Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2023; 20(11):1479-1491. doi:10.7150/ijms.85224 This issue Cite
Research Paper
The Protective Effects of Eicosapentaenoic Acid for Stress-induced Accelerated Senescence in Vascular Endothelial Cells
Min Jeong Kwon1, Hye Sook Jung2, Seon Mee Kang3, Soon Hee Lee1, Jeong Hyun Park1,2 ![]()
1. Department of Internal Medicine, Busan Paik Hospital, College of Medicine, Inje University, Busan, South Korea.
2. Paik Institute for Clinical Research, Inje University, Busan, South Korea.
3. Department of Internal Medicine, Kangwon National University Hospital, School of Medicine, Kangwon National University, Chuncheon, South Korea.
Received 2023-4-12; Accepted 2023-9-6; Published 2023-9-11
Abstract

Background: Eicosapentaenoic acid (EPA) is an omega-3 fatty acid that protects against cardiovascular diseases in patients with hypertriglyceridemia and may have pleotropic effects beyond lowering triglycerides. Many degenerative diseases, such as atherosclerosis and diabetes, are related to cellular senescence as a pathophysiological mechanism. We aimed to examine whether EPA could protect vascular endothelial cells under stress conditions against stress-induced accelerated senescence (SIAS).
Methods: Cultured human umbilical vein endothelial cells (HUVECs) were exposed to H2O2 as oxidative stress and a high glucose concentration with palmitate as a glucolipotoxic condition. Changes in cell viability, apoptosis, lactate dehydrogenase release, and cell cycle analysis were measured by cell counting kit-8 assay, annexin V/ propidium iodide staining, and enzyme-linked immunosorbent assay, respectively. EPA was applied in stress conditions. The degree of senescence was measured by senescence-associated beta-galactosidase staining and p16 staining using immunofluorescence. Apoptosis and cellular senescence-related proteins were measured by Western blotting.
Results: Cultured HUVECs under oxidative and glucolipotoxic stresses revealed accelerated senescence and increased apoptosis. These changes were markedly reversed by EPA administration, and the expressions of apoptosis and cellular senescence-related proteins were reversed by EPA treatment.
Conclusion: EPA effectively protects HUVECs against SIAS, which may be one of its pleotrophic effects.
Keywords: eicosapentaenoic acid, oxidative stress, stress-induced accelerated senescence
Introduction
Atherosclerosis is a progressive chronic inflammatory disease that causes major cardiovascular diseases such as ischemic heart disease, peripheral vascular disease, and cerebral infarction and is closely related to aging (1). Diabetes mellitus also causes atherosclerosis, and cardiovascular disease is a main cause of death in patients with diabetes. Diabetes is evaluated as a coronary artery disease risk equivalent, which means an equivalent risk level as already having cardiovascular disease in terms of the risk of developing cardiovascular disease. Cellular senescence is a process in which various stressors permanently stop cell division, resulting in cell changes that appear as senescent-associated secretory phenotype (SASP) (2). SASP is involved in the secretion of substances such as growth factors, cytokines, and chemokines and the generation of reactive oxygen species (ROS) (3). Diabetes prevalence increases with age; growth factors, cytokines, and ROS are some of the mechanisms of diabetic complications (4). Glucotoxicity and lipotoxicity are also potential mechanisms for diabetic complications, in addition to beta cell dysfunction. Hyperglycemia and hyperlipidemia, common in patients with uncontrolled diabetes, cause oxidative and endoplasmic reticulum stress, causing complications (5).
The beneficial effect of omega-3 fatty acids on atherosclerosis has been suggested to involve several mechanisms such as to induce anti-inflammatory and anti-arrhythmic effects, to improve the lipid profile by lowering triglyceride levels, to prevent plaque development, to promote plaque stabilization, and to inhibit platelet aggregation (6). Additionally, they may have the benefit of improving endothelial cell dysfunction caused by blunted endothelium-dependent vasodilatation mostly due to a reduced bioavailability of endothelium-derived nitric oxide (NO), and, often, endothelium-derived hyperpolarization in an early step of atherogenesis. The eicosapentaenoic acid (EPA) has been shown to cause endothelium-dependent relaxations of isolated sheep pulmonary arteries, and to increase endothelial NO synthase (eNOS) activity and NO formation in cultured endothelial cells (7-9). In addition, EPA but not docosahexaenoic acid (DHA) has also been shown to cause cerebral microvascular vasodilatation involving arachidonic acid metabolites (10). A study showed that highly purified EPA:DHA 6:1 and 9:1 formulations demonstrated greater endothelium-dependent NO-mediated relaxation of porcine coronary artery rings than other ratios (11).
Several clinical studies, but not all, have suggested that omega-3 polyunsaturated fatty acids, in particular EPA and DHA, improve the endothelial function and reduce cardiovascular events in patients with cardiovascular risk factors (12-14). In a recently reported study, Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial (REDUCE-IT), icosapent ethyl (IPE), which is a highly purified and stable eicosapentaenoic acid (EPA) ethyl ester, significantly lowered the risk of ischemic events, including cardiovascular death in patients with elevated triglyceride levels despite the use of statins (15). However, the observed reduction in low-density lipoprotein cholesterol (LDL-C), triglycerides, and non-high-density lipoprotein cholesterol (non-HDL-C) did not fully account for the positive outcomes, indicating that other properties of EPA were likely involved in the benefits (16).
Hence, we aimed to investigate the impact of oxidative stress or glucolipotoxicity at the cellular level on atherosclerosis in patients with diabetes in relation to SASP and explore whether EPA responds to SASP in these environment.
Materials and Methods
Reagents
EPA (E2011, 99% purity, ethyl ester) was purchased from Sigma (MO, USA) and dissolved in dimethyl sulfoxide.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were obtained from Promocell (Heidelberg, Germany). HUVECs were cultured in endothelial growth medium (EGM) with 5 ng/mL epidermal growth factor, 10 ng/mL basic fibroblast growth factor, 20 ng/mL insulin-like growth factor, 0.5 ng/mL vascular endothelial growth factor 165, 1 μg/mL ascorbic acid, 22.5 μg/mL heparin and 0.2 μg/mL hydrocortisone (Promocell, Heidelberg, Germany) in all experiments. All cells were incubated at 37°C with 5% CO2 and used between the third and seventh passage.
Cell viability using cell counting kit (CCK)-8 assay
HUVECs were seeded in 96-well plates at 7×103 cells per well and incubated for 24 hours in EGM. To confirm cell viability by H2O2 (Sigma, MO, USA) or high glucose concentration with palmitate (HGP) with/without EPA (Sigma, MO, USA), the culture medium was aspirated and replaced with fresh medium containing varying concentrations of EPA for 2 hours, followed by incubation with H2O2 (300 μM) or HGP (glucose 25 mM and palmitate 300 μM) for 48 hours. The medium was then replaced with CCK-8 reaction buffer (Dojindo, Munich, Germany), and the cells were incubated for 2 hours at 37°C. The optical density was measured at a wavelength of 450 nm using a microplate reader to determine cell viability.
Cell apoptosis using annexin V/ propidium iodide (PI) staining and lactate dehydrogenase (LDH) release assay
HUVECs were seeded in 12-well plates at 7×104 cells per well and incubated for 24 hours. To confirm the anti-apoptotic effect of H2O2 or HGP with/without EPA, culture mediums were aspirated and replaced with new mediums containing various concentrations of EPA for 2 hours and then incubated in 300 μM H2O2 or HGP (glucose 25 mM and palmitate 300 μM) for 48 hours. To obtain single cells, cells were treated with trypsin-EDTA 200 μL and centrifuged at 1500 rpm for 5 minutes at 4°C. After supernatant aspiration, cells were washed with 1 mL of annexin V binding buffer (140 mM NaCl, 10 mM HEPES, pH 7.4, 2.5 mM CaCl2) and centrifuged at 1,500 rpm for 5 minutes at 4°C. Supernatants were removed, and 3 μL of annexin V-FITC (BD, CA, USA) and 10 μL of PI (BD, CA, USA) was added. After incubation for 15 minutes in the dark, 300 μL of fluorescence activated cell sorting (FACS) buffer (1% FBS, 0.1% NaN3) was added, and analysis was performed using FACSort (BECTON DICKINSON, BD bioscience). After treatment with various concentrations of EPA for 2 hours and 300 μM H2O2 or HGP for 48 hours, the supernatant was transferred into an e-tube. Isolated protein by radioimmunoprecipitation assay (RIPA) buffer was quantified by bicinchoninic Acid (BCA) kits (Pierce, CA, USA). The serum was separated by centrifugation at 300 rpm for 20 minutes. LDH was measured by LDH enzyme-linked immunosorbent assay kit (Abcam, Cambridge, UK).
Cell cycle analysis
HUVECs were washed with phosphate-buffered saline (PBS) and slowly drop-chilled with 70% ethanol to fix the cells. After 30 minutes of incubation, cells were re-washed with PBS and re-suspended with 50 μg/mL PI (BD, CA, USA) and 50 μg/mL RNase A at room temperature. After incubation for 30 minutes in the dark, 300 μL of FACS buffer (1% FBS, 0.1% NaN3) was added, and analysis was performed using FACSort (BECTON DICKINSON, BD bioscience).
Senescence-associated beta-galactosidase (SA-β-Gal) staining
After incubation under appropriate conditions in H2O2 or HGP with/without EPA, HUVECs were washed with PBS, and then SA-β-Gal working solution (pH 6.0) (Cell signaling, MA, USA) was added. After 24 hours, cells were observed under an inverted microscope. The percentages of SA-β-Gal-stained cells per total cells were compared quantitatively.
p16 expression by immunofluorescent staining
Immunofluorescent staining was performed to determine the cellular distribution of p16. HUVECs were plated onto a cover slide. After incubation under appropriate conditions, cells were fixed and permeabilized with cytoperm/cytofix (BD, CA, USA) for 15 minutes and incubated with blocking buffer (5% serum and 0.3% triton X-100 in PBS) for 1 hour and then incubated in anti-p16 antibody (Abcam, Cambridge, UK) for 1 hour at room temperature. Then the cells were incubated in mouse-anti Alexa 488 FluorⓇ as a secondary antibody and 4′,6-diamidino-2-phenylindole (DAPI, 1 μg/mL) for 1 hour at room temperature. Fluorescence in situ hybridization (FISH) analysis was performed using a green fluorescence microscope, Olympus BX-51 (Olympus, Tokyo, Japan).
Western blotting analysis
HUVECs were washed with PBS and lysed using mammalian tissue lysis/extraction reagent (Sigma, MO, USA), including protease inhibitor (Roche, IN, USA). Protein concentrations were determined by BCA protein assay kit (Pierce, CA, USA), and 1× sodium dodecyl sulfate (SDS) sample buffer (50 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 50 mM dithiothreitol, and 0.01% bromophenol blue) was added. Proteins were separated in 10-15% SDS-polyacrylamide gel electrophoresis, transferred onto polyvinylidene difluoride membranes, and immunoblotted with anti-silent information regulator type 1 (Sirt1) (Abcam, Cambridge, UK), anti-phospho-p38 mitogen-activated protein kinase (MAPK) (Cell signaling, MA, USA), anti-p38 MAPK (Cell signaling, MA, USA), anti-p16 (Abcam, Cambridge, UK), anti-Bcl2 (Cell signaling, MA, USA), anti-cleaved caspase3 (Cell signaling, MA, USA), anti-phospho-AMP-activated protein kinase (AMPK) (Cell signaling, MA, USA), anti-AMPK (Cell signaling, MA, USA), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell signaling, MA, USA) at 4°C overnight. Secondary antibodies (goat anti-rabbit or anti-mouse conjugated alkaline phosphatase (Bethyl, TX, USA)) were added for 1 hour at room temperature, and membranes were developed via alkaline phosphatase-conjugated development kit (Bio-rad, CA, USA). Developed protein bands were quantified by Image J program (NIH, Bethesda, MD, USA).
Statistical analysis
Data were expressed as the mean ± standard deviation, and statistical analyses were performed using SPSS software (version 20.0; IBM Corp., NY, USA). Differences between the groups of results were determined using a one-way ANOVA, followed by a Tukey's post hoc test. In all analyses, P<0.05 was considered statistically significant.
Results
Effects of EPA treatment on cell viability and apoptosis of HUVECs under stress conditions
We conducted a CCK-8 experiment to confirm whether the cell viability was decreased by H2O2 or glucolipotoxicity and if EPA had a protective effect (Figure 1A, 1B). When assessed 48 hours after EPA treatment, the protective effect of EPA on cell viability was observed at 25, 50, and 100 μM concentrations in the H2O2 group, and similar results were obtained in the control group at 100 μM of EPA. In the group exposed to HGP (25 mM glucose and 300 μM palmitate), the cell viability increased at 50 and 100 μM of EPA compared to the HGP-treated group without EPA.
Effects of EPA on cell viability and apoptosis of HUVECs under stress conditions. (A) Cell viability measured using the CCK-8 assay was decreased by oxidative stress induced by 300 μM of H2O2, which was attenuated by treatment with 25, 50, and 100 μM EPA. (B) Cell viability measured using CCK-8 assay was decreased by HGP (25 mM glucose and 300 μM palmitate), which was attenuated by treatment with 50 and 100 μM EPA treatment. (C) Cellular apoptosis detected using Annexin V-FITC and PI double staining was increased by oxidative stress, which was attenuated by treatment with 25, 50, and 100 μM EPA. (D) Cellular apoptosis detected using Annexin V/PI staining was increased by HGP, which was attenuated by treatment with 100 μM EPA. These experiments were performed in triplicate. Values are means ± SD. C: control, EPA: eicosapentaenoic acid, CCK: cell counting kit, HGP: high glucose concentration with palmitate, PI: propidium iodide *Significantly different from the stress group, P<0.05.
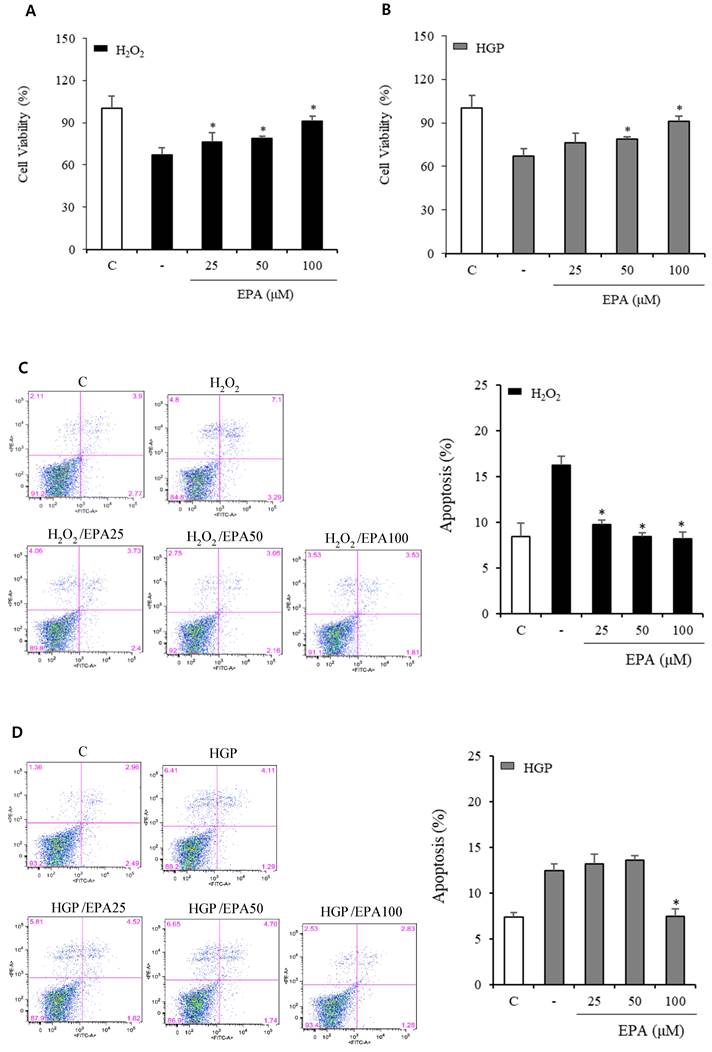
At 100 μM EPA, similar results to that of the control group were obtained, but no protective effect was observed at 25 μM of EPA. After the reaction, cells were stained with annexin V/PI to observe the anti-apoptotic effect of EPA (Figure 1C, 1D). In the control group, apoptosis was approximately 8%, whereas, in the H2O2 treatment group, it was increased by approximately 16% (a two-fold increase). Apoptosis was increased by H2O2 and was decreased by EPA. The anti-apoptotic effect was confirmed at approximately 10% at 25 μM and 8% at 50 and 100 μM. The HGP-treated group exhibited 14% apoptosis, which was increased by approximately 7% compared to the control group. At 100 μM, EPA reduced the apoptosis increased by HGP as much as the control, but no difference was observed with 25 and 50 μM EPA.
In the H2O2 exposed group, LDH release indicating cellular death increased after H2O2 and was reduced by treatment with 50 and 100 μM EPA (Figure 2A). LDH markedly increased over 10 times in the HGP-treated group compared to the control group, and LDH was reduced by half after treatment with 100 μM EPA in the control group compared to the HGP-treated group. However, there was no effect at 50 μM of EPA (Figure 2B).
Effects of EPA on markers related to cellular death and apoptosis in HUVECs under stress conditions (A) LDH release, indicating cellular death, was significantly increased after H2O2 treatment and reduced by treatment with 50 and 100 μM EPA. (B) LDH release was significantly increased after HGP treatment and reduced by 100 μM EPA treatment. (C, D) Cleaved caspase-3 to caspase-3 ratio, a proapoptotic marker increased by H2O2 or HGP treatment, was decreased by treatment with 50 and 100 μM EPA. The Bcl2, an antiapoptotic marker decreased by H2O2 or HGP treatment, was increased by treatment with 50 and 100 μM EPA. These experiments were performed in triplicate. Values are means ± SD. C: control, C.C3: cleaved caspase-3, EPA: eicosapentaenoic acid, HGP: high glucose concentration with palmitate *Significantly different from the stress group, P<0.05.
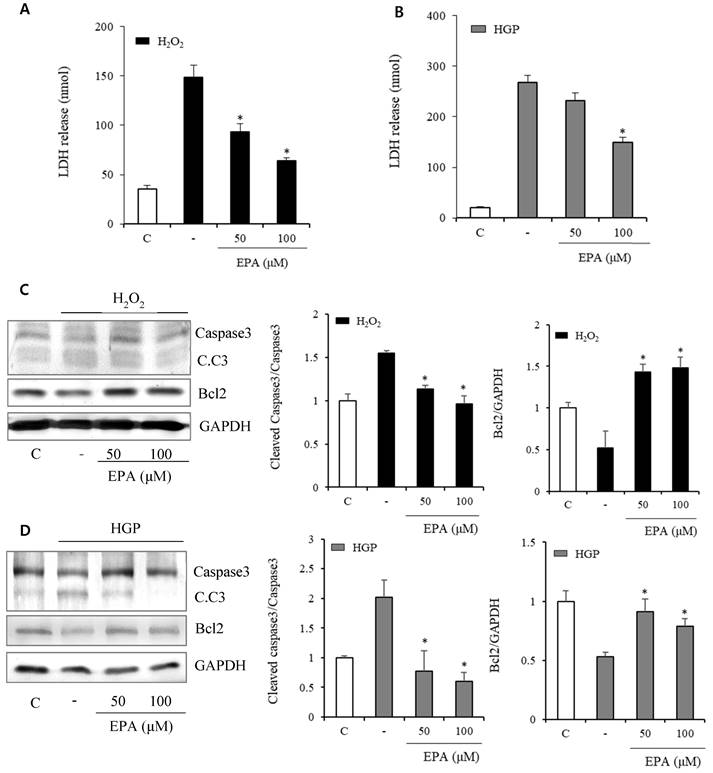
Anti-apoptosis-related proteins were identified (Figure 2C, 2D). Cleaved caspase3, which was increased by H2O2 treatment, was decreased by EPA. Bcl2, an anti-apoptosis protein, was also reduced by H2O2 treatment but restored by EPA. Similar results were obtained in the HGP-treated group.
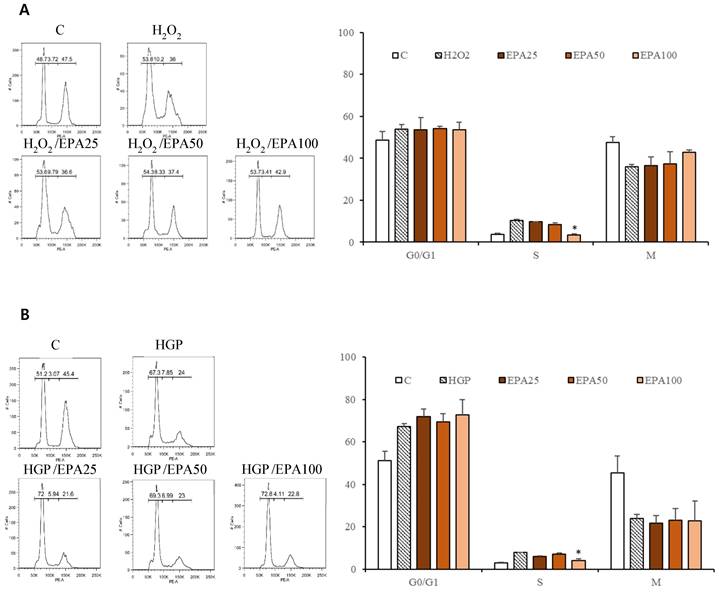
The cell cycle was investigated (Figure 3). HUVECs that were stagnant in the s phase due to H2O2 application (10.2%) were recovered by EPA in a dose-dependent manner (25 μM; 9.79%, 50 μM; 8.33%, 100 μM; 3.41%), although only the 100 μM concentration was significant. Cells that were stagnant in the s phase by HGP (7.85%) were recovered by treatment with 100 μM EPA (4.11%).
The effect of the EPA on the cell cycle in HUVECs under stress conditions. (A) HUVECs that were significantly stagnant in the s phase by H2O2 treatment (10.2%) were recovered by EPA in a dose-dependent manner (25 μM; 9.79%, 50 μM; 8.33%, 100 μM; 3.41%), although only the 100 μM concentration was significant. (B) The cells that were significantly stagnant in the s phase by HGP treatment (7.85%) were recovered by treatment with 100 μM EPA (3.41%). These experiments were performed in triplicate. Values are means ± SD. C: control, EPA: eicosapentaenoic acid, HGP: high glucose concentration with palmitate *Significantly different from the stress group, P<0.05.
Effects of EPA on cellular senescence of HUVECs under stress conditions
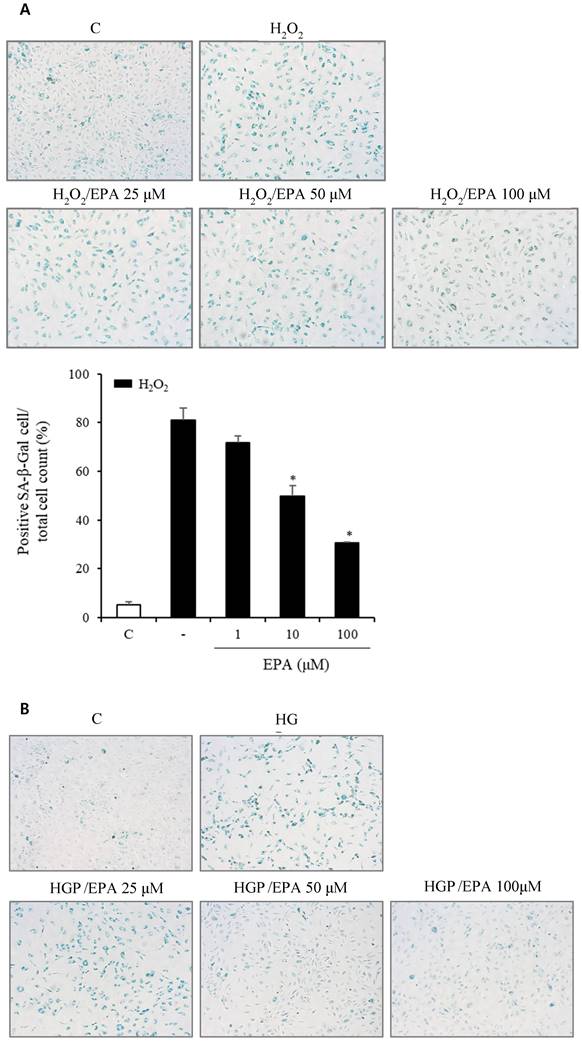
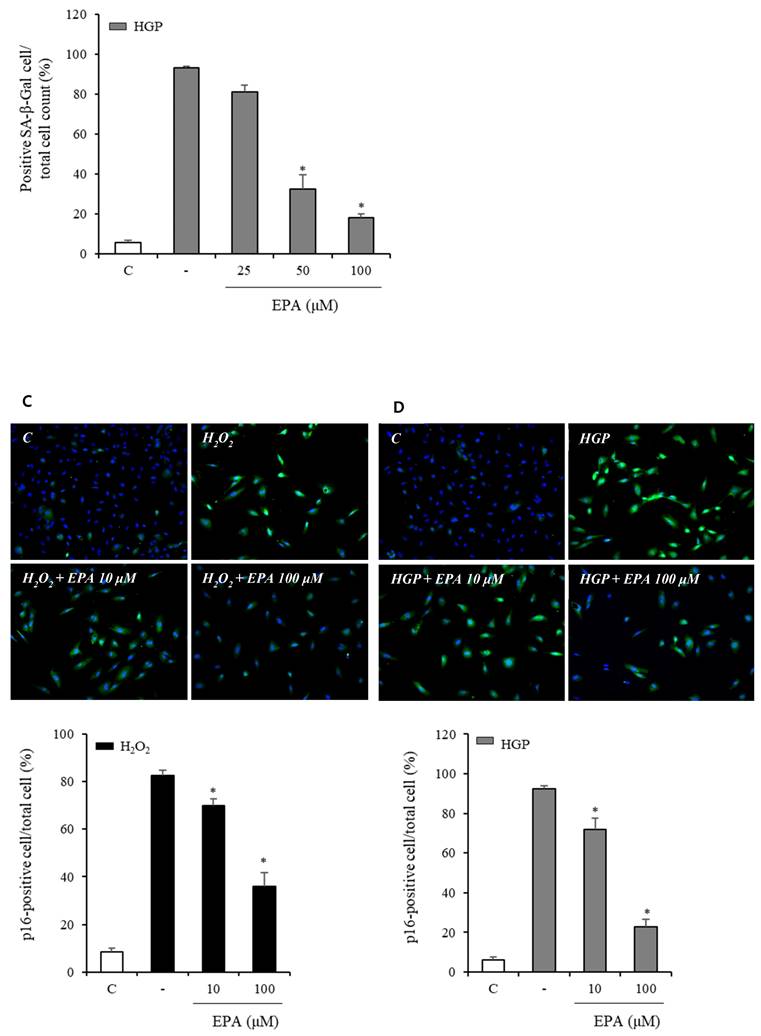
To observe the aging effects of H2O2 and the anti-aging effects of EPA, we conducted an SA-β-Gal stain experiment (Figure 4A, 4B). HUVECs were incubated with various concentrations of EPA for 24 hours and then treated with H2O2 for 48 hours. SA-β-Gal staining increased dramatically in the H2O2 group compared to the control group, whereas EPA inhibited aging, with a more pronounced effect at higher concentrations. The 100 μM EPA treatment group demonstrated similar aging inhibition as the control group. We also tested HGP under the same conditions as H2O2 and discovered that SA-β-Gal staining increased similarly. However, EPA effectively decreased the SA-β-Gal staining increased by HGP, and the anti-aging effect was greater at higher concentrations of EPA. We confirmed the change in the p16 protein through cell fluorescence staining, as p16 protein increases with aging in cells (Figure 4C, 4D). We observed a significant increase in the p16 protein in the H2O2 group compared to the control group.
Effects of EPA on cellular senescence of HUVECs under stress conditions. (A, B) Senescent (SA-β-Gal-positive, green) cells increased after H2O2 or HGP treatment and decreased by treatment with 25, 50, and 100 μM EPA in a dose-dependent manner. (C, D) p16 protein expression (green) detected by immunofluorescent staining was increased after H2O2 or HGP treatment and decreased by treatment with 10 and 100 μM EPA, respectively. C: control, EPA: eicosapentaenoic acid, HGP: high glucose concentration with palmitate.
Anti-senescence mechanism of EPA under stress conditions
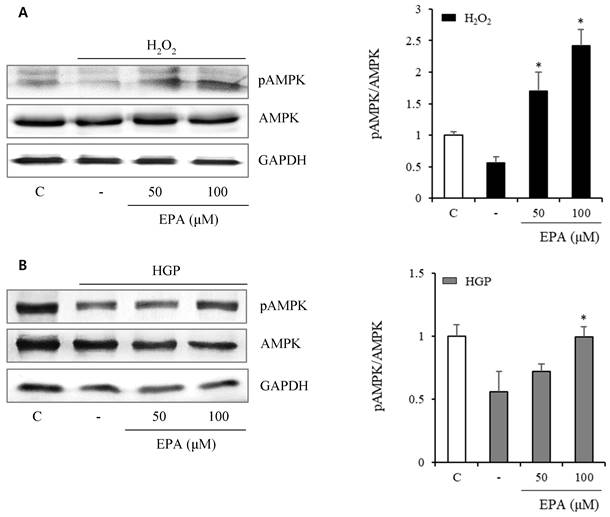
To investigate the anti-senescence mechanism, we observed changes in senescence-related proteins. AMPK phosphorylation (Figure 5) was reduced by 50% and 25% under H2O2 or HGP stress conditions, respectively, compared to the control group. However, in the H2O2-treated group, AMPK phosphorylation was recovered at 50 and 100 μM EPA. In the HGP-treated group, AMPK phosphorylation was increased at 100 μM EPA, similar to the control group.
The effect of EPA on AMP‑activated protein kinase (AMPK) phosphorylation under stress conditions The AMPK phosphorylation was reduced by 50% and 25%, respectively, compared to the control under H2O2 or HGP stress conditions, which was recovered by treatment with 50 and 100 μM EPA in the H2O2 treated group, and 100 μM EPA in the HGP treated group, similar to the control group. These experiments were performed in triplicate. Values are means ± SD. C: control, EPA: eicosapentaenoic acid, HGP: high glucose concentration with palmitate *Significantly different from the stress group, P<0.05.
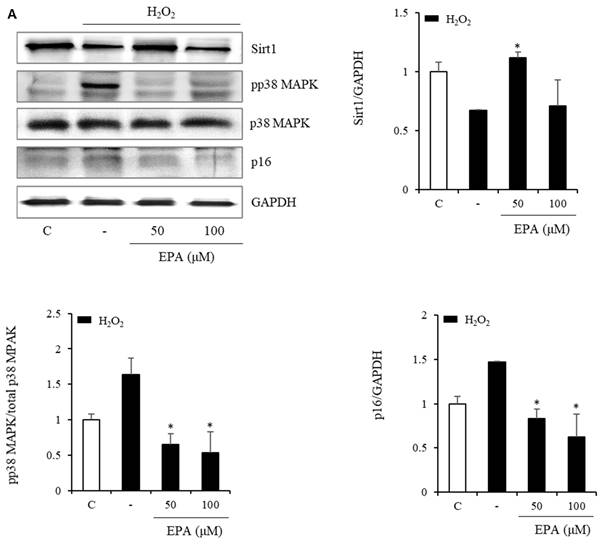
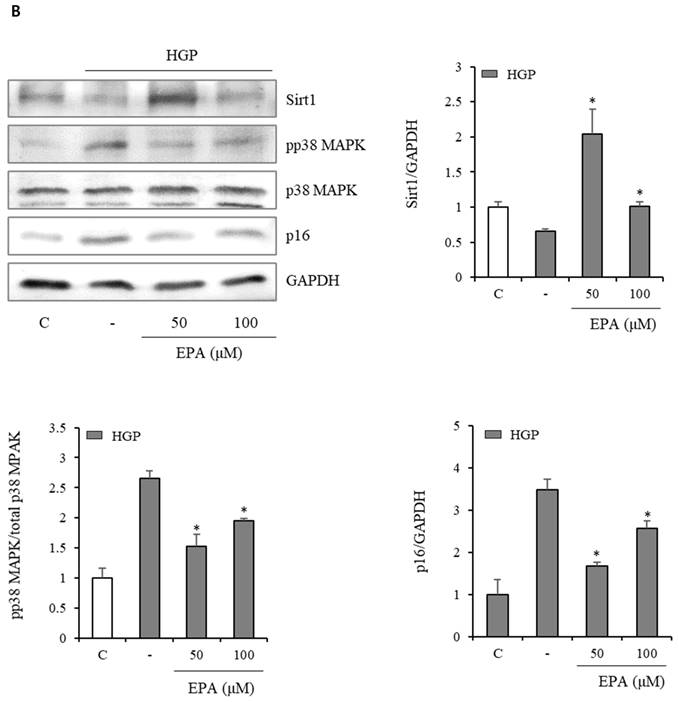
We also observed changes in the Sirt1/p38 MAPK/p16 mechanism (Figure 6). In the H2O2 treatment group, Sirt1 decreased by 50%, whereas p38 MAPK phosphorylation increased more than three-fold, and p16 protein increased two-fold. However, the EPA administration led to a recovery similar to that of the control group. This effect was also observed in the HGP treatment group.
Effects of EPA on the SIRT1/p38 MAPK/p16 pathway in senescent HUVECs under stress conditions. In the H2O2 or HGP treatment group, Sirt1 was decreased, and phosphorylation of p38 MAPK and p16 protein were increased compared to the control and was recovered by EPA similar to the control group. These experiments were performed in triplicate. Values are means ± SD. C: control, EPA: eicosapentaenoic acid, HGP: high glucose concentration with palmitate *Significantly different from the stress group, P<0.05.
Discussion
Aging is a major risk factor for chronic diseases that increase with age, such as neurodegenerative disease, malignancy, metabolic disease, and cardiovascular disease. Aging and age-related diseases have common biological mechanisms, such as inflammation, the accumulation of macromolecular damage, adaptation to stressors, epigenetic changes, metabolic dysfunction, loss of proteostasis, and defective stem cell function. Major age-related diseases include type 2 diabetes, atherosclerosis, and certain cancers (17).
Atherosclerotic cardiovascular disease is one of the leading causes of mortality in patients with diabetes. Blood sugar control is important to prevent complications of diabetes. However, in large-scale clinical trials, it was difficult to confirm the short-term efficacy of strict blood sugar control in preventing cardiovascular disease complications. Rather, extremely strict glycemic control increased mortality due to the risk of hypoglycemia (18). Therefore, managing multiple risk factors, such as blood pressure and cholesterol control, is emphasized over glucose control alone (19).
Cholesterol control is important in preventing cardiovascular disease. However, even if LDL-C is sufficiently lowered using statins, there is a residual risk of cardiovascular disease of approximately 65% (20-22), and hypertriglyceridemia is a known cause (23, 24). A recent REDUCE-IT study revealed that when highly purified EPA called IPE (Vascepa®) was added to statins, atherosclerotic cardiovascular disease was reduced by 25% (15). Thus, various academic societies and related organizations have changed treatment guidelines (25-27). However, in the REDUCE-IT study, the reduction in triglyceride, non-HDL-C, and LDL-C due to IPE could not fully explain the cardiovascular disease prevention effect. It is believed that pleiotropic effects other than reduction may have contributed to cardiovascular disease prevention (16). These include anti-inflammatory action, antithrombotic action, cell membrane stabilizing effect, antioxidant activity, and improving the function of vascular endothelial cells and preventing arrhythmia; however, the mechanism of action has not been completely elucidated.
Cellular senescence is defined as irreversible cell cycle arrest and is activated by multiple stressors such as DNA damage, oxidative stress, and cytotoxic environments, which is stress-induced accelerated senescence (SIAS). These stimuli activate various signaling pathways through p16, p21, and p53 and activate cell cycle inhibitors. Morphologically senescent cells are usually bulky and have increased SA-β-Gal activity due to mitochondrial dysfunction and lysosomal biogenesis (28). Therefore, p16 expression and SA-β-Gal are used as biomarkers of cellular senescence. H2O2-induced oxidative stress or HGP-induced glucolipotoxicity mimicking SIAS increased the SA-β-Gal stain positivity in HUVECs, and the number of SA-β-Gal positive cells significantly decreased after EPA treatment. The expression of p16 in HUVECs was increased under oxidative stress or glucolipotoxic conditions, which decreased after EPA treatment. These results suggest that EPA protects HUVECs from SIAS.
In previous reports of normal human epidermal keratinocytes or human bone marrow-derived mesenchymal stem cells subjected to H2O2, increased ratios of cells were exposed to oxidative stress in the G0/G1 phase (29, 30). However, in this study, the percentage of cells exposed to oxidative stress in the G0/G1 phase did not increase significantly. The G0/G1 phase ratio of HUVECs exposed to glucolipotoxic conditions increased but was unaffected by EPA. The s phase was significantly increased by oxidative stress and glucolipotoxic conditions and was decreased by a high EPA concentration. There may be a difference in experimental conditions; however, additional research on cell cycle-related proteins is needed. During the s phase, the cell continuously scrutinizes its genome for abnormalities. Detection of DNA damages by oxidative and glucolipotoxic stresses induces activation of three canonical s phase "checkpoint pathways" that delay or arrest further cell cycle progression and may lead to the stagnation of cells in the s phase (31). In addition to the actions of these canonical checkpoints, recent evidence suggested that the abnormalities in histone supply and nucleosome assembly can also alter the s phase progression (32). So, the increased number of cells in the s phase after exposure to oxidative and glucolipotoxic stresses in our experiment could be interpreted as the stagnation of cells in the s phase.
On the molecular level, cellular senescence has been associated with two major intracellular signaling pathways: the p38 MAPK/p16INK4α pathway and the p53/p21 pathway (33). The protein p21, a cyclin-dependent kinase inhibitor, triggers the onset of cell cycle arrest, which can be induced by p53 (34). Recently, human SIRT1, a member of the nicotinamide adenine dinucleotide-dependent deacetylase protein family, has been demonstrated to recover the senescence process by increasing cell proliferation and reducing p16INK4α expression in human diploid fibroblasts (35).
Our results revealed that Sirt1 expression was decreased in HUVECs, whereas phospho-p38 MAPK and p16 protein expressions were increased under oxidative stress or glucolipotoxic conditions, and the addition of EPA opposed the senescence marker (36).
In regulating aging and mammalian senescence, the reciprocal regulations of SIRT1 and AMPK pathways were studied (37). A decreased SIRT1 expression and activity accompany the progression from pre-senescence to irreversible senescence and an augmented AMPK function. From our study, it is difficult to interpret the relationship between AMPK phosphorylation and SIRT1 reciprocally, similar to previous studies. Therefore, additional research is needed.
This study had several limitations. First, there are apparent differences between senescence and SIAS. p16 protein and SA-β-Gal staining are considered biomarkers of senescence; however, they could be elevated under other conditions, such as inflammation. Second, we used only HUVECs in our study. However, atherosclerotic plaque builds up in arteries and is produced by the interaction of vascular endothelial cells, vascular smooth muscle cells, and inflammatory molecules (38). Third, we did not experiment with EPA-induced changes under non stressful conditions to compare oxidative stress or glucolipotoxic conditions. And EPA is a potent inducer of NO, and this may contribute to the findings of EPA's cell protection efficacies in our experiments. Additional experiments are needed.
Conclusions
In conclusion, the senescence-related process is partially involved in the development of atherosclerosis and diabetic complications. EPA acts by ameliorating SIAS of HUVECs in oxidative stress or glucolipotoxic environment. These findings suggest that the antisenescence mechanism could be one of the pleotrophic effects of EPA.
Abbreviations
SASP: senescent-associated secretory phenotype; REDUCE-IT: Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial; IPE: icosapent ethyl; ROS: reactive oxygen species; EPA: eicosapentaenoic acid; LDL-C: low-density lipoprotein cholesterol; non-HDL-C: non-high-density lipoprotein cholesterol; HUVECs: human umbilical vein endothelial cells; HGP: high glucose concentration with palmitate; PI: propidium iodide; LDH: lactate dehydrogenase; PBS: phosphate-buffered saline; SA-β-Gal: senescence-associated beta-galactosidase; SDS: sodium dodecyl sulfate; MAPK: mitogen-activated protein kinase; SIAS: stress-induced accelerated senescence.
Acknowledgements
Funding: This work was supported by a grant from Inje University Busan Paik Hospital in 2021.
Author contributions
(I) Conception and design: HS Jung, SM Kang, JH Park; (II) Administrative support: None; (III) Provision of study materials or patients: HS Jung; (IV) Collection and assembly of data: MJ Kwon, HS Jung; (V) Data analysis and interpretation: MJ Kwon, JH Park; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Machado-Oliveira G, Ramos C, Marques ARA. et al. Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells. 2020;9:2146
2. Passos JF, Simillion C, Hallinan J. et al. Cellular senescence: unravelling complexity. Age (Dordr). 2009;31:353-63
3. Childs BG, Durik M, Baker DJ. et al. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424-35
4. Kitada M, Zhang Z, Mima A. et al. Molecular mechanisms of diabetic vascular complications. J Diabetes Investig. 2010;1:77-89
5. Cerf ME. Cardiac Glucolipotoxicity and Cardiovascular Outcomes. Medicina (Kaunas). 2018;54:70
6. Kromhout D, Yasuda S, Geleijnse JM. et al. Fish oil and omega-3 fatty acids in cardiovascular disease: do they really work? European heart journal. 2012;33(4):436-43
7. Boulanger C, Schini VB, Hendrickson H. et al. Chronic exposure of cultured endothelial cells to eicosapentaenoic acid potentiates the release of endothelium-derived relaxing factor (s). British journal of pharmacology. 1990;99(1):176-80
8. Omura M, Kobayashi S, Mizukami Y. et al. Eicosapentaenoic acid (EPA) induces Ca2+-independent activation and translocation of endothelial nitric oxide synthase and endothelium-dependent vasorelaxation. FEBS letters. 2001;487(3):361-6
9. Singh TU, Kathirvel K, Choudhury S. et al. Eicosapentaenoic acid-induced endothelium-dependent and-independent relaxation of sheep pulmonary artery. European journal of pharmacology. 2010;636(1-3):108-13
10. Ellis EF, Police RJ, Yancey L. et al. Effect of fish oil n-3 fatty acids on cerebral microcirculation. American Journal of Physiology-Heart and Circulatory Physiology. 1990;258(6):H1780-H5
11. Zgheel F, Alhosin M, Rashid S. et al. Redox-sensitive induction of Src/PI3-kinase/Akt and MAPKs pathways activate eNOS in response to EPA: DHA 6: 1. PloS one. 2014;9(8):e105102
12. Delgado-Lista J, Perez-Martinez P, Lopez-Miranda J. et al. Long chain omega-3 fatty acids and cardiovascular disease: a systematic review. British Journal of Nutrition.
13. DiNicolantonio JJ, Niazi AK, McCarty MF. et al. Omega-3s and cardiovascular health. Ochsner Journal. 2014;14(3):399-412
14. Maki KC, Palacios OM, Bell M. et al. Use of supplemental long-chain omega-3 fatty acids and risk for cardiac death: An updated meta-analysis and review of research gaps. Journal of Clinical Lipidology. 2017;11(5):1152-60 e2
15. Bhatt DL, Steg PG, Miller M. et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med. 2019;380:11-22
16. Mason RP, Libby P, Bhatt DL. Emerging Mechanisms of Cardiovascular Protection for the Omega-3 Fatty Acid Eicosapentaenoic Acid. Arterioscler Thromb Vasc Biol. 2020;40:1135-47
17. Sabbatinelli J, Prattichizzo F, Olivieri F. et al. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front Physiol. 2019;10:1523
18. Gerstein HC, Miller ME, Byington RP. et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545-59
19. Yudkin JS, Richter B, Gale EA. Intensified glucose lowering in type 2 diabetes: time for a reappraisal. Diabetologia. 2010;53:2079-85
20. Sever PS, Dahlöf B, Poulter NR. et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149-58
21. Downs JR, Clearfield M, Weis S. et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615-22
22. Colhoun HM, Betteridge DJ, Durrington PN. et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364:685-96
23. Libby P. Triglycerides on the rise: should we swap seats on the seesaw. Eur Heart J. 2015;36:774-6
24. Ganda OP, Bhatt DL, Mason RP. et al. Unmet Need for Adjunctive Dyslipidemia Therapy in Hypertriglyceridemia Management. J Am Coll Cardiol. 2018;72:330-43
25. Mach F, Baigent C, Catapano AL. et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111-88
26. Orringer CE, Jacobson TA, Maki KC. National Lipid Association Scientific Statement on the use of icosapent ethyl in statin-treated patients with elevated triglycerides and high or very-high ASCVD risk. J Clin Lipidol. 2019;13:860-72
27. Skulas-Ray AC, Wilson PWF, Harris WS. et al. Omega-3 Fatty Acids for the Management of Hypertriglyceridemia: A Science Advisory From the American Heart Association. Circulation. 2019;140:e673-e691
28. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482-96
29. Sasaki M, Kajiya H, Ozeki S. et al. Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16(INK4a.). Biochem Biophys Res Commun. 2014;452:622-8
30. Zhou L, Chen X, Liu T. et al. Melatonin reverses H2 O2 -induced premature senescence in mesenchymal stem cells via the SIRT1-dependent pathway. J Pineal Res. 2015;59:190-205
31. Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5(10):792-804
32. Günesdogan U, Jäckle H, Herzig A. Histone supply regulates S phase timing and cell cycle progression. Elife. 2014;3:e02443
33. Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal. 2009;11:59-98
34. Cheng H, Qiu L, Ma J. et al. Replicative senescence of human bone marrow and umbilical cord derived mesenchymal stem cells and their differentiation to adipocytes and osteoblasts. Mol Biol Rep. 2011;38:5161-8
35. Huang J, Gan Q, Han L. et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS One. 2008;3:e1710
36. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403-17
37. Wang Y, Liang Y, Vanhoutte PM. SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett. 2011;585:986-94
38. Basatemur GL, Jørgensen HF, Clarke MCH. et al. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. 2019;16:727-44
Author contact
![]() Corresponding author: Jeong Hyun Park, Department of Internal Medicine, Busan Paik Hospital, College of Medicine, Inje University, 75 Bokji-ro, Busanjin-gu, Busan, 47392, Republic of Korea. Tel: +82-51-890-6074, Fax: +82-51-892-0273, Email: pjhdoccom.
Corresponding author: Jeong Hyun Park, Department of Internal Medicine, Busan Paik Hospital, College of Medicine, Inje University, 75 Bokji-ro, Busanjin-gu, Busan, 47392, Republic of Korea. Tel: +82-51-890-6074, Fax: +82-51-892-0273, Email: pjhdoccom.