTCAAGACGGGAAGTG | 21 | 1 | 4.7 | 6464.2 | 52.4 | 60.0 |
Immunoblotting analysis
The cells and tissues that were exposed to CKD-581 (1 µM, 10 µM) for a period of 24 hours were lysed using RIPA lysis buffer (EpiZyme, China, PC101), which includes inhibitors for protease and phosphatase. Bichincinchoninic acid (BCA) protein kits (EpiZyme, China, ZJ102) were used to ascertain protein quantities. Prior to being transferred to PVDF membranes, twenty micrograms of protein were electrophoresed on sodium dodecyl sulfate-polyacrylamide gels at 10% or 15% concentrations (SDS-PAGE). Following an hour of blocking with 1× Protein Free Fast Blocking Solution (EpiZyme, China, PS108) at room temperature (18-25°C), the membranes were incubated with primary antibodies overnight at 4°C. Antibodies used included: P21 (AP021), P27 (AP027), cyclin A2 (AF2524), cyclin B1 (AF6627), cyclin D1 (AF1183) from Beyotime Biotechnology Co. Ltd (Shanghai, China), and cyclin E1 (11554-1-AP) from Proteintech Group (Wuhan, China). For P21, the primary antibody dilution was 1:500, whereas for P27 and cyclins A2, B1, D1, and E1, it was 1:1000. The membranes were immersed in secondary antibody for one hour at room temperature after three washes with TBS-Tween (TBST) buffer. The next step was to visualize the results using enhanced chemiluminescence reagents.
Immunohistochemistry
From the fixed tissue samples, 4 µm thick sections were cut. Following dewaxing, rehydration, and antigen recovery, slices were incubated with primary antibodies at 4°C for the night. Antibodies including HDAC1 (GB11333-100), HDAC4 (GB115576-100), and SIRT7 (GB11355-100) were sourced from Wuhan Sevier Biotechnology Co.; HDAC5 (29342-1-AP), HDAC11 (67949-1-Ig), and SIRT6 (13572-1-AP) were obtained from Wuhan Sanying Biotechnology Co. The next step was a PBS wash, followed by incubation with a secondary antibody (Zhongsui Jinqiao, Beijing, China) at 37°C for 60 minutes, followed by another wash. Color development was achieved using DAB for 2 minutes, followed by hematoxylin restaining, and microscopy images were captured.
Immunofluorescence analysis
After cell digestion, 5 × 104 cells/well were plated on a 24-well plate, subjected to CKD-581 treatment, and then left to incubate overnight at 37°C with 5% CO2. Using 4% paraformaldehyde, the cells were adhered on creep plates after three washes with PBS. They become permeable after 20 minutes. After that, fluorescent primary antibodies were added to the cells and left to overnight at 4°C. For P21 and P27 as well as cyclins A2, B1, D1, and E1, we utilized the identical primary antibodies diluted to a 1:200 ratio as in the immunoblotting study. The next step was to let the cells sit at room temperature for an hour while a secondary fluorescent antibody was added. After that, DAPI was applied in the dark for 5 minutes. Images were captured after sealing.
In vivo tumor xenograft analysis
Hepa1-6 cells (1.75 × 106 cells in 100 µL saline) were subcutaneously injected into the right forelimb of eight-week-old C57BL/6J mice (n = 21). Once over 60% of mice developed subcutaneous tumors up to 50 mm3, they were divided into two groups: a control group (n = 11) and a treatment group (n = 10; 30 mg/kg). The treatment group received intraperitoneal injections of CKD-581 in 0.9% saline once weekly for two weeks, while the control group received no treatment. Using Vernier calipers, the size of the tumors were measured, and the mice were weighed three times weekly. The formula for tumor volume was (length) × (width)2 × π/6. A two-week interval followed the intraperitoneal injection of 2% pentobarbital sodium (100 mg/kg) that put the mice to sleep. Checks on vital signs were performed ten minutes later. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) serum levels were tested using a kit (Nanjing Jiancheng Corp. Nanjing, China) following centrifugation of five separate 0.1 ml blood samples taken from the eye. Tumor, liver, and spleen were collected, and no adverse effects such as ulceration or suppuration of tumors were observed during the experiment.
Statistical analysis
Our statistical methods included two-sample t-tests for pairwise comparisons and one-way ANOVA and Kruskal-Wallis tests for multiple group comparisons. When the P<0.05, statistical significance was established (P<0.05, P<0.01, P<0.001). This is how the data is displayed: mean ± standard deviation (SD). Software packages such as R (4.1.3), PERL (5.30.1), SPSS (25), and GraphPad Prism (8) were utilized.
Results
Pan-cancer analysis and mutational mapping of the HDAC family of genes
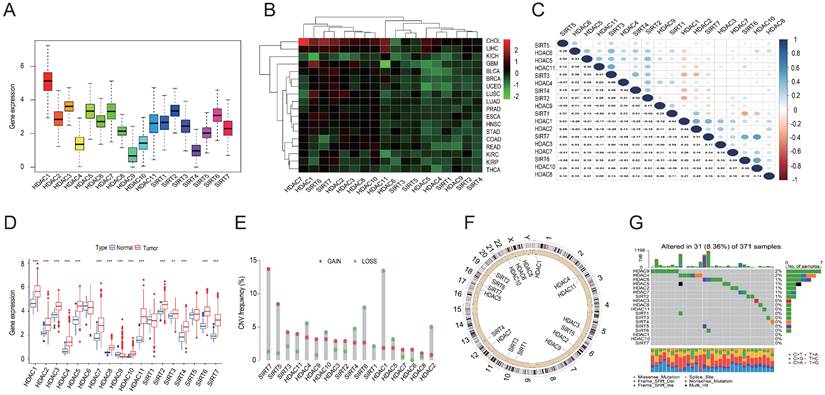
Expression data for 33 tumors were obtained from the UCSC Xena database, leading to pan-cancer analyses. Subsequently, 18 HDAC genes were analyzed further. Differential analysis revealed high overall expression of HDAC genes in all tumor samples. HDAC1 was the most highly expressed, while HDAC4, HDAC9, and SIRT4 were expressed at relatively lower levels (Figures 1A and 1B). With the exception of HDAC1, SIRT6, and SIRT7, most HDAC genes were down-regulated in various tumor types. Pearson correlation analysis indicated correlations between certain HDAC genes, such as HDAC10 and SIRT7 (r = 0.52), HDAC10 and SIRT6 (r = 0.5), HDAC11 and SIRT3 (r = 0.47), and SIRT1 and SIRT6 (r = -0.36) (Figure 1C). Furthermore, with the exception of HDAC9, the majority of HDAC family genes were substantially elevated in tumor tissues when comparing mRNA expression in LIHC tissues to normal tissues (Figure 1D). CNV analysis in LIHC indicated increased CNV in SIRT7, SIRT5, and SIRT3, and decreased CNV in HDAC1, SIRT6, and HDAC2 (Figure 1E). Chromosomal locations of HDAC family gene copy number variants were displayed (Figure 1F), with mutations found in 31 of the 371 LIHC samples (8.36%). The highest mutation rates were observed in HDAC9 and HDAC4 (2%), followed by HDAC6, HDAC5, HDAC2, HDAC7, and SIRT2 (1% each) (Figure 1G).
Pan-cancer and mutation analyses of the HDAC gene family. (A) Average expression levels of HDAC genes across 33 tumor types in TCGA. (B) Differential expression of HDAC genes across various tumor types, with red denoting up-regulation and green indicating down-regulation. (C) Correlation analysis of HDAC genes employing Spearman's correlation coefficient. (D) Comparative expression of HDAC genes in tumor versus normal tissues. (E, F) Copy number variations of HDAC genes in LIHC tissues and their chromosomal localization. (G) Somatic mutation analysis of HDAC genes. **P<0.01, ***P<0.001
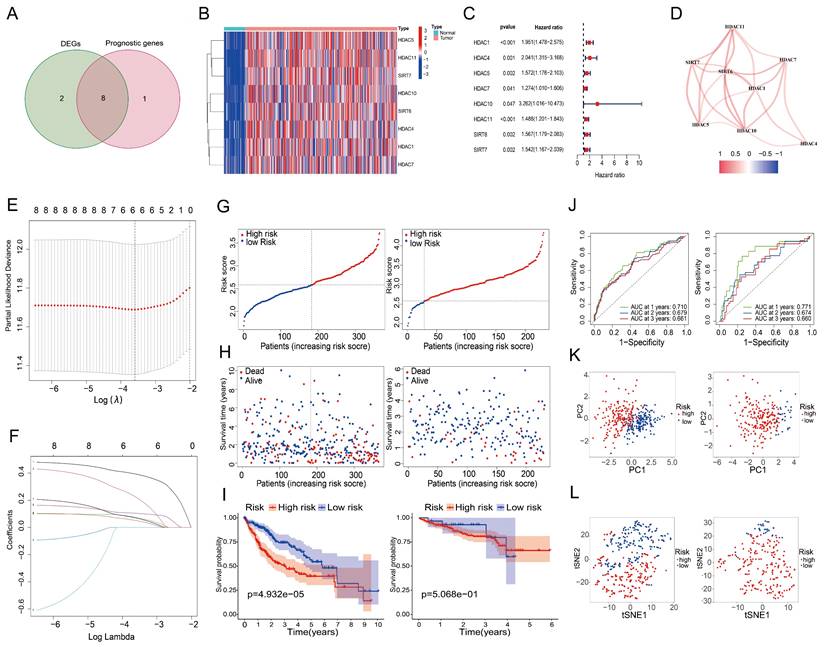
Screening of prognosis-related HDAC genes in LIHC and construction of a risk score prognostic model
An investigation of 18 HDAC genes was conducted to determine the potential involvement of HDACs in the prognosis of LIHC. The analysis identified 10 genes as differentially expressed (P < 0.05) (Table 2). Furthermore, 9 of these genes were found to have prognostic value in the univariate Cox regression analysis (P < 0.05) (Table 3). Of these, eight genes were found at the intersection of differentially expressed and prognosis-related genes (Figure 2A). Heat maps confirmed that all intersecting genes were upregulated in LIHC tissues (Figure 2B). Univariate Cox regression of the intersecting genes demonstrated a negative correlation between their expression and survival rates in LIHC patients (Figure 2C). Correlations among the intersecting genes are depicted in Figure 2D. After excluding patients with a survival duration of 0 months, LASSO regression was utilized to analyze the expression profiles of the eight intersecting genes, resulting in the identification of six genes most suitable for constructing the prognostic model (Figures 2E and 2F), thereby eliminating overfitting genes. Risk scores were calculated as follows using the 6 HDAC genes:
Differentially expressed genes in LIHC.
| Gene | conMean | treatMean | logFC | pValue | fdr |
|---|---|---|---|---|---|
| HDAC1 | 10.14285 | 20.77175384 | 1.03416 | 5.37E-19 | 1.01E-18 |
| HDAC4 | 0.247534 | 0.787871827 | 1.670334 | 5.63E-23 | 2.39E-22 |
| HDAC5 | 3.517294 | 8.262054521 | 1.232035 | 5.92E-22 | 1.68E-21 |
| HDAC7 | 1.132222 | 3.061601049 | 1.435129 | 4.44E-15 | 6.86E-15 |
| HDAC8 | 0.18237 | 0.377620457 | 1.050068 | 5.57E-22 | 1.68E-21 |
| HDAC10 | 0.051058 | 0.166399178 | 1.704439 | 4.77E-21 | 1.16E-20 |
| HDAC11 | 0.930702 | 4.98238694 | 2.420446 | 1.83E-27 | 3.11E-26 |
| SIRT4 | 1.093688 | 2.267897671 | 1.052154 | 7.13E-16 | 1.21E-15 |
| SIRT6 | 2.532166 | 6.132379268 | 1.276075 | 1.88E-23 | 1.07E-22 |
| SIRT7 | 1.171316 | 3.695575534 | 1.657669 | 7.68E-27 | 6.53E-26 |
Prognostic screening of HDAC genes in the TCGA dataset. (A) Venn diagram identifying genes at the intersection of differentially expressed and prognosis-related genes. (B) Expression levels of eight intersecting genes in LIHC versus normal tissues. (C) Forest plot depicting the association between the expression of the eight intersecting genes and OS. (D) Network of correlations among intersecting genes. (E, F) Validation of the prognostic model using LASSO regression to determine the requisite number of genes. Panel E displays the cross-validation curve, which shows how the model's mean squared error varies with changes in Log (λ), indicating that reducing the candidate HDACs to 6 provides the optimal prognostic model; Panel F represents the regression coefficient path diagram, which demonstrates the process of reducing variable numbers and adjusting coefficients in the LASSO regression model. (G) High and low-risk scores, (H) survival status of patients, (I) survival curves, and (J) ROC curves for the test and validation cohorts. (K, L) Principal component and t-SNE analysis of the test and validation cohorts.
Univariate independent prognostic analysis.
| Id | HR | 95% Low | 95% High | P value |
|---|---|---|---|---|
| HDAC1 | 1.9506136 | 1.47785493 | 2.574605486 | 2.38E-06 |
| HDAC2 | 2.2413876 | 1.654272032 | 3.03687559 | 1.91E-07 |
| HDAC4 | 2.04100885 | 1.314764087 | 3.168414127 | 0.001475036 |
| HDAC5 | 1.5724753 | 1.1760063 | 2.102606566 | 0.002260432 |
| HDAC7 | 1.27370289 | 1.010233712 | 1.605884883 | 0.040749063 |
| HDAC10 | 3.26220209 | 1.016148118 | 10.47284573 | 0.046935458 |
| HDAC11 | 1.48783175 | 1.200947157 | 1.843247886 | 0.000277569 |
| SIRT6 | 1.56739156 | 1.179448881 | 2.082935808 | 0.001951356 |
| SIRT7 | 1.54247809 | 1.166652403 | 2.039372352 | 0.002351649 |
Risk score = 0.392 × ExpHDAC1 + 0.221 × ExpHDAC4 + 0.074 × ExpHDAC5 + 0.104 × ExpHDAC11 + 0.069 × ExpSIRT6 + 0.05 × ExpSIRT7
To validate the model, we used the ICGC dataset, and to test it, we used the TCGA dataset. Figure 2G (left) shows the results of the analysis that classified the test group patients as either high-risk or low-risk based on their median risk scores. Figure 2H (left) shows that compared to low-risk patients, high-risk patients died earlier. Based on the Kaplan-Meier curves, the high-risk group had a significantly lower OS (Figure 2I, left). Based on the ROC curves (Figure 2J, left), the 1-year OS, 2-year OS, and 3-year OS AUCs were 0.710, 0.679, and 0.661, respectively. Principal component analysis and t-SNE analysis effectively distinguished patients into two distinct subgroups within the risk categories (Figure 2K and 2L, left). The same analyses were performed on the validation group data, with results paralleling those of the test group, affirming the predictive reliability of the constructed risk score prognostic model (Figures 2G to 2L, right).
Analysis of LIHC prognosis and tumor microenvironment
Figures 3A and 3B show the findings of the test and validation groups' univariate and multivariate Cox regression analysis for OS, respectively. Because of its strong correlation with both survival duration and clinical outcomes, the risk score seems to function as a standalone predictive variable in LIHC patients. When examining the correlation between the risk score and clinical characteristics in the test group, it was shown that patients with stage III-IV LIHC had much higher risk scores than those with stage I-II (P < 0.05) (Figure 3C). Figure 3D shows similar findings in the validation group. To explore possible alterations in immune responses, we used the ssGSEA approach to compare the enrichment scores of thirteen immune-related pathways and sixteen immune cells between the low-risk and high-risk cases grouped based on the 6 HDAC genes. Increases in neutrophil and natural killer (NK) cell counts, as well as scores of type II IFN responses, were observed in the low-risk group (Figures 3E and 3F).
Prognostic and tumor microenvironment analysis in the test and validation groups. (A, B) Univariate and multivariate Cox regression analyses of prognostic factors in the test and validation groups. (C, D) Correlations between clinical characteristics and risk scores in LIHC patients within the test and validation cohorts. (E, F) ssGSEA scores for immune cells and immune-related pathways in both groups. (G) Associations between risk score and various scores (RNAss, DNAss, stromal, and immune). (H) Risk scores across different immune infiltration subtypes. ns, not significant; *P<0.05, **P<0.01, ***P<0.001
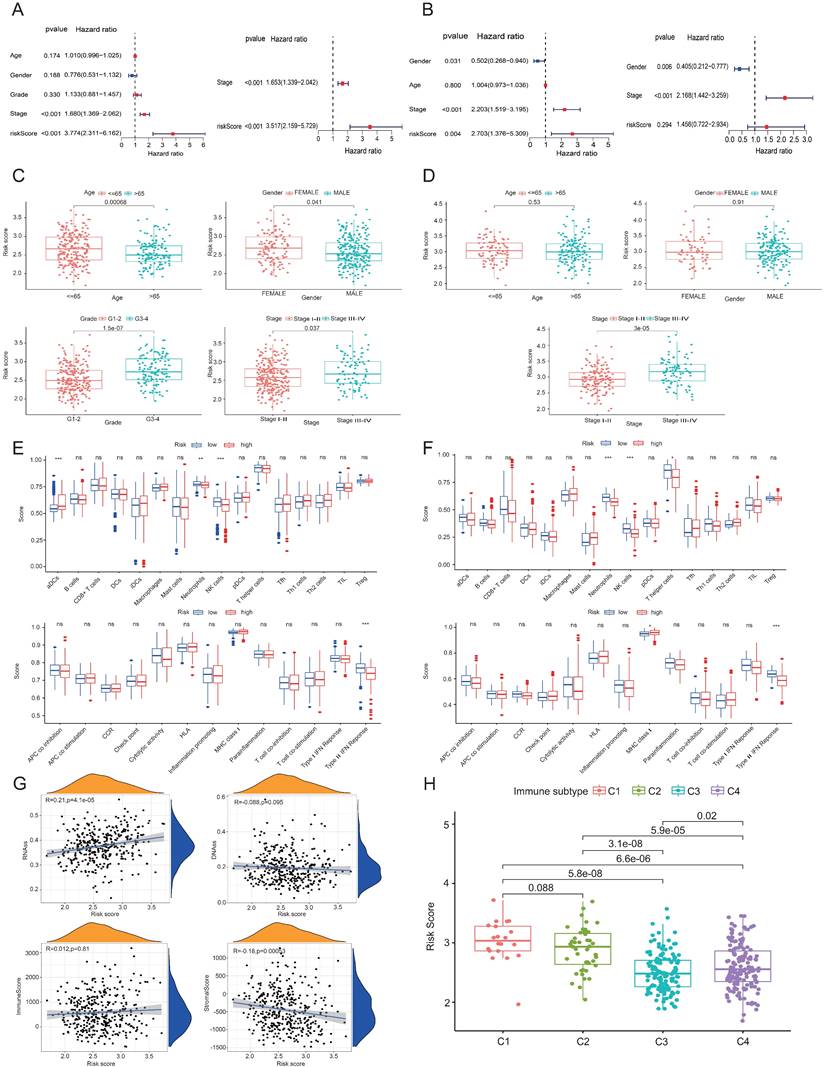
The findings across the test and validation groups showed high consistency (Figures 3E and 3F). An RNA stem cell score (RNAss) based on messenger RNA expression and a DNA stem cell score (DNAss) based on DNA methylation patterns were utilized to determine tumor stem cell scores [19]. Two measures, the immune score and the matrix score, were utilized to assess the tumor's immunological microenvironment. The risk score (based on the 6 HDAC genes) has a positive correlation (P < 0.01) with RNAss and a negative correlation (P < 0.01) with matrix score, as shown in Figure 3G. A considerable correlation between C1 and high-risk scores and C3 and low-risk scores was seen when using TCGA-LIHC data for immune infiltration analysis in LIHC (Figure 3H).
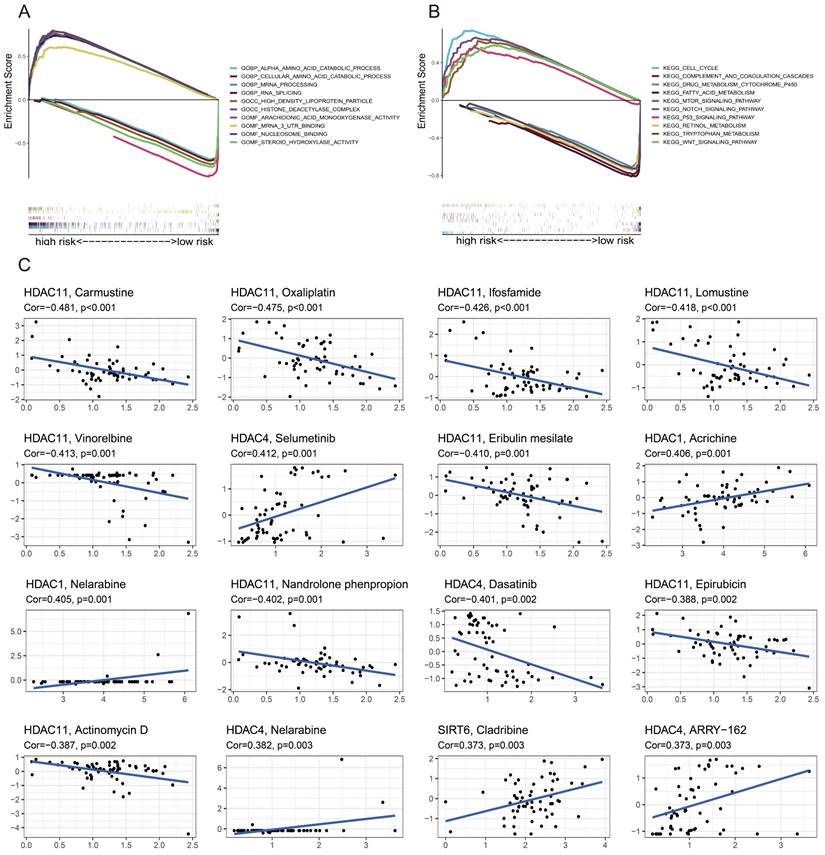
GSEA and drug sensitivity analysis
In order to compare the two groups based on risk, GSEA software was used to perform functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment studies, as well as Gene Ontology (GO) analyses. Figure 4A shows the results of the GO enrichment analysis, which determined that the high-risk group was connected with RNA splicing, the HDAC complex, and mRNA 3' untranslated regions (UTR) binding. According to KEGG enrichment analysis, the low-risk group was associated with pathways involving tryptophan metabolism, fatty acid metabolism, and retinol metabolism (Figure 4B), whereas the high-risk group was linked to the cell cycle, Notch signaling route, and P53 signaling pathway. The correlation between drug sensitivity and gene expression levels was examined in a study that found that higher levels of HDAC11 were associated with increased resistance to carmustine, oxaliplatin, ifosfamide, lomustine, vinorelbine, eribulin mesylate, nandrolone phenylpropionate, actinomycin D, and epirubicin. Figure 4C shows that, on the other hand, selumetinib, nelarabine, and bimetinib sensitivity increased with increasing HDAC4 expression levels.
GSEA and drug sensitivity analysis. (A) GO and KEGG (B). (C) Analyses linking prognostic gene expression to drug sensitivity.
Validation of clinical samples
Apart from HDAC5, the other five HDACs displayed higher staining and more positive regions in cancer tissues, according to immunohistochemistry results from 3 sets of LIHC tissues and corresponding normal paracancerous tissues (Figures 5A to 5F). RT-PCR results supported this observation (Figures 5G and 5K). From the preliminary prognostic model, it was noted that HDAC1 was the most highly expressed among the six model genes and contributed the most to the model's weight. Thus, HDAC1 expression in 64 LIHC cases was assessed by RT-PCR. High HDAC1 expression was associated with a decreased OS after patients with missing clinical data were excluded (Figure 5H). High HDAC1 expression was linked to a poor prognosis in both univariate and multivariate Cox analyses, indicating that it could be a standalone prognostic factor in LIHC (Figures 5I and 5J). Furthermore, correlation analyses between HDAC1 expression, tumor size, and age showed a positive correlation between HDAC1 mRNA levels and tumor size (P<0.05) (Figures 5L and 5M).
Validation of clinical samples. (A-F) Immunohistochemical staining of six HDACs across 6 samples of LIHC tissue (A, C, E; n=3) and paired paracancerous tissue (B, D, F; n=3). (G) The mRNA expression levels of six model genes in human LIHC tissues versus paracancerous tissues. (H) Kaplan-Meier analysis of OS based on HDAC1 expression. (I, J) Univariate and multivariate Cox analyses. (K) The mRNA expression levels of six model genes in mouse tumor and normal liver tissues. (L) Correlation analysis between mRNA levels of HDAC1 and tumor size. (M) Correlation analysis of mRNA levels of HDAC1 with age. LIHC, liver hepatocellular carcinoma; ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001
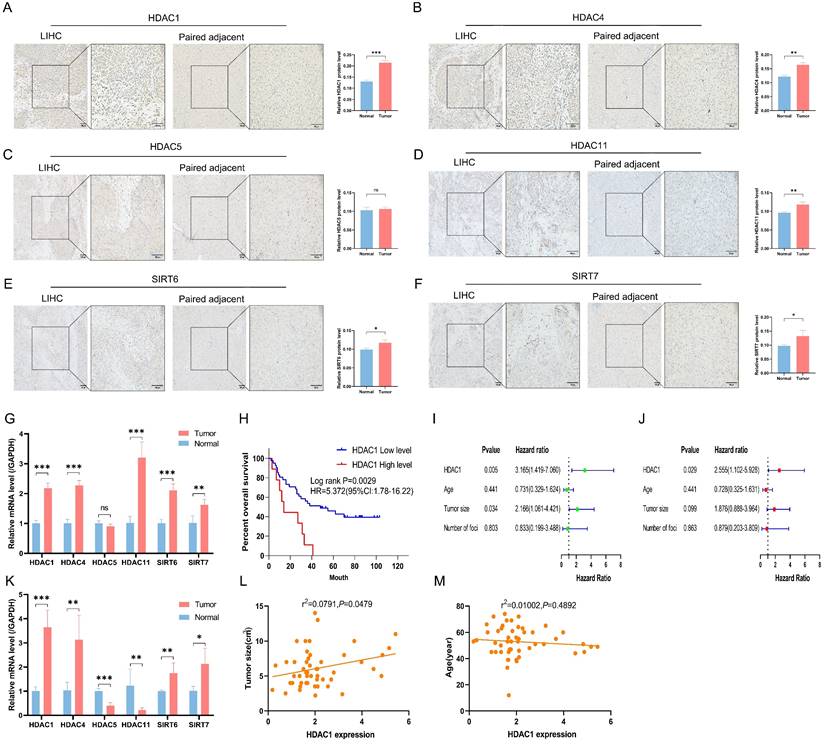
Effect of endogenous HDAC expression inhibition on the biological functions of LIHC cell lines
Bel7402 (IC50 = 56.48 µM), Huh-7 (IC50 = 53.95 µM), and Hepa1-6 (IC50 = 404.1 µM) cell lines were exposed to varying concentrations of CKD-581 for 24 hours, resulting in inhibited growth and proliferation. The antitumor effect was found to increase with the extension of treatment to 48 and 72 hours (Figures 6A to 6C). Figures 6D to 6F show that CKD-581 inhibited the migration and invasion capabilities of these LIHC cell lines, as confirmed by the dramatically reduced number of cells that moved to the lower surface of the chamber in the Transwell assay. Figure 6G shows that CKD-581 inhibited LIHC cell colony growth in a concentration-dependent way in the 14-day plate clone experiment. The use of flow cytometry revealed an augmented cell death rate in CKD-581-treated hepatocellular carcinoma cells (Figures 6H to 6J). Taken together, our results indicate that LIHC cell proliferation, migration, and invasion are inhibited and apoptosis is enhanced when CKD-581 inhibits endogenous HDAC expression.
CKD-581 inhibits the proliferation, migration, and invasion of LIHC cells. CKD-581 significantly reduces the proliferation of (A) Bel7402 cells, (B) Huh-7 cells, and (C) Hepa1-6 cells. IC50 was calculated at 24h after treated with CKD-581. (D-F) Transwell assay for evaluating the migration and invasion capabilities of LIHC cells. (G) Clonogenic assay for LIHC cells. LIHC, liver hepatocellular carcinoma. (H-J) Flow cytometry analysis of apoptosis in Bel7402, Huh-7, and Hepa1-6 cells at 12h after treated with CKD-581. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001
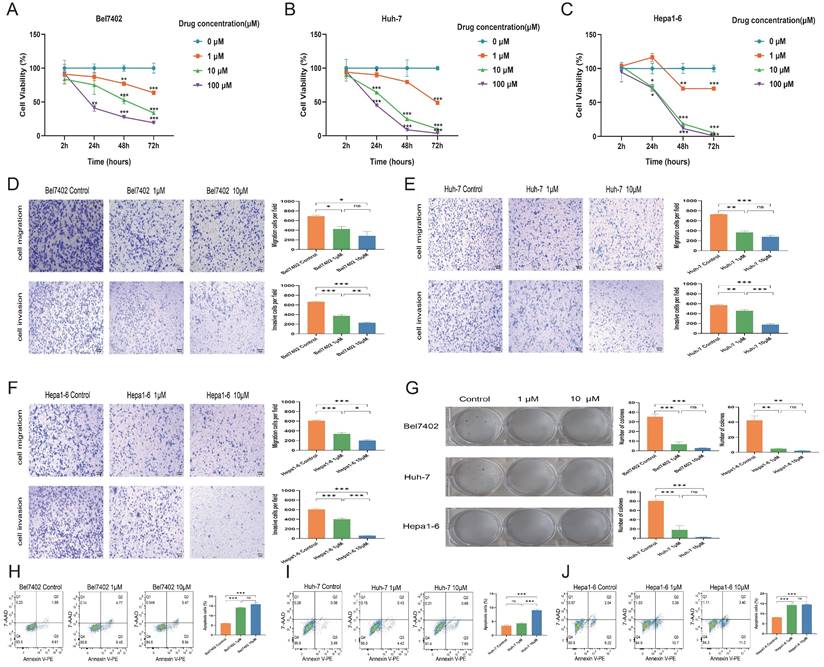
Endogenous HDAC expression inhibition inhibits tumor growth in mice in vivo
Following the in vitro experiments, the anti-tumor efficacy of CKD-581 was assessed in vivo in mice. In the treatment group, notable anti-tumor effects were observed starting from day 13 post-administration (Figure 7A). The tumor volume of the treatment group was considerably less than that of the control group after two weeks of treatment (P < 0.05) (Figures 7B and 7C). Assessment of CKD-581's hepatic safety was done by measuring ALT and AST levels in urine. The levels of ALT and AST did not differ significantly between the treatment and control groups, as shown in Figures 7D and 7E. Furthermore, there was no discernible variation in total body mass index (Figure 7F) between the two sets of data.
Inhibition of tumor growth by CKD-581 in vivo. (A) Changes in tumor volume over the treatment period in control and treated mice. (B) Large tumor specimens from mice at the end of the treatment period. (C) Tumor volumes in the control and treatment groups at the end of the treatment period. (D) Serum ALT and (E) AST levels in mice. (F) Changes in body weight of mice over the treatment period. ns, not significant; *P < 0.05
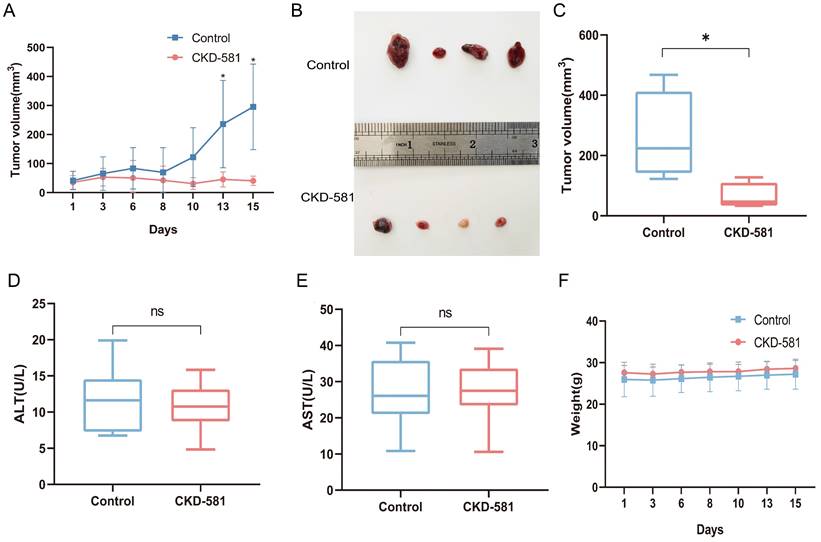
CKD-581 exerts anti-tumor effects by regulating the cell cycle
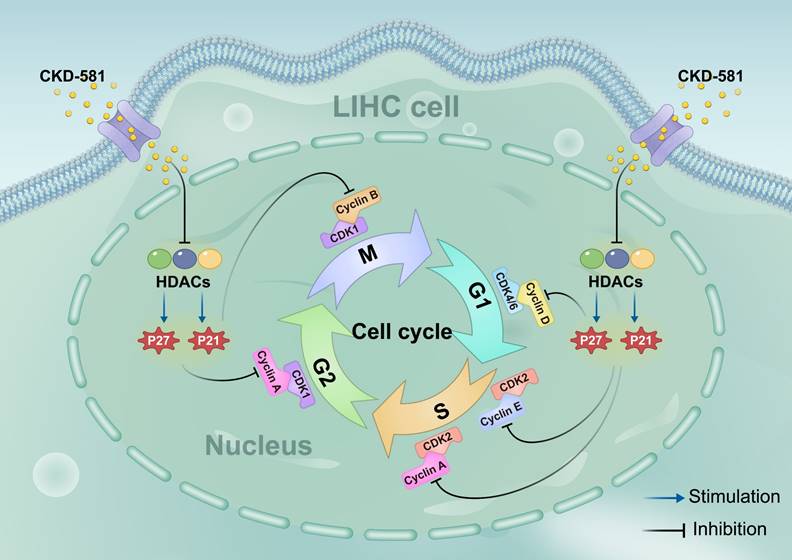
Based on KEGG enrichment analysis results, the impact of CKD-581 on cell cycle-related genes/proteins in LIHC was investigated. In LIHC cells, CKD-581 was observed to decrease cyclin A2, B1, and D1 mRNA levels while increasing P21 and P27 mRNA levels (Figures 8A to 8C). Immunoblotting revealed that in LIHC cells, P21 and P27 protein expression was up after 24 hours of treatment with CKD-581 (1 µM or 10 µM), but cyclins A2, B1, D1, and E1 expression was decreased (Figures 8D to 8I). Furthermore, it was observed that these cell cycle regulatory proteins were predominantly located in the cytoplasm and highly expressed in the nuclei of mitotically active cells, supporting their vital roles in cell proliferation (Supplementary Figures 1-6). These findings suggest that CKD-581 may exert its anticancer effects through modulation of the cell cycle, as illustrated in Figure 9.
CKD-581 modulates anti-tumor effects by regulating the cell cycle. (A-C) Effects of various concentrations of CKD-581 on mRNA expression levels of cyclin proteins, P21, and P27 in Bel7402, Huh-7, and Hepa1-6 cell lines. (D-I) Impact of various concentrations of CKD-581 on protein expression levels of P21, P27, and cyclins A2, B1, D1, and E1 in these cell lines. (K) Expression and subcellular localization of P21, P27, cyclins A2, B1, D1, and E1 in Bel7402, Huh-7, and Hepa1-6 cell lines following CKD-581 treatment. LIHC, liver hepatocellular carcinoma. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001
Schematic diagram of the hypothesis that CKD-581 inhibits HDACs, thereby inducing cell cycle arrest in LIHC cells. LIHC, liver hepatocellular carcinoma.
Discussion
Research has demonstrated that the HDAC family of genes is involved in tumor growth and drug resistance. This includes chemoresistance and anti-apoptosis mechanisms [20]. By hyperacetylating substrates that are not histones but histone-related, HDACs bring about a wide range of cellular and molecular consequences. They also promote tumor growth by changing important molecules to influence oncogenic cell signaling pathways or by suppressing tumor suppressor gene expression [21]. There has been a lack of investigation into the function of HDAC in LIHC.
Understanding the biological roles and possible molecular pathways of endogenous HDACs, as well as their prognostic value in LIHC, were the objectives of this investigation. A risk score prognostic model was constructed using six genes (HDAC1, HDAC4, HDAC5, HDAC11, SIRT6, and SIRT7), selected from a pool of 18 HDAC genes strongly associated with LIHC. Patients were classified as high-risk or low-risk based on the median values of the risk score. The high-risk group had a significantly lower OS when clinical staging was higher. Multiple independent prognostic studies have shown that the risk score is an independent prognostic factor. Based on the ROC curves, the risk score has a good predictive accuracy for LIHC and can be used as a prognostic marker. The 1-, 2-, and 3-year OS AUCs were 0.710, 0.679, and 0.661, respectively.
The risk model was constructed based on six HDAC genes: HDAC1, HDAC4, HDAC5, HDAC11, SIRT6, and SIRT7, all of which were upregulated in LIHC tissues. It has been demonstrated that the absence or downregulation of HDAC1 leads to cell cycle arrest at the G1 phase or G2/M transition, thereby inhibiting cell growth and increasing the percentage of apoptotic cells [22]. High HDAC1 expression in LIHC patients, on the other hand, is linked to worse tissue differentiation, more advanced tumor lymph node metastatic staging, lower survival rates, and an increased incidence of cancer cells invading the portal vein [23]. These findings align with the outcomes of our study. Inhibition of HDAC4 enhances the cytotoxic effects of cisplatin and impedes tumor cell growth [24]. Similarly, downregulation of HDAC5, like HDAC1, triggers a G1 phase block in the cell cycle and promotes apoptosis [25]. The presence of HDAC5, which is abundant in cancerous epithelial cells' cytoplasm, is positively associated with lymph node and distant metastasis [26, 27]. A high correlation between HDAC11 and tumor development, microvascular invasion, tumor differentiation, and clinical staging in patients with LIHC suggests that it may have a regulatory role in maintaining cancer stemness [28]. In LIHC cells, SIRT6 has been found to promote cell migration, invasion, and epithelial-mesenchymal transition [29]. Additionally, it plays a role in supporting tumor development by protecting against DNA damage and cellular senescence [30]. Reducing SIRT7 levels causes cell cycle arrest and slows cell growth [31]. By blocking SIRT7, adriamycin-induced P53 activation is amplified in mice xenografts, leading to apoptosis and tumor growth suppression [32]. These results substantiate the high clinical utility of our risk score prognostic model based on these genes.
Our investigation into the correlation between risk score and immune infiltration revealed that the high-risk group exhibited reduced numbers of NK cells and neutrophils. The innate immune system's cytotoxic lymphocytes known for their ability to kill cancer cells, known as NK cells, were shown to be correlated with a decline in anti-tumor immunity in high-risk LIHC patients [33]. Neutrophils, which participate in various carcinogenic stages including tumor initiation, growth, proliferation, and metastasis [34], were unexpectedly found in lower proportions in the high-risk group, suggesting a disruption in the tumor microenvironment, though the exact cause requires further investigation. Furthermore, a reduced type II IFN response—which normally aids in host defense and immunological surveillance and promotes tumor cell apoptosis—was linked to a high-risk score [35]. C1 was associated with high-risk scores according to immunophenotype analysis, but C3 and C4 were associated with low-risk scores; C1 is conducive to tumor development, whereas C3 and C4 act as effective protective factors. This aligns with previous findings that higher immunophenotypes, indicative of increased cytotoxic cells, are associated with better patient survival [36].
Furthermore, we analyzed the expression differences of 6 HDACs in LIHC tissues compared to adjacent normal tissues and observed that five HDACs, with the exception of HDAC5, were upregulated in LIHC. Patients whose HDAC1 expression was high had a much worse survival rate compared to those whose expression was low, according to survival analysis focusing on HDAC1. Univariate and multivariate Cox analyses showed that patients with LIHC with a high level of HDAC1 expression had a poorer prognosis. The strong correlation between HDAC1 mRNA expression and tumor size further supports its central role in the preconstructed prognostic model.
There are currently four HDAC inhibitors that have been approved by the US Food and Drug Administration for cancer treatment [37]. However, their effectiveness in treating solid tumors has been limited. Our study demonstrates that CKD-581, a novel HDAC inhibitor, exhibits strong antitumor activity against LIHC cells both in vivo and in vitro. CKD-581 has been demonstrated to effectively hinder the growth, proliferation, migration, and invasion of LIHC cell lines, as well as induce apoptosis at certain doses, according to biological function experiments. In vivo, CKD-581 not only reduces tumor size in mice but also displays potent anti-tumor effects without significant hepatotoxic or adverse reactions, suggesting a favorable safety profile for CKD-581.
Based on KEGG enrichment analysis results, we explored how HDAC inhibitors mediate their antitumor effects. By reducing cyclin A2, B1, and D1 expression and increasing P21 and P27 expression, CKD-581 was discovered to suppress LIHC cell growth. There are four separate phases in the cell cycle: G1, S, G2, and M [38]. Cell cycle protein-dependent kinases (CDKs), cyclins, and CDK inhibitors (CDKIs) are all involved in cell cycle regulation. CDK activation happens upon binding to a cyclin [39]. Cell cycle progression relies on cyclins and CDKs (1, 2, 4, and 6). Cell cycle protein D, the first to respond to mitotic signals, activates CDK4 and CDK6 during the G1 phase, acting as a growth factor sensor [40]. CDK2 activation by cell cycle protein E1 facilitates the G1 to S phase transition. In the S phase, cell cycle protein A interacts with CDK1/2 to promote transition to the M phase. By the end of the G2 phase, CDK1 associates with cyclin B. Uncontrolled CDK activation can lead to continuous cell division, contributing to tumor development; this activity is regulated by CDKIs, such as the CIP/KIP family (P21, P27, P57) and the INK family (P15, P16, P18, P19) [41]. CIP/KIP family members inhibit the activity of A, B, D, and E/CDK complexes, leading to cell cycle arrest. P21, a widely recognized cell cycle inhibitor, suppresses tumor growth by causing G1 phase arrest [42]. Normally, P27 levels are strictly regulated, and its upregulation prevents cyclin binding to CDK, thus blocking the G1 to S phase transition [43]. Low levels of P21 and P27, which act as tumor suppressors, can lead to unrestrained cell division and proliferation. In this study, CKD-581 was shown to block the G1/S phase by markedly upregulating P21 and P27 and downregulating cyclins A2, B1, and D1, thereby inhibiting cell proliferation and promoting cell death in LIHC cells (Figure 9). This finding supports our KEGG analysis results.
To summarize, this study examined the expression and prognostic significance of HDAC genes in LIHC, establishing a model based on six HDACs (HDAC1, HDAC4, HDAC5, HDAC11, SIRT6, and SIRT7). This model proved effective in predicting OS in LIHC patients, with HDAC1 identified as a key component. Additionally, CKD-581, an HDAC inhibitor, was shown to exert antitumor effects by regulating the cell cycle. Despite its insights, this study is limited by the absence of experiments on exogenous HDAC genes to confirm the cellular functions attributed to HDACs. Moreover, the enrollment of limited number of LIHC cases in the ICGC database resulted in the non-significant OS prediction in the validation cohort. Finally, the TCGA test set and the ICGC validation set arises due to the clinical grading information is absent in the ICGC database, hence lacking related analyses and resulting in weak correlation results in the ICGC validation set. These limitations are intended to be addressed in future research.
Abbreviations
ALT: alanine aminotransferase; AST: aspartate aminotransferase; ANOVA: one-way analysis of variance; CNV: copy number variation; DNAss: DNA stem cell scores; GSEA: gene set enrichment analysis; HDACs: histone deacetylases; ICGC: International Cancer Genome Consortium; IFN: interferon; LASSO: least absolute shrinkage and selection operator; LIHC: liver hepatocellular carcinoma; NK: natural killer; OS: overall survival; TCGA: The Cancer Genome Atlas; t-SNE: t-distributed stochastic neighbor embedding; RT-qPCR: Real-time quantitative PCR; RNAss: RNA stem cell scores; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gels electrophoresis; SD: standard deviation; ssGSEA: single sample gene set enrichment analysis; UCSC: University of California Santa Cruz.
Supplementary Material
Supplementary figures.
Acknowledgements
Funding
This study was supported by the Provincial Natural Science Fund of Fujian (Grant numbers: 2021J01439, 2023J011294), Fujian Provincial Health Technology Project (Grant number: 2021GGA047), and the Outstanding Young Talent Program of Fujian Cancer Hospital (2020YNYQ07).
Ethics approval and consent to participate
The study was approved by Ethics Review from Branch from the Ethics Committee of the Fujian Cancer Hospital (Approval No. SQ2015-049-01). For the animal study, all mice received humane care, and the study protocol was approved by the Animal Ethics Center of Fujian Medical University (ethics number: IACUC FJMU 2022-0723).
Availability of data and materials
The data that support the findings of this study are available on request from the first author (cuileidizi@fjmu.edu.cn).
Author contributions
ZL. Cui: Conceptualization, Funding acquisition, Writing - original draft. CQ. Zheng: Formal analysis, Methodology, Writing - original draft. YQ. You: Investigation, Data curation, Formal analysis. SJ. He, and S. Jiang: Data curation, Software. Y. Chen: Project administration, Validation. ZZ. Xiao, and YY. Lin Conceptualization, Supervision, Writing - review & editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2024;74:229-63
2. Long S, Chen Y, Wang Y, Yao Y, Xiao S, Fu K. Identification of Ferroptosis-related molecular model and immune subtypes of hepatocellular carcinoma for individual therapy. Cancer medicine. 2023;12:2134-2147
3. King J, Patel M, Chandrasekaran S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites. 2021;11:792
4. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harbor perspectives in biology. 2014;6:a018713
5. Ramaiah MJ, Tangutur AD, Manyam RR. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life sciences. 2021;277:119504
6. Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacological research. 2010;62:18-34
7. Shen L, Li Y, Li N, Shen L, Li Z. Comprehensive analysis of histone deacetylases genes in the prognosis and immune infiltration of glioma patients. Aging. 2022;14:4050-68
8. Cheng F, Zheng B, Wang J, Zhao G, Yao Z, Niu Z. et al. Comprehensive analysis of a new prognosis signature based on histone deacetylases in clear cell renal cell carcinoma. Cancer medicine. 2021;10:6503-14
9. Rahbari R, Rasmi Y, Khadem-Ansari MH, Abdi M. The role of histone deacetylase 3 in breast cancer. Medical oncology (Northwood, London, England). 2022;39:84
10. Schizas D, Mastoraki A, Naar L, Tsilimigras DI, Katsaros I, Fragkiadaki V. et al. Histone Deacetylases (HDACs) in Gastric Cancer: An Update of their Emerging Prognostic and Therapeutic Role. Current medicinal chemistry. 2020;27:6099-111
11. Bouyahya A, El Hachlafi N, Aanniz T, Bourais I, Mechchate H, Benali T. et al. Natural Bioactive Compounds Targeting Histone Deacetylases in Human Cancers: Recent Updates. Molecules (Basel, Switzerland). 2022;27:2568
12. Du L, Li Y, Kang M, Feng M, Ren Y, Dai H. et al. USP48 Is Upregulated by Mettl14 to Attenuate Hepatocellular Carcinoma via Regulating SIRT6 Stabilization. Cancer research. 2021;81:3822-34
13. Hu B, Xu Y, Li YC, Huang JF, Cheng JW, Guo W. et al. CD13 promotes hepatocellular carcinogenesis and sorafenib resistance by activating HDAC5-LSD1-NF-κB oncogenic signaling. Clinical and translational medicine. 2020;10:e233
14. Tsai CL, Liu WL, Hsu FM, Yang PS, Yen RF, Tzen KY. et al. Targeting histone deacetylase 4/ubiquitin-conjugating enzyme 9 impairs DNA repair for radiosensitization of hepatocellular carcinoma cells in mice. Hepatology (Baltimore, Md). 2018;67:586-99
15. Xiang J, Zhang N, Sun H, Su L, Zhang C, Xu H. et al. Disruption of SIRT7 Increases the Efficacy of Checkpoint Inhibitor via MEF2D Regulation of Programmed Cell Death 1 Ligand 1 in Hepatocellular Carcinoma Cells. Gastroenterology. 2020;158:664-78.e24
16. Zhou H, Cai Y, Liu D, Li M, Sha Y, Zhang W. et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21(Waf1/Cip1) and p19(INK4d) upregulation in hepatocellular carcinoma. Cell proliferation. 2018;51:e12447
17. Cho H, Yoon DH, Kim KP, Bae KS, Kim WS, Eom HS. et al. Phase I study of CKD-581, a pan-histone deacetylase inhibitor, in patients with lymphoma or multiple myeloma refractory to standard therapy. Investigational new drugs. 2018;36:877-85
18. He Y, Jiang Z, Chen C, Wang X. Classification of triple-negative breast cancers based on Immunogenomic profiling. Journal of experimental & clinical cancer research: CR. 2018;37:327
19. Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN. et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell. 2018;173:338-54.e15
20. Freese K, Seitz T, Dietrich P, Lee SML, Thasler WE, Bosserhoff A. et al. Histone Deacetylase Expressions in Hepatocellular Carcinoma and Functional Effects of Histone Deacetylase Inhibitors on Liver Cancer Cells In Vitro. Cancers. 2019;11:1587
21. Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harbor perspectives in medicine. 2016;6:a026831
22. Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A. et al. Role for histone deacetylase 1 in human tumor cell proliferation. Molecular and cellular biology. 2007;27:4784-95
23. Rikimaru T, Taketomi A, Yamashita Y, Shirabe K, Hamatsu T, Shimada M. et al. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology. 2007;72:69-74
24. Spaety ME, Gries A, Badie A, Venkatasamy A, Romain B, Orvain C. et al. HDAC4 Levels Control Sensibility toward Cisplatin in Gastric Cancer via the p53-p73/BIK Pathway. Cancers. 2019;11:1747
25. Fan J, Lou B, Chen W, Zhang J, Lin S, Lv FF. et al. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:11523-32
26. Gu H, Fang Z, Cai X, Song R, Lin M, Ye J. et al. Highly expressed histone deacetylase 5 promotes the growth of hepatocellular carcinoma cells by inhibiting the TAp63-maspin pathway. American journal of cancer research. 2018;8:462-75
27. Levidou G, Arsenakis D, Bolovis DI, Meyer R, Brucker CVM, Papadopoulos T. et al. Clinical Significance of the Immunohistochemical Expression of Histone Deacetylases (HDACs)-2, -4, and -5 in Ovarian Adenocarcinomas. Biomedicines. 2024;12:947
28. Bi L, Ren Y, Feng M, Meng P, Wang Q, Chen W. et al. HDAC11 Regulates Glycolysis through the LKB1/AMPK Signaling Pathway to Maintain Hepatocellular Carcinoma Stemness. Cancer research. 2021;81:2015-28
29. Han LL, Jia L, Wu F, Huang C. Sirtuin6 (SIRT6) Promotes the EMT of Hepatocellular Carcinoma by Stimulating Autophagic Degradation of E-Cadherin. Molecular cancer research: MCR. 2019;17:2267-80
30. Lee N, Ryu HG, Kwon JH, Kim DK, Kim SR, Wang HJ. et al. SIRT6 Depletion Suppresses Tumor Growth by Promoting Cellular Senescence Induced by DNA Damage in HCC. PloS one. 2016;11:e0165835
31. Yanai M, Kurata M, Muto Y, Iha H, Kanao T, Tatsuzawa A. et al. Clinicopathological and molecular analysis of SIRT7 in hepatocellular carcinoma. Pathology. 2020;52:529-37
32. Zhao J, Wozniak A, Adams A, Cox J, Vittal A, Voss J. et al. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. Journal of experimental & clinical cancer research: CR. 2019;38:252
33. Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nature reviews Clinical oncology. 2021;18:85-100
34. Ocana A, Nieto-Jiménez C, Pandiella A, Templeton AJ. Neutrophils in cancer: prognostic role and therapeutic strategies. Molecular cancer. 2017;16:137
35. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Frontiers in immunology. 2018;9:847
36. Tamborero D, Rubio-Perez C, Muiños F, Sabarinathan R, Piulats JM, Muntasell A. et al. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clinical cancer research: an official journal of the American Association for Cancer Research. 2018;24:3717-28
37. Singh AK, Bishayee A, Pandey AK. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients. 2018;10:731
38. Piezzo M, Cocco S, Caputo R, Cianniello D, Gioia GD, Lauro VD. et al. Targeting Cell Cycle in Breast Cancer: CDK4/6 Inhibitors. International journal of molecular sciences. 2020;21:6479
39. Abbastabar M, Kheyrollah M, Azizian K, Bagherlou N, Tehrani SS, Maniati M. et al. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA repair. 2018;69:63-72
40. Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. Journal of molecular medicine (Berlin, Germany). 2016;94:1313-26
41. Roy A, Banerjee S. p27 and leukemia: cell cycle and beyond. Journal of cellular physiology. 2015;230:504-9
42. Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA repair. 2016;42:63-71
43. Razavipour SF, Harikumar KB, Slingerland JM. p27 as a Transcriptional Regulator: New Roles in Development and Cancer. Cancer research. 2020;80:3451-8
Author contact
![]() Corresponding author: Yingying Lin (E-mail: linyingying226121edu.cn), Zhenzhou Xiao (E-mail: xiaozhenzhou250272edu.cn). Address: No. 420 Fuma Road, Jin'an District, Fuzhou 350014, Fujian Province, China. Tel.: +8659162752638; Fax: +8659162752638.
Corresponding author: Yingying Lin (E-mail: linyingying226121edu.cn), Zhenzhou Xiao (E-mail: xiaozhenzhou250272edu.cn). Address: No. 420 Fuma Road, Jin'an District, Fuzhou 350014, Fujian Province, China. Tel.: +8659162752638; Fax: +8659162752638.