Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2023; 20(3):329-345. doi:10.7150/ijms.80358 This issue Cite
Review
Genomic Fingerprint Associated with Familial Idiopathic Pulmonary Fibrosis: A Review
Dongyan Ding1, Rong Gao1, Qianfei Xue2, Rumei Luan1, Junling Yang1 ![]()
1. Department of Respiratory Medicine, The Second Hospital of Jilin University, Changchun, China.
2. Hospital of Jilin University, Changchun, China.
Received 2022-10-31; Accepted 2023-1-12; Published 2023-1-31
Abstract

Idiopathic pulmonary fibrosis (IPF) is a severe interstitial lung disease; although the recent introduction of two anti-fibrosis drugs, pirfenidone and Nidanib, have resulted in a significant reduction in lung function decline, IPF is still not curable. Approximately 2-20% of patients with IPF have a family history of the disease, which is considered the strongest risk factor for idiopathic interstitial pneumonia. However, the genetic predispositions of familial IPF (f-IPF), a particular type of IPF, remain largely unknown. Genetics affect the susceptibility and progression of f-IPF. Genomic markers are increasingly being recognized for their contribution to disease prognosis and drug therapy outcomes. Existing data suggest that genomics may help identify individuals at risk for f-IPF, accurately classify patients, elucidate key pathways involved in disease pathogenesis, and ultimately develop more effective targeted therapies. Since several genetic variants associated with the disease have been found in f-IPF, this review systematically summarizes the latest progress in the gene spectrum of the f-IPF population and the underlying mechanisms of f-IPF. The genetic susceptibility variation related to the disease phenotype is also illustrated. This review aims to improve the understanding of the IPF pathogenesis and facilitate his early detection.
Keywords: Familial idiopathic pulmonary fibrosis, Sporadic idiopathic pulmonary fibrosis, Telomerase-associated gene, Mucin 5B, Surfactant-related gene.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a form of idiopathic interstitial pneumonia (IIP) characterized by progressive lung scarring, persistent decline in lung function, and poor prognosis (1). Among all IIPs, IPF is the most common and fatal type with the worst prognosis. Approximately 80% of all patients with familial IIP are diagnosed with IPF (2). Patients with IPF have a shorter disease course than those with other types of IIP. The median survival of patients with IPF after diagnosis is only 2-4 years (3), and the clinical course and survival rates vary greatly (4). Pirfenidone and Nidanib are anti-fibrotic drugs approved by the US Food and Drug Administration (5), which can delay, but not prevent or improve, the decline of lung function. To date, lung transplantation is the only known life-prolonging intervention (6). The etiology of PF has not been fully established; however, it is thought to be caused by environmental factors and genetic susceptibility (7).
IPF occurs in sporadic, familial, or syndromic forms (8). The familial form accounts for 20% of all IPF cases, hereafter referred to as f-IPF (9). However, 80% of occurrences of IPF (the great majority) have not been divided among the two other forms. One of them could be rare, or they could be equally common. Sporadic IPF (s-IPF) is multifactorial; in addition to environmental risk factors, it is occasionally associated with polymorphic variants of various genes. Risk factors include smoking, inhaling metal and wood dust particles, and exposure to particular medications, such as methotrexate (2). Moreover, males have been identified to be more at risk than females. The clinical symptoms of f-IPF and s-IPF are similar; however, f-IPF is caused by gene variation and characterized by the earlier onset of symptoms (8). An increasing number of reports on PF in familial clusters and some rare genetic diseases suggest that genetic factors play an important role in IPF (7). The incidence of IPF and rate of disease progression differ in patients with the same levels of exposure, indicating the influence of genetic background heterogeneity.
Furthermore, IPF may occur in a familial setting, and patients with IPF show familial aggregation in different environments, suggesting a genetic cause (10). IPF has been reported in monozygotic twins raised in different environments from an early age (11). Additionally, IPF has been detected in successive generations within families (12) and family members separated at an early age (13). The known familial incidence of IPF provides evidence of the genetic background of the disease (14). Genetic testing may reveal the necessity of screening for other etiopathogenesis factors of IPF (15), including multisystem genetic disorders such as dyskeratosis congenita (DC) (16), neuroendocrine cell hyperplasia of infancy (17), Niemann-Pick disease (18), Hermansky-Pudlak syndrome, and short telomere syndrome (14).
Family cases of IPF were first described in 1907 by Sandoz (19) in Germany, who described a pair of 18-year-old twin sisters who both died of slowly progressive respiratory failure and whose autopsies revealed terminal honeycomb. Between 2% and 20% of patients with IPF have a first-degree relative with DPLD (20). Since the earliest description of IPF, familial clusters of similar cases have been identified as familial PF or f-IPF, which is defined by at least two biological family members with clinical and histological features of IPF (21). The occurrence of familial cases of PF raises the possibility of a genetic basis, and the search for the underlying genetic determinants of this disease through familial aggregates can better elucidate the pathogenesis of familial and more common sporadic diseases.
However, it must first be speculated whether the sporadic and familial forms of IPF are indeed distinct manifestations of a single clinical entity, or whether they represent two distinct disease processes. Among relatives with f-IPF, the most common phenotype of interstitial lung disease (ILD) is usual interstitial pneumonia (UIP); however, in at least 50% of known familial cases, affected family members develop more than one ILD phenotype (14). The Mayo Clinic series further concluded that f-IPF and s-IPF share the same clinical, radiological, and pathological features (12), except that the average age of onset of f-IPF (55 years) is lower than that of s-IPF (68 years). In addition, the average age of diagnosis for f-IPF (58 years) is lower than that for s-IPF (65 years) (22).
Genetic factors play an important role in both s-IPF and f-IPF (23), especially in patients with f-IPF. This finding suggests that a single autosomal dominant mechanism of reduced penetration cannot exclude autosomal recessive inheritance or heterogeneous coexistence of genetic traits (24); however, the genetic pattern of f-IPF remains unclear. Current data suggest that at least one-third of the risks of s-IPF or f-IPF can be attributed to common genetic variants (25), some of which have prognostic significance for patients with IPF (26). In addition, more rare genetic variants that affect susceptibility to f-IPF have been identified (23).
Susceptibility gene variation is informative for clarifying the pathogenesis of f-IPF and determining its key disease-causing targets. To date, there are no clinical guidelines on when and which gene tests should be performed in patients with IPF. Previous research has examined genetic associations, common single nucleotide polymorphisms (SNP) and rare gene mutations are associated with their development (25). Rare mutations in genes telomerase reverse transcriptase (TERT) (27), telomerase RNA component (TERC) (28), dyskeratosis congenita 1 (DKC1) (29), telomere repeat binding factor 1-interacting nuclear factor 2 (TINF2) (30), regulator of telomere length 1 (RTEL1), zinc finger CCHC-type containing 8 (ZCCHC8), poly(A)-specific ribonuclease (PARN) (31), surfactant protein C (SFTPC) (32), surfactant protein A2 (SFTPA2) (33), the ATP-binding cassette-type family A member 3 transporter (ABCA3) (34)), and common variants of 10 loci (3q26, 4q22, 5p15, 6p24, 7q22, 10q24, 11p15, 13q34, 15q14-15, and 19p13) (25) are associated with f-IPF.
Telomere-associated genes
Telomeres are cap-like structures at the ends of chromosomes comprising multiple repeated 5ʹ-TTAGGG-3ʹ DNA sequences that protect chromosome integrity and stability during cell division (35). As the cells divide, the telomere length progressively shrinks (36). Patients with familial and, less commonly, sporadic IPF have extremely short telomeres. TERC and TERT are the two main components that constitute the telomerase complex (37), a specialized ribonucleic protein and major component of the mechanism that maintains telomere lengthening (38). Telomerases are found in germlines, stem cells, tissue cells, and most cancer cells, with the ability to proliferate actively, but are scarce or absent in normal somatic cells (39). Mutations in genes that regulate telomere lengthening can negatively affect their maintenance, which can lead to gradual telomere shortening. When telomeres are shortened to a certain critical point, DNA damage reaction is activated, mitochondria cannot divide, and apoptosis ensues (40) (Fig. 1). Telomere shortening is related to aging and senile diseases (41), as telomere length decreases with age (36). Telomere mutations are found in 25% of patients with f-IPF and 1-3% of those with s-IPF (42).
Role of short telomeres in PF.
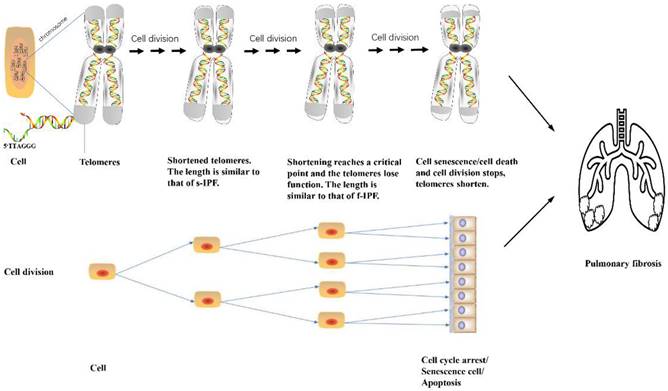
Telomere shortening occurs gradually throughout the life cycle as cells undergo continuous mitosis, especially in individuals genetically susceptible to familial IPF who have significantly shorter telomeres than patients with sporadic IPF. The mutation of the telomerase complex accelerates telomere shortening. When telomere shortening reaches a critical value, DNA damage is activated, and mitochondria fail to divide, resulting in cell senescence, apoptosis, or cell division cycle termination.
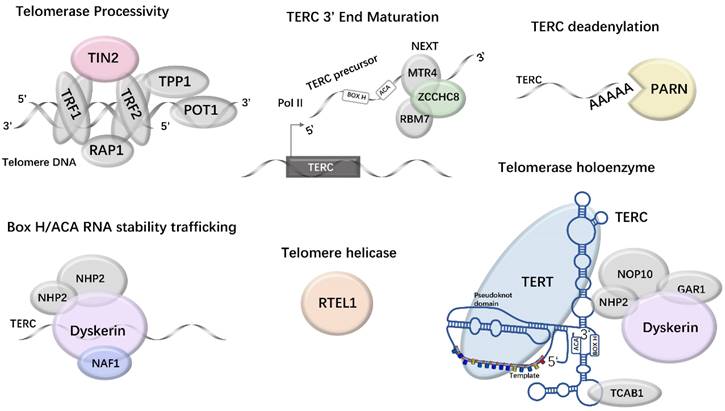
Half of IPF families with known mutations carry TERT or TERC mutations (43). Half member might carry mutations in one or more of the other telomere maintenance genes, such as RTEL1, PARN, nuclear assembly factor 1 (NAF1), TINF2, ZCCHC8, and DKC1(Fig. 2). Four genes that affect TERC mRNA deadenylation, TERC trafficking, and stability are PARN, NAF1, ZCCHC8, and DKC1. Fig. 2 shows how they indirectly affect telomere maintenance.
Eight telomerase and telomere maintenance genes are mutated in f-IPF. The mutated components are shown in color.
Telomerase
Telomerase inactivation is due to the silence of TERT expression (44). TERT mutations are associated with short telomere syndrome and a wide range of clinical phenotypes, such as congenital dyskeratosis, bone marrow failure, liver disease, and PF (45). Age-related PF is usually the first indicator of genetic defects in families with TERT mutations (46), with an age at onset of 60-70 years (47), and mutations in telomerase genes are the most common inherent risk factors for f-IPF (48). Families with telomerase gene mutations for f-IPF show an expected genetic predisposition (49). Germline TERT mutations lead to haploinsufficiency, which in turn leads to telomere shortening (50). These shorter telomeres are inherited over generations. This is why TERT mutations are considered pathogenic; however, it may take over 300 years before the cumulative effect of hereditary telomere shortening leads to PF (51). Ultimately, in these families, the onset of the disease is anticipated. This is followed by successive generations with increased symptoms of an earlier onset (52). This finding explains the clinical complexity of TERT mutations, which are based on randomly occurring mutations and hereditary genetic defects.
The non-coding RNA component of telomerase TERC provides a structural scaffold for the formation and action of telomerase complexes (53). It is used for the synthesis of template regions and progressive telomere repeats (54). TERC RNA is made up of 451 nucleotides, but longer transcripts can be identified by reverse transcription-polymerase chain reaction (55) and cDNA 3' end (56).
Diaz de Leon et al. assessed 134 patients with f-IPF from 21 families with PF (all with telomerase TERT mutations) and found that 18% of the patients had heterozygous TERT mutations, covering 18 mutation sites, and some patients may have several telomerase mutations (57). Telomerase gene mutations have been reported in approximately 10% of patients with s-IPF, and in approximately 25% of patients with f-IPF (48). Armanios et al. observed 73 families with f-IPF, six of whom (8%) had shorter telomeres in peripheral blood leukocytes than those observed in their peers, and TERT/TERC heterozygous mutations encoding RNA ligands of the telomerase complex in an autosomal dominant inheritance pattern (43). TERT and TERC mutations have also been validated in another study in families with f-IPF (58).
RTEL1
RTEL1 is a DNA helicase that unlocks the T-ring structure of telomeres at the end of chromosomes (59). Mutations in RTEL1 result in the cleavage of telomere ends near the T-loop by the structure-specific endonuclease subunit (SLX4), which leads to the release of telomeric repeats (T-circles). These successive DNA cycle mutations gradually generate shortened telomeres (60). RTEL1 mutants have very short telomeres, whose length is similar to those of peripheral blood mononuclear cells (PBMCs) in individuals with TERT, TERC, and DKC1 mutations (61). Damage to RTEL1 also leads to genomic instability and replication defects (62), which result in disruption of cell cycle progression. Short telomeres are present in approximately one-third of patients with familial ILD, and rare variations in RTEL1 are associated with the development of familial ILD, especially in patients with short telomeres in PBMCs (61).
PARN
PARN is a widely expressed enzyme that removes poly(A) tails by cap-dependent deadenylation of mRNAs and, in doing so, typically plays a role in regulating global mRNA levels during development (63), as well as other more specialized functions, including Dicer-independent end pruning of microRNA miR-451 (64) and deadenylation of small nucleolar RNAs (snoRNAs) (65). It contributes to the poly(A)-specific maturation and telomere length maintenance in fibroblasts and induces pluripotent stem cells (66). PARN haploinsufficiency may lead to telomere shortening through a decrease in small nucleolar RNAs of the H/ACA box or TERC RNA composition. PARN mutations cause diseases that have been related to telomere length, including f-IPF (67) and DC (68). Carriers of PARN and RTEL1 mutations have shortened leukocyte telomere lengths, and family members who did not inherit PARN or RTEL1 mutations have shorter mean telomere lengths than unrelated individuals (i.e., spouses) but longer telomere lengths than carriers of PARN or RTEL1 mutations, with epigenetic inheritance of short telomeres observed in family members (67). Together, these genes explain approximately 7% of f-IPF and highlight the link between PF and telomere dysfunction.
DKC1
The DC gene variant, X-linked gene DKC1, is another component of the telomerase complex and is also important for its stability and maintenance (69). In 1995, the first link between DKC1 and pulmonary fibrosis was found in 46 families recruited at Hammersmith Hospital which occurs in 19% of cases and is a congenital telomere disease characterized by skin abnormalities, bone marrow failure, and lung fibrosis (70). DNA analyses of two siblings who died of IPF revealed significantly shortened peripheral blood telomeres, undetectable alveolar epithelial telomeres, and a shared new A-to-G transition near the TERC-binding domain in DKC1; this domain would encode an amino acid substitution for Thr405Ala, a mutation that destabilizes TERC and impairs telomerase function (71). The X-linked DKC1 variant represents a telomere-associated gene for the genetic cause of f-IPF.
TINF2
TINF2 encodes a protein that is involved in telomere protection and maintenance (72). The shelterin complex, made up of six proteins, protects chromosome ends from DNA damage and regulates signaling and repair mechanisms as well as telomerase entry and telomere activity (73). TERF1-interacting nuclear factor 2, the protein product of TINF2, is a subunit of the shelterin complex. Mutations in TINF2 lead to telomere shortening and the impaired recruitment of tripeptidyl-peptidase 1 (TPP1)-dependent telomerase to telomeres (74). Epithelial DNA damage response activation at chromosome ends in TPP1-null mice increases telomere fusion and fragility. This confirms the role of TPP1 in telomere protection, which is required for TERT recruitment to telomeres and telomere elongation in vivo (75). Jonathan et al. identified TINF2 mutations in a family with PF by exome sequencing. They confirmed that TINF2 mutations are a rare cause of f-IPF and explained the genetic risk in approximately 1% of the reported cases (30). However, TINF2 mutations have also been reported in s-IPF (72).
ZCCHC8
ZCCHC8 is a component of the nuclear exosome targeting complex in mammalian cells (76). It drives the decay of predominantly short, unprocessed RNA with non-adenylated 3' termini (77), which is associated with short telomere disease (78). In single cells and model organisms, ZCCHC8 loss leads to the accumulation of short-extended TERC forms while sacrificing mature TERC, possibly at an early stage, before PARN deadenylation (79). A genome-wide analysis identified ZCCHC8 mutations in an adult with IPF who exhibited typical features of short telomere syndrome and a family history consistent with autosomal dominant pulmonary fibrosis; 13 affected family members were deceased.
Their TERC levels could not be measured to determine preclinical status; however, the proband's two children, who had abnormally short telomeres (shorter than those of the proband), had 50% lower TERC levels than healthy controls, and the remaining relatives had similar TERC levels to healthy controls. No ZCCHC8 variants or other short telomere syndrome phenotypes were found in 42 genetically-unidentified families that had also been screened for PF. This suggests that these mutations are rare (1 out of 43.2%). Furthermore, this result is consistent with a loci heterogeneity found in short telomere syndrome (79). ZCCHC8 is essential for TERC 3ʹ-terminal maturation and telomerase function, and a loss in heterozygosity causes f-IPF (79).
NAF1
NAF1 is a non-coding RNA that encodes an essential protein for the biogenesis of H/ACA box small nucleolus RNA. NAF1 affects telomere activity by combining with other proteins (such as TERT) to form telomerase, which plays an important role in telomeric DNA synthesis (80). Mammalian NAF1 is essential, probably because of its conserved role in rRNA modification (81). In contrast to other box H/ACA, NAF1 has its own transcriptional regulation and is not protected by an intron lariat, so TERC may be particularly susceptible (82). Naf1 +/- mice have reduced H/ACA RNA levels, and their telomerase RNA levels were only half those of the control group, but snoRNA-guided rRNA pseudouridylation still occurred (83). Thus, NAF1 deficiency selectively disrupts telomere length homeostasis by reducing telomerase RNA levels and preserving rRNA pseudouridylation. Loss of NAF1 function leads to short telomere syndrome, manifested as PF emphysema and telomere-mediated extrapulmonary disease, which is common in patients with s-IPF and f-IPF (83).
Short telomeres and PF
The association between telomere abnormality and PF is not fully understood (84). Notably, short telomeres can lead to apoptosis, senescence, and a combination of phenotypic DNA damage responses (85). Senescent cells have altered gene expression profiles and secrete a range of cytokines and growth factors (senescence-related secretory phenotypes) that may play a direct role in pathogenesis. It is hypothesized that the loss of regenerative potential of alveolar type II (AT2) epithelial cells after injury underlies telomere-associated PF, accompanied by excessive proliferation of airway cells exhibiting abnormal phenotypes (86). A study using mouse models, in which telomere repeat factor 1 was knocked out in alveolar and bronchiolar epithelial cells, has shown that the preferential activation of cellular senescence leads to inflammatory responses, upregulation of immune signaling pathways, increased mortality, lung remodeling, and spontaneous fibrosis compared with controls (87).
In the absence of telomerase mutations, s-IPF is often associated with telomere shortening; 5.71-68.2% of s-IPF patients present shorter blood leukocyte telomeres than age-matched controls, most of whom had no identifiable telomerase gene mutation, suggesting that the pathways involved in familial disease contribute to sporadic disease (88). The telomere length in PBMCs and alveolar epithelial cells is shorter in patients with f-IPF than in controls (88) and is significantly longer in the non-fibrotic region than in the fibrotic region (89) in patients with f-IPF after controlling for age and ethnicity. There is a significant difference in age at onset between the younger and older generations of patients with IPF (90), probably owing to shorter telomeres across generations. Short telomeres are disproportionately frequent in patients with s-IPF and are associated with worse survival (91).
Telomere length in peripheral blood is considered to be a marker of biological age (92); the survival of patients with IPF may be predicted by the genomic DNA telomere length. Regardless of the underlying diagnosis, faster disease progression has been observed in patients with ILD with fibrotic telomere gene mutations, suggesting that telomere length affects disease severity. Short telomeres are disproportionately present in patients with s-IPF and associated with worse survival in those with IPF (91). Observations in families with telomerase mutations suggest that emphysema is the first manifestation of telomere-associated lung disease in telomerase mutation carriers, either alone or in conjunction with PF presenting as mixed lung injury (93). No significant lung phenotypic abnormalities were found in mice with short telomeres (93). However, when mice with short telomeres were chronically exposed to cigarette smoke, they developed features of emphysema (93).
Like IPF, emphysema is associated with cumulative DNA damage in alveolar epithelial cells, with age and smoking as major risk factors (94). It follows that short telomeres represent a heterogeneous group of age-related lung diseases, of which IPF is the most common, that may be accompanied by some forms of emphysema. Patients with IPF have an annual decline in forced vital capacity of 130-210 mL, short telomeres caused by mutations in telomere-related genes, and forced vital capacity is reduced by an average of 300 mL per year (47). Short telomeres are associated with increased fibrosis, histological patterns, and mortality (95). These data support the notion that telomere function is involved in the development and progression of PF.
Short telomeres and lung transplantation
Regarding lung transplantations, lung epithelial cell renewal rates are high. This leads to accelerated telomere wear, senescent reprogramming of airway progenitor cells, and ultimately, airway remodeling, and fibrosis (96). In a lung transplant study of 20 patients with fibrotic ILD, eight patients presented f-IPF, and telomere shortening was found in five of them. Moreover, four patients with f-IPF had rare mutations in the telomere-related T cell receptor gamma locus (TRG) gene (potential risk of pathogenicity: TERT, TERC, DCK1, PARN, RTEL1, and TINF2), suggesting that telomere dysfunction is associated with adverse outcomes after transplantation. Despite that, the 1-year survival rate after transplantation was above 80%, and the procedure was beneficial to the survival of patients with fibrotic ILD, regardless of telomere dysfunction (97). Conversely, Swaminathan et al. reported a significantly increased risk of death after lung transplantation in patients carrying TERT, RTEL1, or PARN variants (98).
Telomere length can be inherited independently of rare genetic variants, and inheriting short telomeres (and disease risk) without inheriting the mutation is possible; this is called “occult hereditary disease” (57). Among the 75 asymptomatic first-degree relatives of patients with f-IPF, telomere length was shorter than in healthy controls, with 36% having telomere lengths less than the 10th percentile of age (99). One study showed that 97% of patients with IPF have shorter telomeres than age- and sex-matched controls (100), suggesting that short telomeres are not only found in patients with telomerase-related gene mutations associated with f-IPF. PF may be one of the few clinical symptoms of some inherited multisystem diseases, and f-IPF is the only manifestation of the disease in some DC families, members of which lack typical skin and mucosal characteristics (43).
A family history of short telomeres may be associated with neonatal respiratory distress, childhood ILD, aplastic anemia, and cryptogenic cirrhosis (101). Short telomeres and reduced keratinoid-associated undesirable protein expression are found in families with f-IPF (102); however, no mutations in TERT, TERC, DKC1, or other genes known to induce DC were detected (103), suggesting that the abnormal regulation of the telomerase complex is another mechanism of telomere shortening in patients with PF. However, the proportion of telomerase mutations in f-IPF was not high, indicating that other factors contributing to telomere shortening should be studied.
Mucin 5B genes
In normal lungs, the airway secretion of mucus is a defense mechanism that traps pathogens, particles, and toxic chemicals floating in the air. The mucus also assists in the removal of endogenous debris, including dying epithelial cells and leukocytes, which are expelled from the airways by cilia and cough (104). Extracellular gels made of water and mucins (highly glycosylated proteins) are the most important components of mucus. Paradoxically, although the lack of a mucus barrier makes the lungs vulnerable, excess mucus or an impaired clearance thereof is the pathogenesis of many common airway diseases, including PF (104). Excessive mucin secretion or surface fluid volume imbalance may increase the solid concentration by up to 15%, resulting in sticky and elastic mucus that is not easily removed (105). Mucin 5B (MUC5B) encodes major gel-forming mucin, which is mainly secreted by the proximal submucosal gland and distal airway, and plays a key role in mucociliary clearance (106). This protein mainly affects the rheological properties of airway mucus and participates in airway defense.
Expression of MUC5B in IPF
IPF has long been thought to involve mainly the alveolar region (107). However, abnormal distal airways may also contribute to IPF pathogenesis. In addition to consistent abnormal physiological changes in the distal airways, they have been implicated in ILD (108). A recent histological study showed increased bronchiolar and peribronchiolar inflammation, fibrosis, and thickened bronchial walls in patients with IPF compared with controls (109). The ERBB-YAP axis is considered a driver of distal airway epithelial cell obstruction. This signaling pathway dynamically interacts with MUC5B to increase the degree of distal airway obstruction and is specific to IPF. Distal airway epithelial dysfunction is sufficient to drive primary lung fibroblast activation (108). Visually, the characteristic changes of fibrotic lesions can be seen as subpleural honeycomb-like cysts and predominantly reticular infiltrations in the lower region, as well as a microscopic honeycomb, which represents the airway expanded by the traction of the fibrotic process. They are filled with MUC5B protein and chronic inflammatory cells (110).
MUC5B is co-expressed in respiratory bronchiolar airway epithelia (111) and type 2 alveolar epithelial cells (112). In mice, the MUC5B concentration in bronchoalveolar epithelial cells is directly related to the degree and persistence of bleomycin-induced PF and mortality (113). In normal mouse airways that resemble the distal human airway, MUC5B is generated in surface secretory cells in the airways (114). This suggests that it mediates baseline barrier and clearance functions in mice, and that it might have the same role in the human distal airways (115). MUC5B expression was increased in tissue sections of distal airways from subjects with PF (116). These data suggest that the distal airways are involved in the pathogenesis of IPF.
MUC5B promoter single nucleotide polymorphism and IPF
The gain-of-function promoter variant, rs35705950, is 3 kb upstream of the MUC5B transcriptional start site and is the strongest risk factor for the development of f-IPF (117). The conversion from G to T in the 5ʹ region interferes with the binding site of DNA enzymes to transcription factors and upregulates the expression of MUC5B (118). The downstream 32 bp of rs35705950 is a highly conserved forehead box A2 (FOXA2) binding motif, which is differentially methylated in IPF (119), resulting in increased MUC5B expression and rs35705950 being considered a risk allele. This hypermethylation may lead to increased occupancy of FOXA2 in the binding motif, resulting in increased expression of MUC5B (120). The rs35705950 variant enhances MUC5B transcription. The resulting post-transcriptional processing can produce a pro-fibrotic response, induce endoplasmic reticulum (ER) stress, and activate the unfolded protein response (UPR) (121). The UPR attempts to restore normal protein folding through three reactions: IRE1/XBP1, PERK/ATF4, and BiP/ATF6 (122).
When protein homeostasis is abnormal, UP accumulates in the ER, leading to increased ER pressure, which results in apoptosis. ER stress has been observed in patients with IPF (123). In experimental models of PF, the activation of the UPR in alveolar epithelial cells leads to an epithelial-mesenchymal transition (124) and fibroproliferative pulmonary disease (125). ER stress and apoptosis interfere with normal epithelial responses to injury and repair, which are hallmarks of IPF (126). Whole-genome sequencing analysis has identified an SNP in the promoter region of the MUC5B gene, i.e., the rs35705950 variant. It poses a major risk factor for developing IPF, accounting for 30-35% of the risk (127).
Plantier et al. studied the correlation between morphology and transcription factors in the signal transduction system in IPF and found that the uncontrolled expression of MUC5B is involved in PF formation (110). Both the homozygous mutation (T/T) and heterozygous mutation (G/T) of the MUC5B rs35705950 variant significantly increase the risk of developing f-IPF and s-IPF. By scanning the entire gene sequence of the chromosome 11 p-terminal, 19 SNPs were found to be associated with f-IPF. Finally, rs35705950 located in the promoter region of MUC5B was confirmed to be the most strongly associated with familial lung stromal disease and IPF, present in 34% of familial lung stromal disease, 38% of IPF, and only 9% of normal controls, and was considered a risk allele for both diseases (117). The 4-kb promoter region gene of MUC5B has three CpG islands and other regulatory sites, suggesting that the expression of MUC5B is affected by both genetic and non-genetic factors.
In 2011, two laboratories simultaneously reported that the MUC5B promoter variant rs35705950 is the strongest risk factor for the development of f-IPF and s-IPF (128) and appears to be similarly prevalent in patients with f-IPF and s-IPF (117) (approximately 50-60% of individuals with FIP or IPF). This factor increases the risk of heterozygosity by six-fold and that of homozygosity by 20-fold, suggesting that both forms of the disease share common genetic variations. IPF is caused by excessive and continuous lung injury or repair abnormalities (122).
In addition, common exposure and basic biological processes can affect MUC5B expression (Table 1).
Regulation of MUC5B expression in airway epithelial cells.
| Species | Effector | Pathway | Transcriptional factors | Secretion | References |
|---|---|---|---|---|---|
| Bacteria spp | LPS/staphylococcus enterotoxin | MAPKs | CREB/AP1/SP1/ NF-κB | + | (129-137) |
| Cytokines | IL-1 β | NF-κB | + | (132, 138, 139) | |
| IL-17A | NF-κB | + | (132, 139, 140) | ||
| IL-6 | MAPKs | CREB/AP1/SP1/ NF-κB | + | (131-135, 140, 141) | |
| STAT3 | FOX | Decrease | (119, 142, 143) | ||
| IL-13 | STAT6 | FOXA2 | Decrease | (119, 144, 145) | |
| Lipid Mediator | PGD2 | MAPKs | CREB/AP1/SP1/ NF-κB | + | (119, 131-135, 146) |
| Oxidation factors | ROS | MAPKs | NF-κB | + | (147) |
Note: Different stimuli in airway epithelial cells can regulate MUC5B expression, including certain bacterial components, some interleukins, oxidative factors, lipid mediators, which may induce MUC5B overexpression through MAPK, STAT3, or STAT6 pathways. Some important transcription factors are also directly or indirectly involved in MUC5B overexpression.
Pathological role of MUC5B in IPF
In addition to the association between the SNP of the MUC5B promoter and IPF, its protein may play a direct role in the pathogenesis of IPF (117). Persistent bronchiolar epithelial injury and the overproduction of MUC5B by airway progenitor cells lead to honeycomb cysts and IPF (116).
First, excessive expression of MUC5B can impair mucosal host defense and leads to excessive lung injury caused by inhaled substances. Over time, a reduced clearance rate may lead to the formation of scar tissue, which replaces normal lung tissue. It also results in persistent fiber proliferation, which in turn develops into IPF (117). Exposure to cigarette smoke increases MUC5B expression in the lung through the dysregulation of classical signaling pathways (e.g., TGF-β and Wnt), resulting in a fibroblast-like phenotype in alveolar epithelial cells, as demonstrated in mouse (MLE-12) and rat (RLE-6TN) epithelial cells (148). Additionally, several clinical and epidemiological studies have shown that f-IPF and s-IPF are more common in smokers (148). In IPF model mice infected with H1N1, distal airway stem cells proliferate and express keratin 5-positive (Krt5+), ablates the intrabronchial region, and assemble into alveolar structures (149). Persistent notch signaling after H1N1 injury leads to failure of regeneration and formation of cellular cysts. KRT5+ cells are present in the fibrotic stroma of IPF and are closely associated with MUC5B-rich cellular cysts (150).
Alveolar stem cells are mainly derived from the distal airway epithelium (151), and MUC5B and dry/progenitor cells participate in the fiber proliferation response. When stem cells attempt to regenerate damaged bronchioles and alveolar epithelium, MUC5B overexpression interferes with the interaction between AT2 cells and the underlying matrix, inhibiting alveolar basement membrane re-epithelization. This may exacerbate the ongoing alveolar collapse and fibrosis of adjacent bronchoalveolar units, resulting in chronic fiber proliferation and regeneration process disorders and cellular cyst formation (151). These structures inhibit normal alveolar homeostasis and promote fibroproliferative activity in persistent foci.
Second, under normal conditions, mucociliary clearance depends on effective ciliary movement, adequate hydration of the fluid layer around the cilia, and complete cough (152). The overexpression and accumulation of MUC5B prevent efficient mucus hydration and ciliary function (111) and cause excessive retention of inhalants. Endogenous inflammatory fragments at the bronchoalveolar junction are associated with changes in the osmotic gradient, water moving out of the periciliary layer and into the airway lumen (153), and a reduction in mucociliary clearance. They may also physically affect cilial function (150), enhance inhalation retention. Finally, when sticky protein expression relies on the cystic fibrosis transmembrane conductance regulator anion secretion separation, abnormally thick mucus may be beyond the influence of airway dehydration, which can lead to abnormal host defense (154). The common defects of mucous membranes of inhaled particles may exaggerate the host defense. Facilitate recurrent injury/repair/regeneration mechanism disruption (106), and lead to microscopic scarring and progressive fibrous proliferation in the lungs, lung structural destruction, and IPF development over time (3).
Third, IPF is characterized by the co-overexpression of MUC5B and cilium-associated genes (155), which are expressed and operate in ciliated airway epithelial cells of the distal airways and are related to microscopic cellular structures. Matrix metalloproteinase 7 (MMP7) is a WNT/β-catenin target gene encoding for a metalloproteinase, which is overexpressed in IPF proliferative epithelial cells. Based on the discovery that MMP7-deficient mice are protected from bleomycin-induced PF, it is speculated that MMP7 plays a role in promoting fibrosis (156). Plasma MMP7 concentration in IPF was correlated with two single nucleotide polymorphisms in gene promoter regions, suggesting a potential genetic basis for MMP7 upregulation.
A known biomarker of IPF can indicate the presence, severity, and prognosis of the disease (157). Plasma MMP7 has been proposed as a univariate predictor of early interstitial lung disease in first-degree relatives of patients with familial interstitial pneumonia (158). Ciliary MUC5B gene expression is associated with MMP7 concentration and attenuation of ciliary cell differentiation in the airways (159). However, MUC5B promoter mutations cause mucus accumulation in the distal region of the lung and impair mucociliary function. Abnormal repair processes after distal airway injury and failure of terminal airway regeneration are activated by cellular cysts.
All three mechanisms are reasonable and can act alone or together to cause IIP associated with the MUC5B promoter SNP (Fig. 3).
Possible influencing factors of MUC5B expression and its role in airways.
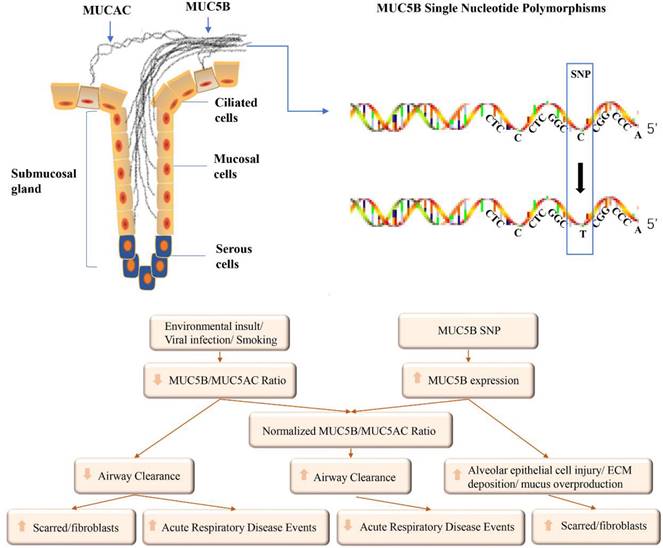
MUC5B is mainly produced by submucosal glands and mucous cells on the surface of the airway. SNP were found in the promoter region of MUC5B. The abnormal expression of MUC5B can be induced by environmental exposure and MUC5B SNP. The mucus in the airways can be adjusted by the ability to clear, eventually leading to acute events or pulmonary fibrosis.
MUC5B and TOLLIP reside at the same genetic locus. If IPF is also associated with the TOLLIP promoter SNP, the risk of death is higher (160). The independence of the association signals at 11p15.5 suggests that multiple variants of this locus may be influencing disease susceptibility and course (127). However, this does not prove a relationship between the two genes.
In a meta-analysis of 28 studies, the overall IIP and FIP risk of the TT genotype and the T allele of rs35705950 was significantly increased across all genetic models. These results suggest that MUC5B rs35705950 may be a predictor for IIP and FIP susceptibility (161). Preclinical PF (PrePF) is frequently reported in smokers and patients with f-IPF (162), and the polymorphism of the MUC5B promoter can be used as an indicator of IPF susceptibility for identifying individuals with PrePF (163) and predicting the radiological progression of PrePF, as well as the prognosis of IPF (164). Participants affected by the IPF variant rs35705950 showed better survival than those who were not (26).
For early asymptomatic f-IPF, the increased MUC5B levels can appear years before the symptoms of PF, and it can be detected in patients with mild PF (165); the variant can serve as an effective new molecular target for intervention during the early stages of IPF. The MUC5B promoter variant rs35705950 is predictive of PrePF; however, rs35705950 is present in approximately 19% of the population (117), and IPF rarely occurs (& LT, 0.1%) (166). Therefore, the observed differential expression of MUC5B in cases and controls cannot be fully explained (117), and additional biomarkers are needed to identify individuals with PrePF in high-risk populations.
Surfactant-related genes
Dysfunction and repair of alveolar epithelial cells are considered important components of the pathogenesis of IPF and childhood ILD (167). Nogee et al. first reported a case of nonspecific interstitial pneumonia with exon 4 and amino acid deletion at position 37, which was caused by mutations in the carboxy-terminal region of the gene encoding the surfactant protein of alveolar epithelial cells; the mother of the patient had desquamative interstitial pneumonia (168). F-IPF may also be present in children, accounting for a larger proportion of children with ILD (169).
The central role of AT2 cells in IPF is to generate pulmonary surfactant (170), and AT2 cells self-renew as progenitor cells and transdifferentiate into AT1 cells to maintain alveolar stability (171). Rare mutations in the components of the surfactant system, including surfactant-associated protein C (SP-C), SP-A, and ABCA3 lipid transporters, provide new clues for IPF caused by AT2 cell dysfunction (Fig. 4). More than 60 rare mutations in the SP-C gene (SFTPC) have been identified in affected children and adults combined (168), two mutations in the SP-A2 gene (SFTPA2), although present in less than 5% of s-IPF, and 150 mutations in ABCA3 (172). Protein aggregation, ER stress, proinflammatory/profibrotic cytokine processing, altered macroautophagy, and apoptosis occur in model systems with SFTPC, SFTPA, and ABCA3 mutations. In humans, they occur in lung and AT2 epithelial cells of patients with f-IPF and s-IPF alike (173).
Core mechanisms and candidate molecular biomarkers of idiopathic pulmonary fibrosis.
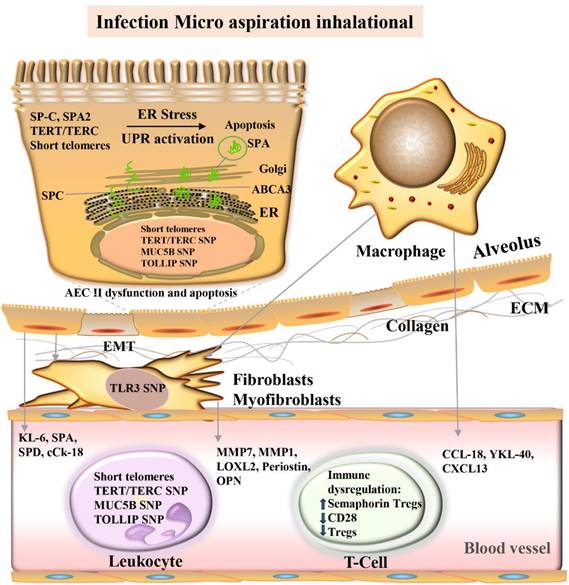
Pictured are the alveolar lumen (top), epithelial cell layer (tan), interstitial space (white), and capillaries (bottom, pink). Damage to alveolar epithelial cells by multiple stimuli (blue boxes) leads to epithelial dysfunction, fibrosis/extracellular matrix (ECM) deposition, and immune dysregulation. Proposed molecular biomarkers for these processes include blood proteins, genomic markers, and blood cells (see light purple/light green boxes). ABCA3, ATP-binding cassette class A3 lipid transporter; AEC, Alveolar epithelial cells; cCK18, cleaved cytokeratin 18; CCL-18, CC chemokine ligand-18; CXCL, chemokine ligand; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; ER, endoplasmic reticulum; MMP, Matrix metalloproteinase; KL-6, Krebs von den Lungen-6; LOXL2, Lysyl oxidase-like 2; MUC5B, mucin 5B; SNP Single Nucleotide Polymorphism; OPN, osteopontin; SPA, surfactant protein A; SPA2, surfactant protein A2; SPC, surfactant protein C; SPD, surfactant protein D; TERT telomere RNA component; TERC, telomere reverse transcriptase; TLR3, Toll-like receptor 3; TOLLIP, Toll-interacting protein; Treg, regulatory T cells; UPR, unfolded protein response.
Rare mutations of surfactant system components (SP-C, SP-A, and ABCA3) cause stress and activate the unfolded protein response, AT2 dysfunction, and apoptosis, thereby promoting epithelial-mesenchymal transformation and the development of PF. ER, endoplasmic reticulum; TERT, telomere reverse transcriptase; TERC, telomere RNA component; MUC5B, Mucin 5B; SNP, single nucleotide polymorphism; TOLLIP, toll-interacting protein.
SP-C
SP-C is a highly hydrophobic protein present in lung surfactants and is produced by ER integration of membrane protein precursors in AT2. The gene encoding SFTPC is located on chromosome 8P and organized into six exons (encoded from I to V, with VI untranslated) and five introns, which produce an mRNA that encodes a 21-kDa proprotein (pro-SP-C 21) at either 191 or 197 amino acids. The pro-SP-C 21 COOH terminal, known as the BRICHOS domain (residue 90-197), is the carboxy-terminal region of the pro-SP-C proprotein. Most SFTPC mutations are located in the intrapeptide disulfide bond within the pro-SP-C COOH terminal (BRICHOS) domain (174). SFTPC BRICHOS mutants are retained by the ER, inhibit the proteasome, and activate the UPR (175), a set of protective biochemical pathways aimed at matching the protein capacity of the ER. The resulting cytokine processing, apoptosis, and functional disruption of progenitor cells lead to PF under AT2 and AEC lumen ER stress (176).
Although SFTPC mutations occur in less than 5% of patients with sporadic IPF, these rare mutants enhance our understanding of AT2 cell and AEC cavity biology in PF. Lawson et al. demonstrated evidence of UPR activation markers (including heavy-chain- binding protein (BiP), ER degradation enhancing α-mannosidase-like proteins (EDEM), and XBP1) in patients with f-IPF expressing the L188Q SFTPC mutation and samples from non-SFTPC f-IPF and s-IPF cases (177). Korfei et al. extended these findings by reporting molecular signatures of multiple UPR pathways and apoptosis in AT2 cells from patients with s-IPF and UIP pathologic features (178). These included activating transcription factor 6 (ATF6), ATF4, XBP1, C/EBP-homologous protein (CHOP), and caspase 3.
Chronic ER stress in the alveolar epithelium may represent a broad mechanism of ILD pathogenesis (125). In a transgenic mouse model, the diphtheria toxin receptor was expressed exclusively in AT2 cells, using the SFTPC promoter. The administration of the toxin induced AT2 cell death and increased lung collagen deposition, leading to spontaneous PF (179).
The missense substitution G.1286T>C, which replaces isoleucine at position 73 in the SP-C preprotein (pre-SP-C I73T) with threonine, is the most common SFTPC mutation associated with ILD (180). The deposition of pre-SP-C I73T in the plasma membrane and subsequent accumulation of misprocessed allotypes result in a dysfunctional cellular phenotype characterized by late macrophage arrest, impaired mitophagy, and defective cellular protein homeostasis (181). In the bleomycin-induced mouse model of PF, immunohistochemistry of pre-SP-C I73T confirmed that the proliferative cells in the fibrotic and tissue deformable areas are type II AEC, similar to human UIP, and that the fibrotic areas in the AEC cavity are positive for the expression of pre-SP-C (182).
Furthermore, the expression of mutant SFTPC alveolar epithelial cells of mice drives the outbreak of a spontaneous FP. This demonstrates that AT2 cells can promote the creation of characteristic endophenotypes of IPF/UIP, including abnormal tissue remodeling to collagen deposition, AT2 cell proliferation, α-smooth muscle actin-positive cells, and limiting lung physiology (183). SP-C null mice are motile at birth and grow normally in the absence of a pulmonary phenotype (184); however, they show a higher sensitivity to bleomycin-induced fibrosis (185) and respiratory syncytial virus infection (186). When the original SP-C-null-mouse strain was backcrossed to a different genetic background, the resulting loss of SP-C was associated with spontaneous inflammation and lung remodeling (187), suggesting that SP-C deficiency is a disease regulator.
SP-A
SP-A, the most abundant surfactant-associated protein, is an intra-luminal multifunctional sialic acid glycoprotein. It has a molecular mass of 28-36 kDa and contains a COOH-terminal C-type lectin motif, triple helix collagen domain, and carbohydrate recognition domain (188). Mutations in SFTPA, encoding one of the two isomers of surfactant proteins, are associated with the heritability of f-IPF (mutations SFTPA1 and SFTPA2, 4.5 kb long, each, and located on chromosome 10) (189). In vivo, the expression of disease-associated SFTPA1 mutations leads to AT2 UPR activation, necrotizing apoptosis, and IPF, which are associated with ER stress-induced c-Jun N-terminal kinase (JNK) signaling (190).
Takezaki et al. detected a homozygous mutation of the SFTPA1 locus in two Japanese patients with f-IPF (190). These patients exhibited a significant decrease in downstream SP-A secretion and an increased susceptibility to the influenza virus, which accelerated the progression of IPF. The overexpression of receptor interacting serine/threonine kinase 3 (RIPK3) and mixed lineage kinase-like (MLKL) was detected in AT2 of SFTPA1 homozygous knockout mice, but both RIPK3 and MLKL could significantly improve the progression of IPF, suggesting that the SFTPA1 homozygous mutant reduced SP-A production.
Furthermore, we found that the loss of SFTPA1 leads to ER stress, which results in the overexpression of inositol requiring enzyme 1α (IRE1α) and increases JNK phosphorylation, thereby upregulating RIPK3 expression. The inhibition of IRE1α and JNK significantly inhibited RIPK3 expression and prevented the progression of PF. In contrast, these processes can be reversed by RIPK3 overexpression; this finding supports the idea that family genetic history can increase the susceptibility of IPF to external stimuli, aggravate the damage caused by the fibrotic process of AT2, and mediate the aggravation of IPF. Thus, RIPK3 may be an important target to treat IPF.
ABCA3
Clinical mutations in ABCA3, encoding for an ATP-dependent transporter that converts phosphatidylcholine and cholesterol into lysosome-associated organelles and is critical for lamellar formation, link lamellar body dysfunction to fibrotic lung disease. Human ABCA3 has been mapped to chromosome 16p13.3 and encodes a protein of 1,704 amino acids (191). Although ABCA3 mRNA has been detected in many tissues, its mRNA is highly expressed in AT2 cells (192). Homozygous ABCA3 mutations lead to lamellar body deletion and neonatal respiratory failure in both humans and mice (193), indicating its importance in the production of pulmonary surfactants. Heterozygous ABCA3 mutations exist with partial loss of function owing to ER retention (type I), improper functioning of the lipid pump (type II), or both (type I/II compound heterozygotes). These mutants act as genetic modifiers of pulmonary disease associated with SFTPC mutations in children (194).
The heterozygous variants, I73T SFTPC and D123N ABCA, increase disease penetrance, suggesting an interaction between these genotypes; however, the molecular mechanism between them remains unclear (195). In addition to the loss of function, several clinical ABCA3 mutations are associated with PF, and cell phenotypes derived from ABCA3 mutations are found in isotypes, ER retention, and misallocation homotypes based on protein behavior (167). However, the mechanistic links among ABCA3 mutants, changes in cell quality control, and fibrosis require further investigation.
Conclusions
F-IPF is rarely identified in patients before disease progression, which limits our ability to study mechanisms. Identifying diagnostic and prognostic biomarkers and mechanisms helps to understand the pathogenesis of early f-IPF. Recent studies have increased the understanding of the underlying genetic susceptibility to PF and the pathogenesis of this disease. Genomic factors can also influence the development of f-IPF and the observed heterogeneity in pathogenicity. Genetically susceptible broncho-alveoli may sustain disproportionate damage at different time points owing to different environmental exposures and can eventually manifest as PF.
A positive gene diagnosis may be significant for the early detection, prognosis, and risk assessment of close relatives, and genetic testing may be beneficial for patients with high FIP and positive gene probability (101). If these changes predict disease progression, they will provide the strongest evidence to date that there is a long but identifiable pre-symptomatic period during which targeted therapies can prevent the progression of PF. There are no standardized guidelines for the timing of genetic tests for patients with IPF (3); however, experts generally agree that individualized genetic testing should be performed case by case.
The relationship between the mechanisms of rare and common genetic variations in FIP is not fully understood; they are thought to act synergistically to modulate disease-related phenotypes, such as telomere shortening. In addition, when considering lung transplantation, a positive genetic diagnosis in patients with f-IPF helps predict disease course and assess the risks. Further research should be aimed at revealing the underlying and pathogenic genetic factors and epigenetic changes in patients with f-IPF to improve the usefulness of precision therapies.
Abbreviations
ABCA: adenosine triphosphate binding box subfamily A3
AT2: alveolar type II
ATF: activating transcription factor 6
BRICHO: bond within the pro-SP-C COOH terminal
CHO: C/EBP-homologous protein
DC: dyskeratosis congenita
DDR: DNA damage response
DKC1: dyskeratosis congenita 1
EDEM: ER degradation-enhancing alpha-mannosidase-like proteins
ER: endoplasmic reticulum
f-IPF: familial idiopathic pulmonary fibrosis
ILD: interstitial lung disease
IIP: idiopathic interstitial pneumonia
IRE1α: inositol requiring enzyme 1α
JNK: c-Jun N-terminal kinase
MLKL: mixed lineage kinase-like
MMP7: matrix metalloproteinase 7
MUC5B: mucin 5B
NAF1: nuclear assembly factor 1
PARN: poly(A)-specific ribonuclease
PBMCs: peripheral blood mononuclear cells
PrePF: preclinical PF
RIPK3: receptor interacting serine/threonine kinase 3
RTEL1: regulator of telomere length 1
snoRNAs: small nucleolar RNAs
SNP: single nucleotide polymorphism
SFTPA2: surface-active protein A2
SFTPC: surface-active protein C
s-IPF: sporadic IPF
SP-A: surfactant protein A
SP-C: surfactant-associated protein C
TERC: telomerase RNA component
TERT: telomerase reverse transcriptase
TINF2: telomere repeat binding factor 1-interacting nuclear factor 2
TPP1: tripeptidyl-peptidase 1
TRG: T cell receptor gamma locus
UIP: usual interstitial pneumonia
UPR: unfolded protein response
ZCCHC8: zinc finger CCHC-type containing 8
Acknowledgements
We thank Editage Ltd. for the editorial work.
Funding
This study was funded by the National Key Technologies Research and Development Program (Grant No. 2021YFC2500700).
Author Contributions
D.Y.D.: Writing—original draft preparation, funding acquisition; J.L.Y.: Writing—review and editing; R.G., Q.F.X., R.M.L.: Figure preparation and proofreading. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Alfaro TM, Moor CC, Alfieri V, Jeny F, Kreuter M, Wijsenbeek MS. et al. Research highlights from the 2018 ERS International Congress: interstitial lung diseases. ERJ Open Res. 2019 5(1)
2. Fernandez BA, Fox G, Bhatia R, Sala E, Noble B, Denic N. et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res. 2012;13(1):64
3. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ. et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68
4. Wells AU, Kouranos V. An IPF-like disease course in disorders other than IPF: how can this be anticipated, recognized, and managed? Expert Rev Clin Immunol. 2021;17(10):1091-101
5. Saito S, Alkhatib A, Kolls JK, Kondoh Y, Lasky JA. Pharmacotherapy and adjunctive treatment for idiopathic pulmonary fibrosis (IPF). J Thorac Dis. 2019;11(Suppl 14):S1740-s54
6. Glass DS, Grossfeld D, Renna HA, Agarwala P, Spiegler P, DeLeon J. et al. Idiopathic pulmonary fibrosis: Current and future treatment. Clin Respir J. 2022;16(2):84-96
7. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378(19):1811-23
8. Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB. et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59(11):977-80
9. Cutting CC, Bowman WS, Dao N, Pugashetti JV, Garcia CK, Oldham JM. et al. Family History of Pulmonary Fibrosis Predicts Worse Survival in Patients With Interstitial Lung Disease. Chest. 2021;159(5):1913-21
10. Spagnolo P, Cottin V. Genetics of idiopathic pulmonary fibrosis: from mechanistic pathways to personalised medicine. J Med Genet. 2017;54(2):93-9
11. Javaheri S, Lederer DH, Pella JA, Mark GJ, Levine BW. Idiopathic pulmonary fibrosis in monozygotic twins. The importance of genetic predisposition. Chest. 1980;78(4):591-4
12. Lee HL, Ryu JH, Wittmer MH, Hartman TE, Lymp JF, Tazelaar HD. et al. Familial idiopathic pulmonary fibrosis: clinical features and outcome. Chest. 2005;127(6):2034-41
13. Swaye P, Van Ordstrand HS, McCormack LJ, Wolpaw SE. Familial Hamman-Rich syndrome. Report of eight cases. Dis Chest. 1969;55(1):7-12
14. Kropski JA. Familial Interstitial Lung Disease. Semin Respir Crit Care Med. 2020;41(2):229-37
15. Grant-Orser AG-O, Avitzur N, Morisset J, Fell CD, Johannson KA. Perceptions of Genetic Testing: A Mixed-methods Study of Patients with Pulmonary Fibrosis and their First-degree Relatives. Ann Am Thorac Soc. 2022
16. Wang P, Xu Z. Pulmonary fibrosis in dyskeratosis congenita: a case report with a PRISMA-compliant systematic review. BMC Pulm Med. 2021;21(1):279
17. Wakamatsu I, Yatomi M, Uno S, Oishi Y, Ikeuchi H, Hanazato C. et al. A case of a patient with neurofibromatosis type I who developed pneumothorax and eosinophilic pleural effusion after suffering from COVID-19 pneumonia. Radiol Case Rep. 2021;16(11):3504-8
18. Opoka L, Wyrostkiewicz D, Radwan-Rohrenschef P, Roży A, Tylki-Szymańska A, Tomkowski W. et al. Combined Emphysema and Interstitial Lung Disease as a Rare Presentation of Pulmonary Involvement in a Patient with Chronic Visceral Acid Sphingomyelinase Deficiency (Niemann-Pick Disease Type B). Am J Case Rep. 2020;21:e923394
19. Thomas H, Costabel U. [Progressive course of idiopathic pulmonary fibrosis in 2 monozygotic twin sisters]. Pneumologie. 1996;50(9):679-82
20. Kaur A, Mathai SK, Schwartz DA. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front Med (Lausanne). 2017;4:154
21. Borie R, Kannengiesser C, Nathan N, Tabèze L, Pradère P, Crestani B. Familial pulmonary fibrosis. Rev Mal Respir. 2015;32(4):413-34
22. Krauss E, Gehrken G, Drakopanagiotakis F, Tello S, Dartsch RC, Maurer O. et al. Clinical characteristics of patients with familial idiopathic pulmonary fibrosis (f-IPF). BMC Pulm Med. 2019;19(1):130 -
23. Kropski JA, Blackwell TS, Loyd JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(6):1717-27
24. Al-Mutairy EA, Imtiaz FA, Khalid M, Al Qattan S, Saleh S, Mahmoud LM. et al. An atypical pulmonary fibrosis is associated with co-inheritance of mutations in the calcium binding protein genes S100A3 and S100A13. The European respiratory journal. 2019;54(1):1802041
25. Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP. et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613-20
26. Biondini D, Cocconcelli E, Bernardinello N, Lorenzoni G, Rigobello C, Lococo S. et al. Prognostic role of MUC5B rs35705950 genotype in patients with idiopathic pulmonary fibrosis (IPF) on antifibrotic treatment. Respiratory research. 2021;22(1):98 -
27. Guzmán-Vargas J, Ambrocio-Ortiz E, Pérez-Rubio G, Ponce-Gallegos MA, Hernández-Zenteno RdJ, Mejía M. et al. Differential Genomic Profile in TERT, DSP, and FAM13A Between COPD Patients With Emphysema, IPF, and CPFE Syndrome. Frontiers in medicine. 2021;8:725144 -
28. Stock CJW, Renzoni EA. Telomeres in Interstitial Lung Disease. J Clin Med. 2021 10(7)
29. Gaysinskaya V, Stanley SE, Adam S, Armanios M. Synonymous Mutation in DKC1 Causes Telomerase RNA Insufficiency Manifesting as Familial Pulmonary Fibrosis. Chest. 2020;158(6):2449-57
30. Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. 2015;147(5):1361-8
31. Philippot Q, Kannengiesser C, Debray MP, Gauvain C, Ba I, Vieri M. et al. Interstitial lung diseases associated with mutations of poly(A)-specific ribonuclease: A multicentre retrospective study. Respirology. 2022;27(3):226-35
32. Gooptu B. Surfactant protein C mutations and familial pulmonary fibrosis: stuck in a loop on the scenic route. Eur Respir J. 2022 59(1)
33. Liu L, Qin J, Guo T, Chen P, Ouyang R, Peng H. et al. Identification and functional characterization of a novel surfactant protein A2 mutation (p.N207Y) in a Chinese family with idiopathic pulmonary fibrosis. Mol Genet Genomic Med. 2020;8(9):e1393
34. Manali ED, Kannengiesser C, Borie R, Ba I, Bouros D, Markopoulou A. et al. Genotype-Phenotype Relationships in Inheritable Idiopathic Pulmonary Fibrosis: A Greek National Cohort Study. Respiration. 2022:1-13
35. Stock CJW, Renzoni EA. Telomeres in Interstitial Lung Disease. Journal of clinical medicine. 2021;10(7):1384
36. Turner KJ, Vasu V, Griffin DK. Telomere Biology and Human Phenotype. Cells. 2019;8(1):73
37. Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276(5312):561-7
38. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB. et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349-52
39. Blasco MA, Rizen M, Greider CW, Hanahan D. Differential regulation of telomerase activity and telomerase RNA during multi-stage tumorigenesis. Nat Genet. 1996;12(2):200-4
40. van Batenburg AA, Kazemier KM, van Oosterhout MFM, van der Vis JJ, Grutters JC, Goldschmeding R. et al. Telomere shortening and DNA damage in culprit cells of different types of progressive fibrosing interstitial lung disease. ERJ Open Res. 2021 7(2)
41. Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350(6265):1193-8
42. Kannengiesser C, Borie R, Ménard C, Réocreux M, Nitschké P, Gazal S. et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J. 2015;46(2):474-85
43. Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317-26
44. Ulaner GA, Hu JF, Vu TH, Giudice LC, Hoffman AR. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998;58(18):4168-72
45. Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA. et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 2005;102(44):15960-4
46. Erratum. Parry EM, Alder JK, Qi X, Chen JJ-L, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117(21):5607-5611. Blood. 2016;127(14):1837
47. Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C. et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710-20
48. Hoffman TW, van Moorsel CHM, Borie R, Crestani B. Pulmonary phenotypes associated with genetic variation in telomere-related genes. Current Opinion in Pulmonary Medicine. 2018 24(3)
49. Dai J, Cai H, Zhuang Y, Wu Y, Min H, Li J. et al. Telomerase gene mutations and telomere length shortening in patients with idiopathic pulmonary fibrosis in a Chinese population. Respirology. 2015;20(1):122-8
50. Strong MA, Vidal-Cardenas SL, Karim B, Yu H, Guo N, Greider CW. Phenotypes in mTERT⁺/⁻ and mTERT⁻/⁻ mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol Cell Biol. 2011;31(12):2369-79
51. van der Vis JJ, van der Smagt JJ, Hennekam FAM, Grutters JC, van Moorsel CHM. Pulmonary Fibrosis and a TERT Founder Mutation With a Latency Period of 300 Years. Chest. 2020;158(2):612-9
52. Alder JK, Cogan JD, Brown AF, Anderson CJ, Lawson WE, Lansdorp PM. et al. Ancestral mutation in telomerase causes defects in repeat addition processivity and manifests as familial pulmonary fibrosis. PLoS Genet. 2011;7(3):e1001352
53. Zappulla DC, Cech TR. Yeast telomerase RNA: a flexible scaffold for protein subunits. Proc Natl Acad Sci U S A. 2004;101(27):10024-9
54. Blackburn EH, Collins K. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol. 2011 3(5)
55. Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP. et al. The RNA component of human telomerase. Science. 1995;269(5228):1236-41
56. Tseng CK, Wang HF, Burns AM, Schroeder MR, Gaspari M, Baumann P. Human Telomerase RNA Processing and Quality Control. Cell Rep. 2015;13(10):2232-43
57. Diaz de Leon A, Cronkhite JT, Katzenstein AL, Godwin JD, Raghu G, Glazer CS. et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 2010;5(5):e10680
58. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC. et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552-7
59. Cogan JD, Kropski JA, Zhao M, Mitchell DB, Rives L, Markin C. et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015;191(6):646-55
60. Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149(4):795-806
61. Vannier JB, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends Cell Biol. 2014;24(7):416-25
62. Petrovski S, Todd JL, Durheim MT, Wang Q, Chien JW, Kelly FL. et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. American journal of respiratory and critical care medicine. 2017;196(1):82-93
63. Dehlin E, Wormington M, Körner CG, Wahle E. Cap-dependent deadenylation of mRNA. Embo j. 2000;19(5):1079-86
64. Yoda M, Cifuentes D, Izumi N, Sakaguchi Y, Suzuki T, Giraldez AJ. et al. Poly(A)-specific ribonuclease mediates 3'-end trimming of Argonaute2-cleaved precursor microRNAs. Cell Rep. 2013;5(3):715-26
65. Berndt H, Harnisch C, Rammelt C, Stöhr N, Zirkel A, Dohm JC. et al. Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. Rna. 2012;18(5):958-72
66. Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P. et al. Poly(A)-specific ribonuclease (PARN) mediates 3'-end maturation of the telomerase RNA component. Nat Genet. 2015;47(12):1482-8
67. Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R. et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512-7
68. Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S. et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125(5):2151-60
69. Vulliamy TJ, Dokal I. Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie. 2008;90(1):122-30
70. Knight S, Vulliamy T, Copplestone A, Gluckman E, Mason P, Dokal I. Dyskeratosis Congenita (DC) Registry: identification of new features of DC. Br J Haematol. 1998;103(4):990-6
71. Kropski JA, Mitchell DB, Markin C, Polosukhin VV, Choi L, Johnson JE. et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest. 2014;146(1):e1-e7
72. Hoffman TW, van der Vis JJ, van Oosterhout MF, van Es HW, van Kessel DA, Grutters JC. et al. TINF2 Gene Mutation in a Patient with Pulmonary Fibrosis. Case Rep Pulmonol. 2016;2016:1310862
73. Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM. et al. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol. 2010;30(12):2971-82
74. Frank AK, Tran DC, Qu RW, Stohr BA, Segal DJ, Xu L. The Shelterin TIN2 Subunit Mediates Recruitment of Telomerase to Telomeres. PLoS Genet. 2015;11(7):e1005410
75. Tejera AM, Stagno d'Alcontres M, Thanasoula M, Marion RM, Martinez P, Liao C. et al. TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev Cell. 2010;18(5):775-89
76. Puno MR, Lima CD. Structural basis for RNA surveillance by the human nuclear exosome targeting (NEXT) complex. Cell. 2022;185(12):2132-47.e26
77. Garland W, Müller I, Wu M, Schmid M, Imamura K, Rib L. et al. Chromatin modifier HUSH co-operates with RNA decay factor NEXT to restrict transposable element expression. Mol Cell. 2022;82(9):1691-707.e8
78. Savage SA, Niewisch MR. Dyskeratosis Congenita and Related Telomere Biology Disorders. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al. GeneReviews. Seattle (WA): University of Washington, Seattle. 1993
79. Gable DL, Gaysinskaya V, Atik CC, Talbot CC Jr, Kang B, Stanley SE. et al. ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 2019;33(19-20):1381-96
80. Massenet S, Bertrand E, Verheggen C. Assembly and trafficking of box C/D and H/ACA snoRNPs. RNA Biol. 2017;14(6):680-92
81. Dez C, Noaillac-Depeyre J, Caizergues-Ferrer M, Henry Y. Naf1p, an essential nucleoplasmic factor specifically required for accumulation of box H/ACA small nucleolar RNPs. Mol Cell Biol. 2002;22(20):7053-65
82. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77-94
83. Stanley SE, Gable DL, Wagner CL, Carlile TM, Hanumanthu VS, Podlevsky JD. et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. 2016;8(351):351ra107
84. Grill S, Nandakumar J. Molecular mechanisms of telomere biology disorders. J Biol Chem. 2021;296:100064
85. Armanios M. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest. 2013;123(3):996-1002
86. Chilosi M, Doglioni C, Murer B, Poletti V. Epithelial stem cell exhaustion in the pathogenesis of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2010;27(1):7-18
87. Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM. et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A. 2015;112(16):5099-104
88. Planas-Cerezales L, Arias-Salgado EG, Buendia-Roldán I, Montes-Worboys A, López CE, Vicens-Zygmunt V. et al. Predictive factors and prognostic effect of telomere shortening in pulmonary fibrosis. Respirology. 2019;24(2):146-53
89. Snetselaar R, van Batenburg AA, van Oosterhout MFM, Kazemier KM, Roothaan SM, Peeters T. et al. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS One. 2017;12(12):e0189467
90. Ravaglia C, Tomassetti S, Gurioli C, Piciucchi S, Dubini A, Gurioli C. et al. Features and outcome of familial idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(1):28-36
91. Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS. et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. 2014;2(7):557-65
92. Fasching CL. Telomere length measurement as a clinical biomarker of aging and disease. Crit Rev Clin Lab Sci. 2018;55(7):443-65
93. Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI. et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med. 2011;184(8):904-12
94. Tuder RM, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc. 2006;3(6):503-10
95. Cecchini MJ, Tarmey T, Ferreira A, Mangaonkar AA, Ferrer A, Patnaik MM. et al. Pathology, Radiology, and Genetics of Interstitial Lung Disease in Patients With Shortened Telomeres. Am J Surg Pathol. 2021;45(7):871-84
96. Naikawadi RP, Green G, Jones KD, Achtar-Zadeh N, Mieleszko JE, Arnould I. et al. Airway Epithelial Telomere Dysfunction Drives Remodeling Similar to Chronic Lung Allograft Dysfunction. Am J Respir Cell Mol Biol. 2020;63(4):490-501
97. Planas-Cerezales L, Arias-Salgado EG, Berastegui C, Montes-Worboys A, González-Montelongo R, Lorenzo-Salazar JM. et al. Lung Transplant Improves Survival and Quality of Life Regardless of Telomere Dysfunction. Front Med (Lausanne). 2021;8:695919
98. Swaminathan AC, Neely ML, Frankel CW, Kelly FL, Petrovski S, Durheim MT. et al. Lung Transplant Outcomes in Patients With Pulmonary Fibrosis With Telomere-Related Gene Variants. Chest. 2019;156(3):477-85
99. Kropski JA, Pritchett JM, Zoz DF, Crossno PF, Markin C, Garnett ET. et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. American journal of respiratory and critical care medicine. 2015;191(4):417-26
100. Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD. et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051-6
101. Kropski JA, Young LR, Cogan JD, Mitchell DB, Lancaster LH, Worrell JA. et al. Genetic Evaluation and Testing of Patients and Families with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2017;195(11):1423-8
102. Calado RT. Telomeres in lung diseases. Prog Mol Biol Transl Sci. 2014;125:173-83
103. Parry EM, Alder JK, Lee SS, Phillips JA 3rd, Loyd JE, Duggal P. et al. Decreased dyskerin levels as a mechanism of telomere shortening in X-linked dyskeratosis congenita. J Med Genet. 2011;48(5):327-33
104. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233-47
105. Lai SK, Wang YY, Wirtz D, Hanes J. Micro- and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61(2):86-100
106. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N. et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. 2018;9(1):5363
107. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN. et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103(35):13180-5
108. Stancil IT, Michalski JE, Davis-Hall D, Chu HW, Park JA, Magin CM. et al. Pulmonary fibrosis distal airway epithelia are dynamically and structurally dysfunctional. Nat Commun. 2021;12(1):4566
109. Figueira de Mello GC, Ribeiro Carvalho CR, Adib Kairalla R, Nascimento Saldiva PH, Fernezlian S, Ferraz Silva LF. et al. Small airway remodeling in idiopathic interstitial pneumonias: a pathological study. Respiration. 2010;79(4):322-32
110. Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A. et al. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax. 2011;66(8):651-7
111. Nakano Y, Yang IV, Walts AD, Watson AM, Helling BA, Fletcher AA. et al. MUC5B Promoter Variant rs35705950 Affects MUC5B Expression in the Distal Airways in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2016;193(4):464-6
112. Zhang Q, Wang Y, Qu D, Yu J, Yang J. The Possible Pathogenesis of Idiopathic Pulmonary Fibrosis considering MUC5B. Biomed Res Int. 2019;2019:9712464
113. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N. et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nature communications. 2018;9(1):5363 -
114. Evans CM, Williams OW, Tuvim MJ, Nigam R, Mixides GP, Blackburn MR. et al. Mucin is produced by clara cells in the proximal airways of antigen-challenged mice. Am J Respir Cell Mol Biol. 2004;31(4):382-94
115. Casalino-Matsuda SM, Monzon ME, Day AJ, Forteza RM. Hyaluronan fragments/CD44 mediate oxidative stress-induced MUC5B up-regulation in airway epithelium. Am J Respir Cell Mol Biol. 2009;40(3):277-85
116. Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK. et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8(3):e58658
117. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE. et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503-12
118. Gally F, Sasse SK, Kurche JS, Gruca MA, Cardwell JH, Okamoto T. et al. The MUC5B-associated variant rs35705950 resides within an enhancer subject to lineage- and disease-dependent epigenetic remodeling. JCI insight. 2021;6(2):e144294
119. Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L. et al. Regulation of MUC5B Expression in Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2017;57(1):91-9
120. Stock CJ, Conti C, Montero-Fernandez Á, Caramori G, Molyneaux PL, George PM. et al. Interaction between the promoter MUC5B polymorphism and mucin expression: is there a difference according to ILD subtype? Thorax. 2020;75(10):901-3
121. Chen G, Ribeiro CMP, Sun L, Okuda K, Kato T, Gilmore RC. et al. XBP1S Regulates MUC5B in a Promoter Variant-Dependent Pathway in Idiopathic Pulmonary Fibrosis Airway Epithelia. Am J Respir Crit Care Med. 2019;200(2):220-34
122. Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. The Journal of clinical investigation. 2020;130(10):5088-99
123. Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG. et al. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol. 2012;46(6):731-9
124. Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE. et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2015;290(6):3277
125. Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302(8):L721-9
126. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949-61
127. Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y. et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1(4):309-17
128. Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM. et al. Muc5b is required for airway defence. Nature. 2014;505(7483):412-6
129. Smirnova MG, Guo L, Birchall JP, Pearson JP. LPS up-regulates mucin and cytokine mRNA expression and stimulates mucin and cytokine secretion in goblet cells. Cell Immunol. 2003;221(1):42-9
130. McNamara N, Basbaum C. Signaling networks controlling mucin production in response to Gram-positive and Gram-negative bacteria. Glycoconj J. 2001;18(9):715-22
131. Wu DY, Wu R, Chen Y, Tarasova N, Chang MM. PMA stimulates MUC5B gene expression through an Sp1-based mechanism in airway epithelial cells. Am J Respir Cell Mol Biol. 2007;37(5):589-97
132. Preciado D, Lin J, Wuertz B, Rose M. Cigarette smoke activates NF kappa B and induces Muc5b expression in mouse middle ear cells. Laryngoscope. 2008;118(3):464-71
133. Lee JW, Kim YI, Im CN, Kim SW, Kim SJ, Min S. et al. Grape Seed Proanthocyanidin Inhibits Mucin Synthesis and Viral Replication by Suppression of AP-1 and NF-κB via p38 MAPKs/JNK Signaling Pathways in Respiratory Syncytial Virus-Infected A549 Cells. J Agric Food Chem. 2017;65(22):4472-83
134. Li XM, Sun SZ, Wu FL, Shi T, Fan HJ, Li DZ. Study on JNK/AP-1 signaling pathway of airway mucus hypersecretion of severe pneumonia under RSV infection. Eur Rev Med Pharmacol Sci. 2016;20(5):853-7
135. Luo J, Phan TX, Yang Y, Garelick MG, Storm DR. Increases in cAMP, MAPK activity, and CREB phosphorylation during REM sleep: implications for REM sleep and memory consolidation. J Neurosci. 2013;33(15):6460-8
136. Bae CH, Jeon BS, Choi YS, Song SY, Kim YD. Delphinidin Inhibits LPS-Induced MUC8 and MUC5B Expression Through Toll-like Receptor 4-Mediated ERK1/2 and p38 MAPK in Human Airway Epithelial Cells. Clin Exp Otorhinolaryngol. 2014;7(3):198-204
137. Song SY, Chi DH, Bae CH, Kim YD. Staphylococcus enterotoxin A induces MUC5B expression via Toll-like receptor 2, extracellular signal-regulated kinase 1/2, and p38 mitogen-activated protein kinase in human airway epithelial cells. Am J Rhinol Allergy. 2014;28(1):e25-30
138. Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune Mechanisms in Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2016;55(3):309-22
139. Fujisawa T, Chang MM, Velichko S, Thai P, Hung LY, Huang F. et al. NF-κB mediates IL-1β- and IL-17A-induced MUC5B expression in airway epithelial cells. Am J Respir Cell Mol Biol. 2011;45(2):246-52
140. Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278(19):17036-43
141. Sun H, Jiang Y, Song Y, Zhang X, Wang J, Zhang J. et al. The MUC5B Mucin Is Involved in Paraquat-Induced Lung Inflammation. Oxid Med Cell Longev. 2020;2020:7028947
142. Kobayashi T, Tanaka K, Fujita T, Umezawa H, Amano H, Yoshioka K. et al. Bidirectional role of IL-6 signal in pathogenesis of lung fibrosis. Respir Res. 2015;16(1):99
143. Le TT, Karmouty-Quintana H, Melicoff E, Le TT, Weng T, Chen NY. et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J Immunol. 2014;193(7):3755-68
144. Hao Y, Kuang Z, Jing J, Miao J, Mei LY, Lee RJ. et al. Mycoplasma pneumoniae modulates STAT3-STAT6/EGFR-FOXA2 signaling to induce overexpression of airway mucins. Infect Immun. 2014;82(12):5246-55
145. Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z. et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8(8):885-9
146. Choi YH, Lee SN, Aoyagi H, Yamasaki Y, Yoo JY, Park B. et al. The extracellular signal-regulated kinase mitogen-activated protein kinase/ribosomal S6 protein kinase 1 cascade phosphorylates cAMP response element-binding protein to induce MUC5B gene expression via D-prostanoid receptor signaling. J Biol Chem. 2011;286(39):34199-214
147. Song SY, Jung EC, Bae CH, Choi YS, Kim YD. Visfatin induces MUC8 and MUC5B expression via p38 MAPK/ROS/NF-κB in human airway epithelial cells. J Biomed Sci. 2014;21(1):49
148. Checa M, Hagood JS, Velazquez-Cruz R, Ruiz V, García-De-Alba C, Rangel-Escareño C. et al. Cigarette Smoke Enhances the Expression of Profibrotic Molecules in Alveolar Epithelial Cells. PLoS One. 2016;11(3):e0150383
149. Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D. et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147(3):525-38
150. Chakraborty A, Mastalerz M, Ansari M, Schiller HB, Staab-Weijnitz CA. Emerging Roles of Airway Epithelial Cells in Idiopathic Pulmonary Fibrosis. Cells. 2022;11(6):1050
151. Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y. et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106(31):12771-5
152. Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86(1):245-78
153. Anderson WH, Coakley RD, Button B, Henderson AG, Zeman KL, Alexis NE. et al. The Relationship of Mucus Concentration (Hydration) to Mucus Osmotic Pressure and Transport in Chronic Bronchitis. Am J Respir Crit Care Med. 2015;192(2):182-90
154. Solomon GM, Francis R, Chu KK, Birket SE, Gabriel G, Trombley JE. et al. Assessment of ciliary phenotype in primary ciliary dyskinesia by micro-optical coherence tomography. JCI Insight. 2017;2(5):e91702
155. Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J. et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68(12):1114-21
156. Pardo A, Cabrera S, Maldonado M, Selman M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respiratory research. 2016;17:23 -
157. Inchingolo R, Varone F, Sgalla G, Richeldi L. Existing and emerging biomarkers for disease progression in idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2019;13(1):39-51
158. Tzouvelekis A, Herazo-Maya JD, Slade M, Chu JH, Deiuliis G, Ryu C. et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology. 2017;22(3):486-93
159. Gharib SA, Altemeier WA, Van Winkle LS, Plopper CG, Schlesinger SY, Buell CA. et al. Matrix metalloproteinase-7 coordinates airway epithelial injury response and differentiation of ciliated cells. Am J Respir Cell Mol Biol. 2013;48(3):390-6
160. Sterclova M, Kishore A, Sikorova K, Skibova J, Petrek M, Vasakova M. Effect of genotype on the disease course in idiopathic pulmonary fibrosis despite antifibrotic treatment. Biomed Rep. 2021;15(5):87
161. Lou H-Q, Huang C-X, Li G-Y, Li P, Zhang S-M, Li H-G. et al. The Association between MUC5B Rs35705950 and Risks of Idiopathic Interstitial Pneumonia, Systemic Sclerosis Interstitial Lung Disease, and Familial Interstitial Pneumonia: A Meta-Analysis. Iran J Public Health. 2020;49(12):2240-50
162. Putman RK, Rosas IO, Hunninghake GM. Genetics and early detection in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;189(7):770-8
163. Schwartz DA. Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways. Annals of the American Thoracic Society. 2018;15(Suppl 3):S192-S7
164. Budzikowski JD, Foy JJ, Rashid AA, Chung JH, Noth I, Armato SG 3rd. Radiomics-based assessment of idiopathic pulmonary fibrosis is associated with genetic mutations and patient survival. J Med Imaging (Bellingham). 2021;8(3):031903 -
165. Kropski JA, Pritchett JM, Zoz DF, Crossno PF, Markin C, Garnett ET. et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am J Respir Crit Care Med. 2015;191(4):417-26
166. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK. et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824
167. Mulugeta S, Nureki S-I, Beers MF. Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. American journal of physiology Lung cellular and molecular physiology. 2015;309(6):L507-L25
168. Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344(8):573-9
169. Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG. et al. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013;188(3):376-94
170. Beers MF, Moodley Y. When Is an Alveolar Type 2 Cell an Alveolar Type 2 Cell? A Conundrum for Lung Stem Cell Biology and Regenerative Medicine. Am J Respir Cell Mol Biol. 2017;57(1):18-27
171. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR. et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025-36
172. Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med. 2005;172(8):1026-31
173. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C. et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125(2):521-38
174. Willander H, Askarieh G, Landreh M, Westermark P, Nordling K, Keränen H. et al. High-resolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc Natl Acad Sci U S A. 2012;109(7):2325-9
175. Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ. et al. An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI insight. 2019;4(6):e126125
176. Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018;68-69:355-65
177. Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB. et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119-26
178. Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M. et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838-46
179. Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A. et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(3):254-63
180. Cameron HS, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE. et al. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr. 2005;146(3):370-5
181. Hawkins A, Guttentag SH, Deterding R, Funkhouser WK, Goralski JL, Chatterjee S. et al. A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am J Physiol Lung Cell Mol Physiol. 2015;308(1):L33-47
182. Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB. et al. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L442-52
183. Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S. et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest. 2018;128(9):4008-24
184. Glasser SW, Burhans MS, Korfhagen TR, Na CL, Sly PD, Ross GF. et al. Altered stability of pulmonary surfactant in SP-C-deficient mice. Proc Natl Acad Sci U S A. 2001;98(11):6366-71
185. Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB. et al. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol. 2005;167(5):1267-77
186. Glasser SW, Witt TL, Senft AP, Baatz JE, Folger D, Maxfield MD. et al. Surfactant protein C-deficient mice are susceptible to respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L64-72
187. Glasser SW, Detmer EA, Ikegami M, Na CL, Stahlman MT, Whitsett JA. Pneumonitis and emphysema in sp-C gene targeted mice. J Biol Chem. 2003;278(16):14291-8
188. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5(1):58-68
189. Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B. et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet. 2016;25(8):1457-67
190. Takezaki A, Tsukumo SI, Setoguchi Y, Ledford JG, Goto H, Hosomichi K. et al. A homozygous SFTPA1 mutation drives necroptosis of type II alveolar epithelial cells in patients with idiopathic pulmonary fibrosis. J Exp Med. 2019;216(12):2724-35
191. Connors TD, Van Raay TJ, Petry LR, Klinger KW, Landes GM, Burn TC. The cloning of a human ABC gene (ABC3) mapping to chromosome 16p13.3. Genomics. 1997;39(2):231-4
192. Mulugeta S, Gray JM, Notarfrancesco KL, Gonzales LW, Koval M, Feinstein SI. et al. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J Biol Chem. 2002;277(25):22147-55
193. Rindler TN, Stockman CA, Filuta AL, Brown KM, Snowball JM, Zhou W. et al. Alveolar injury and regeneration following deletion of ABCA3. JCI Insight. 2017 2(24)
194. Bullard JE, Nogee LM. Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein C gene (SFTPC) mutation. Pediatr Res. 2007;62(2):176-9
195. Crossno PF, Polosukhin VV, Blackwell TS, Johnson JE, Markin C, Moore PE. et al. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest. 2010;137(4):969-73
Author contact
![]() Corresponding author: Junling Yang; The Second Hospital of Jilin University, 218 Ziqiang Street, Nanguan District, Changchun, Jilin Province 130041, P. R. China; Phone: (+86) 431-8113-6820; E-mail: junlingedu.cn.
Corresponding author: Junling Yang; The Second Hospital of Jilin University, 218 Ziqiang Street, Nanguan District, Changchun, Jilin Province 130041, P. R. China; Phone: (+86) 431-8113-6820; E-mail: junlingedu.cn.