Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and materials
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2023; 20(3):307-317. doi:10.7150/ijms.80942 This issue Cite
Research Paper
Nicotinamide mononucleotides alleviated neurological impairment via anti-neuroinflammation in traumatic brain injury
Xiaolu Zhu1#, Jin Cheng1#, Jiangtao Yu1, Ruining Liu1, Haoli Ma2 ![]() , Yan Zhao1,3
, Yan Zhao1,3 ![]()
1. Emergency Center, Zhongnan Hospital of Wuhan University, Wuhan, China.
2. Department of Biological Repositories, Zhongnan Hospital of Wuhan University, Wuhan, China.
3. Hubei Clinical Research Center for Emergency and Resuscitation, Zhongnan Hospital of Wuhan University, Wuhan, China.
# These authors contributed equally to this work.
Received 2022-11-7; Accepted 2023-1-20; Published 2023-1-31
Abstract

Traumatic brain injury (TBI) is one of the main factors of death and disability in adults with a high incidence worldwide. Nervous system injury, as the most common and serious secondary injury after TBI, determines the prognosis of TBI patients. NAD+ has been confirmed to have neuroprotective effects in neurodegenerative diseases, but its role in TBI remains to be explored. In our study, nicotinamide mononucleotides (NMN), a direct precursor of NAD+, was used to explore the specific role of NAD+ in rats with TBI. Our results showed that NMN administration markedly attenuated histological damages, neuronal death, brain edema, and improved neurological and cognitive deficits in TBI rats. Moreover, NMN treatment significantly suppressed activated astrocytes and microglia after TBI, and further inhibited the expressions of inflammatory factor. Besides, RNA sequencing was used to access the differently expressed genes (DEGs) and their enriched (Kyoto Encyclopedia of Genes and Genomes) KEGG pathways between Sham, TBI, and TBI+NMN. We found that 1589 genes were significantly changed in TBI and 792 genes were reversed by NMN administration. For example, inflammatory factor CCL2, toll like receptors TLR2 and TLR4, proinflammatory cytokines IL-6, IL-11 and IL1rn which were activated after TBI and were decreased by NMN treatment. GO analysis also demonstrated that inflammatory response was the most significant biological process reversed by NMN treatment. Moreover, the reversed DEGs were typically enriched in NF-Kappa B signaling pathway, Jak-STAT signaling pathway and TNF signaling pathway. Taken together, our data showed that NMN alleviated neurological impairment via anti-neuroinflammation in traumatic brain injury and the mechanisms may involve TLR2/4-NF-κB signaling.
Keywords: Traumatic brain injury, Nicotinamide mononucleotides, anti-neuroinflammation, neuronal injury, NF-kappa B
Introduction
Traumatic brain injury (TBI), as the most common cause of death in trauma centers, is also one of the major causes of death and disability in adults worldwide [1, 2]. Globally, the annual incidence rate of TBI is about 50 million [3], and more than 700,000 TBI incidents have been reported every year in China, of which severe TBI led to more than a quarter of the mortality rate and half of the adverse consequences [4]. Car crashes, falls, acceleration/deceleration and assaults are common causes of TBI. Therefore, the only possible treatment of primary injury is prevention, such as reducing dependence on motor vehicles and wearing helmets properly [5, 6]. Secondary injury in the seconds, minutes, hours, days is caused by a complex series of pathophysiological reactions, such as excitotoxicity, mitochondrial dysfunction, apoptosis, autophagy and inflammation, which will cause irreversible damage to neurons [7, 8]. There are several follow-up managements for TBI. At present, surgery such as debridement and inhibition of intracranial pressure target for penetrating TBI of acute stage [3]; drugs like diuretics and anticonvulsants used for preventing hydrocephalus and seizures in modest TBI [9]; physical therapy can relieve muscle spasms and contractions in mild TBI [10], but their effects on secondary injuries are limited [11]. Previous study has shown that Aucubin can reduce the inflammatory in mice after TBI to exert neuroprotective effect [12]. Omega-3 can reduce the loss of neurons by reducing the expression of inflammatory and apoptotic factors [13].
NAD+, as a key coenzyme in eukaryotic organisms, determines hundreds of enzymatic reactions [14]. In the most basic energy metabolism process, reduced and oxidized NAD are indispensable factors as hydrogen ion transfer carriers [15]. In addition, NAD+ can also indirectly regulate the process of anti-stress, cell growth, anti-inflammatory and anti-aging processes by regulating the function of Sirtuins factor which has NAD+ dependent protein deacetylation activity [16]. Previous studies have shown that the decline of NAD+ level will also lead to heart failure and Alzheimer's disease [17]. In vivo models of excitotoxic injury, the death of neurons is usually accompanied by a decrease in NAD+ levels [18]. In addition, the study showed that moderate and severe TBI could lead to a decrease in the level of NAD+ in the brain [19].
Nicotinamide mononucleotides (NMN) is the direct precursor of NAD+. It has been used in many medical researches because it can rapidly and effectively increase the level of NAD+ [14]. NMN is currently being used in the research of various neurological diseases, and has been observed to have neuroprotective properties and improve neurological function [20-24]. In cerebral ischemia mice, NMN showed neuroprotective effects though improving mitochondrial metabolism and alleviating oxidative stress injury [24]. Besides, NMN can also reduce the loss of neurons and inhibit the decline of cognitive function in AD mice [25].
Therefore, we propose the hypothesis that NMN may improve the behavior function as well as memory ability and alleviate the neurological damage of moderate-to-severe TBI rats. Through the treatment of Sprague-Dawley rats with TBI, the behavioral and pathological changes of rats were observed, and transcriptome sequencing of hippocampal tissues of rats was conducted to explore the role of NMN in TBI rats and the pathways which might be involved, hoping to find a new treatment for TBI.
Methods and materials
Animals
A total of 66 male Sprague Dawley rats, weighing 280-320g (7 weeks), were purchased from Vital River Laboratory Animal Technology (Beijing, China). The rats were housed in the Animal Experimental Center of Zhongnan Hospital at Wuhan University under 12 hours light/dark cycles with temperature controlled at 25 °C ± 2 °C. Rats had free access to standard laboratory diet and water. All animal experiments were carried out in accordance with the guidelines for experimentation with lab animals established by the Animal Experiment Center and Ethics Committee of Zhongnan Hospital of Wuhan University.
TBI procedure
Controlled cortical impact (CCI) device (Custom Design & Fabrication, USA) was employed to establish the rat model of a moderate-to-severe traumatic brain injury as described previously [26, 27]. After one week of rest, the animals were assigned randomly to one of three groups: sham group, TBI group and TBI+NMN group. Diet was stopped 12 hours before surgery. 3% Pentobarbital sodium (50mg/kg, i.p.) was injected to anesthetize rats. The scalp of rats was shaved and disinfected. The skull was exposed by incision in the middle of the skin of head. A craniotomy with a diameter of 5 mm was drilled in the right parietal region, the caudal side of the coronal and right side of the sagittal suture. The rats were placed on CCI device and their heads were fixed - the impactor was directed at the bone window and contacted with the meninges, and the parameter velocity was 5 m/s, dwell time was 200 ms and impact depth was 3 mm. The sham group received the same treatment except for impact. Analgesics and antibiotics were applied after surgery.
The detailed experimental arrangement
All animals were randomly divided into three groups, Sham, TBI and TBI+NMN (22 rats in each group). One hour after the operation, the TBI group was not given drugs, while the TBI+NMN group were injected with NMN and sham group were injected with vehicle (PBS). The brain tissues of some rats were collected 24 hours after TBI for sequencing, RT-qPCR, brain water content and blood brain barrier tests. The tissues were directly put into liquid nitrogen and then transferred to -80 °C for storage. The neurological deficits of the remaining rats were measured by mNSS score at 1,3,5 and 7 days after TBI. Morris water maze test was conducted from the 3rd to the 8th day. On the 8th day, the brain was removed and stored in 4% paraformaldehyde solution (Fig. 1).
Scheme of the procedures used for experiments.
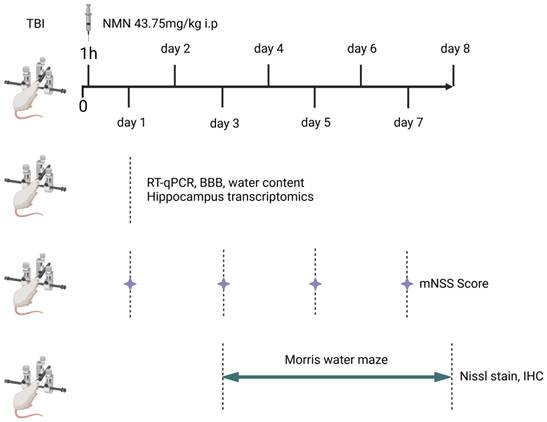
NMN administration
NMN was dissolved in Phosphate Buffer Saline (PBS) with a concentration of 20 mg/ml. Rats were injected with NMN (MCE, USA., HY-F0004, 43.75mg/kg, i.p.) or vehicle (PBS, same volume, i.p.) 1 hours after TBI impact experiment [28].
Nissl staining
After the rats were euthanized 8 days after TBI, normal saline and 4% paraformaldehyde were perfused into the left atrium to flush out the blood and fix the tissues. Three rats brain tissue in each group were obtained and fixed in paraformaldehyde. The paraffin blocks were sliced with a thickness of 10 μm. The brain was cut into three coronal sections to observe the structure of hippocampal CA1, CA3 and DG regions. After dewaxing, the slices were dyed in 0.1% cresol violet solution for 10 minutes, then washed in distilled water for three times, and dehydrated in 95% ethanol. Observe and collect images under Olympus microscope [29]. Image J software was used for processing.
mNSS Score
On the days 1, 3, 5 and 7 days after TBI, the neurological function was scored using modified neurological severity score (mNSS), and 6 rats in each group were tested each time. [30]. mNSS is a comprehensive test including motor, sensory, balance and reflex (normal score, 0, maximum score, 18 points). The final points were 13-18 for severe injury, 7-12 for moderate injury, and 1-6 for mild injury. The higher the score, the more severe the nerve damage of rats. The test was conducted in double blindness and scored by three professional researchers.
Morris water maze test
Morris water maze (MWM) test was performed on 5 rats in sham group, 6 rats in TBI and TBI+NMN groups. Animals were subjected to the Morris water maze test for the purpose of assessing spatial learning and memory [31]. The rats were put into a vat of inky water with a diameter of 2.1 meters. The tank is delimited four quadrants, and a platform is placed in one quadrant, which is 1cm below the water surface. On the 3rd to 7th days after TBI, all rats were tested four times a day. Each time, one rat was placed in a different quadrant, and the time spent to find the platform after it was placed in water was recorded. If it exceeds 120 seconds, guide the rat to find the platform and allow it to stay on the platform for 30 seconds. Remove the platform on the sixth day, and recorded the escape latency of the rats. Each stage of the experiment was recorded by the automatic tracking system (Xmaze™, Xinruan Information Technology Co., Shanghai, China).
Brain water content
Three rats per group were euthanized by giving 3% pentobarbital (150 mg/kg, i.p.). Brain tissues of rats were obtained after 24h post-TBI, removing the olfactory bulb and cerebellum. The brain was divided into ipsilateral (damaged) and contralateral (control) hemispheres, immediately weighed to obtain wet weight. Then drying samples for 24 hours at 100 °C in the oven to obtain the dry weight. The calculation formula of brain water content is as follows, (wet weight - dry weight)/ wet weight*100% [32].
Immunofluorescence staining
The brains of 3 rats in each group used for immunofluorescence were taken from the rats killed after neurological function test. The brain samples were dissected in 4% paraformaldehyde (24h, 4 °C), transferred to 30% sucrose solution for dehydration (72h, 4 °C), and then frozen (- 80 °C). The frozen brain tissues were then sliced at a thickness of 10 μm coronal brain sections with cryostat microtome (Leica Microsystems, Germany). The slices were stored in a -80 °C freezer until analysis. Three sections were taken from each rat for immunofluorescence staining, and one of the sections with the best effect was shown. The brain tissue sections were placed in a repair box filled with antigen repair buffer (0.01M citric acid buffer, pH 6.0), heated to 100 °C and incubated for 15 minutes for antigen repair. Sample washed with PBS (PH 7.4) for three times, and add goat serum at RT for 30 minutes for nonspecific binding sites blocking. Then add GFAP, IBA-1, NEUN (Abcam, ab7260, ab178847, ab177487, 1:500) primary antibody and incubate them in a 4 °C wet box overnight. After PBST washing, sections were incubated with second antibody in a wet box at 37 °C for 1 hour. NEUN incubated sections, with a drip concentration of 20 μg/ml protease K solution was incubated at room temperature for 20 minutes, and then the apoptosis detection kit (Vazyme, A113-03) was used for tunel staining. DAPI (Beyotime, C1002, 1:1000) was used for the nuclear counterstaining. Fluorescence images were captured with a fluorescence microscope (Olympus, Japan) and fluorescence intensity was quantified with Image J.
RNA-seq analysis
RNA-seq analysis was performed on 4 rats in each group. Euthanized rats were perfused 0.9% NaCl, and the brain tissues were removed. Bilateral hippocampus was separated on ice, directly put into liquid nitrogen, and transferred to - 80 °C refrigerator for preservation. Total RNA of hippocampus was extracted and purified by using Trizol reagent (Thermofisher, 15596018) based on the protocol provided by the manufacturer. RNA samples quantity and purity were analysis of Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA, 5067-1511), high-quality RNA samples with RIN number > 7.0 were used to construct cDNA library. Finally, we use illumina Novaseq™ 6000 (LC Bio Technology CO., Ltd. Hangzhou, China) conducts double ended sequencing (PE150) according to the supplier's recommended protocol.
To get high quality clean reads, reads were further filtered by Cutadapt, removing reads containing adapters, poly A and poly G, and unknown nucleotides bases. Then sequence quality was verified using FastQC. The reference genome was mapped using HISAT2 software. StringTie (1.3.1) was used to reconstruct transcripts and calculate the FPKM value of all gene expression levels in each sample. The differentially expressed mRNAs were used for GO enrichment and KEGG enrichment analysis, screened with | log2 (fold change) | > 0.5 and p value < 0.01. OmicStudio (https://www.omicstudio.cn/) was applied to perform the heatmap, venny analysis, GO and KEGG enrichment analysis.
RT-qPCR confirmation
To confirm the accurate RNA sequencing, Real-Time PCR was performed on 6 rats each group. The experiments were performed on a CFX Connect Real-Time PCR System (Bio-Rad; CFX Maestro 1.0 software). The target RNA and TB Green Premix Ex Taq II (Tli RNaseH Plus) (RR820A, TaKaRa, Japan) were subjected to the system. All procedures were performed according to standard protocols. Primers used in this study were listed in Table 1.
Primer sequences used in this study
| Gene | Species | Primer sequence |
|---|---|---|
| Ccl2 | RAT | Forward: 5' -CCACTCACCTGCTGCTACTCATTC -3' |
| Reverse: 5' -CTGCTGCTGGTGATCCTCTTGTAG -3' | ||
| Trl2 | RAT | Forward: 5' - GGAATCAACACAATAGAGGGAG -3' |
| Reverse: 5' -CTGAACCAGGAGGAAGATAAAC -3' | ||
| Trl4 | RAT | Forward: 5' - CCTCCCTGGTGTTGGATTTAC -3' |
| Reverse: 5' -AGATGCTTTCTCCTCTGCTGTA -3' | ||
| Stat3 | RAT | Forward: 5' - CAATACCATTGACCTGCCGAT -3' |
| Reverse: 5' - GAGCGACTCAAACTGCCCT -3' | ||
| Il11 | RAT | Forward: 5' - TGGGGACATGAACTGTGTTTGT -3' |
| Reverse: 5' -TGCAAAGATCCCAGTGTCCC -3' | ||
| Il6 | RAT | Forward: 5' - ACTTCCATCCAGTTGCCTTCTTGG -3' |
| Reverse: 5' - TTAAGCCTCCGACTTGTGAAGTGG -3' | ||
| Il1rn | RAT | Forward: 5' - TCCTTCTCATCCTTCTGTTTCGT -3' |
| Reverse: 5' - AGTGATGTTAACCTCCTCCAGC -3' | ||
| Zfp36 | RAT | Forward: 5' - AGCGGCTCCCAGATCAACT -3' |
| Reverse: 5' - CGAAAGCGAAGGCGTTGTTA -3' | ||
| Casp4 | RAT | Forward: 5' - ACAAACACCCTGACAAACCAC-3' |
| Reverse: 5' - CACTGCGTTCAGCATTGTTAAA -3' | ||
| Hmox1 | RAT | Forward: 5' - CTAAGACCGCCTTCCTGCTC -3' |
| Reverse: 5' -TGCAGAGGTAGTATCTTGAACC -3' | ||
| Hes5 | RAT | Forward: 5' - CCAAGGAGAAAAATCGACTGCG -3' |
| Reverse: 5' -CGAAGGCTTTGCTGTGCTTC -3' | ||
| Ccn1 | RAT | Forward: 5' - GGCGTTGACAGTACGTTTGG-3' |
| Reverse: 5' -AGAGGCTTCCTGTCTTTGGC -3' | ||
| TNFα | RAT | Forward: 5' - CTCCAGGCGGTGCCTATG -3' |
| Reverse: 5' -GGGCCATAGAACTGATGAGAGG -3' | ||
| IL1β | RAT | Forward: 5' - GCACACCCACCCTGCA -3' |
| Reverse: 5' -ACCGCTTTTCCATCTTCTTCTT -3' |
Statistical analysis
All data were expressed as mean ± standard deviation (SD). Difference between two groups was tested by Student's t test, and a one- or two-way analysis of variance (ANOVA) was performed for multiple groups. Values of P < 0.05 were considered to indicate a statistical different. Analyses were performed using the SPSS software (V25.0, IBM, United States).
Results
NMN treatment reduced the neurological damage and improved neurological functions after TBI
To explore the neuroprotective effect of NMN on TBI, we first examined the survival of nerve cells. As shown by Nissl staining (Fig. 2A), neuron structure was clear and Nissl bodies were evenly distributed in sham group, but the structure of neurons was disappeared after TBI operation. However, less loss of neurons combined with normal structure were showed in the TBI+NMN group Compared to TBI group, indicating that NMN treatment reduced the neurological damage in hippocampal CA1 area after TBI. Quantitative statistics also indicated that the numbers of Nissl-positive cells in sham group was 97 ± 14 cells/field, while the TBI group was far less than sham group (P < 0.01), which was 24±17 cells/field. And the numbers of Nissl-positive cells in TBI+NMN group was significantly increased compared to TBI group, which was 71 ± 13 cells/field (P < 0.05) (Fig. 2B).
NMN treatment reduced the neurological damage and improved neurological function after TBI. A and B) Nissl staining at 8 days after TBI in the Brain sections (n = 3 per group). C) Brain water content of damaged hemisphere 1day post-TBI (n = 3 per group). D) mNSS score at 1, 3, 5, and 7 days following TBI/sham (n = 6 per group). E) Escape latency in MWM test 3 to 7 days following TBI/sham (n = 5 sham group, n=6 TBI and TBI + NMN group). F) Escape latency at test day following TBI/sham (*p < 0.05, **p < 0.01, and ***p < 0.001 TBI vs Sham group; #p < 0.05 and ##p < 0.01 TBI vs TBI +NMN group).
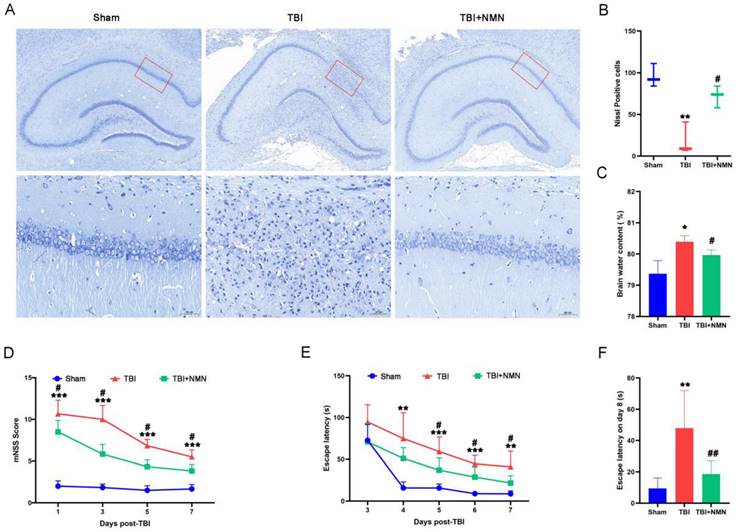
In addition, the measurement of brain water content also demonstrated that the brain edema after TBI was also alleviated by NMN treatment, as the brain water content in the TBI+NMN group was significantly lower than TBI group (TBI+NMN vs TBI, 79.97% ± 0.16% vs 80.41% ± 0.19%, P < 0.05) (Fig. 2C).
In order to observe whether NMN administration affect the neurological functions after TBI, we conducted tests to measure mNSS scores and performed behavioral experiments using the Morris water maze. At 1, 3, 5, and 7 days after TBI, the mNSS scores of TBI group were significantly higher than those of sham group (all P < 0.001). But after NMN treatment, the mNSS scores were significantly reduced. (P < 0.05) (Fig. 2E). We also performed MWM tests at 3-8 days after surgery to investigate the effects of NMN on spatial learning and memory ability after TBI. On the first day of training, there was no evidently difference in the escape latency of the three groups. However, from 4 to 7 days after TBI, the escape latency of the TBI group was significantly higher than the sham group (P < 0.01 on days 4 and 7; P < 0.001 on days 5 and 6). Consistent with the mNSS score results, NMN treatment rats showed a significantly lower escape latency compared to TBI group during this period (P < 0.05) (Fig. 2F). Moreover, the latency in TBI+NMN group was also lower than TBI group on the test day of MWM (P < 0.01) (Fig. 2G). Taken together, these results strongly suggest that NMN has significant therapeutic effect against neurological injury and neurobehavioral damage after TBI.
NMN reduced the numbers of reactive astrocytes and microglia, and inhibited the transcription level of TNF-α and IL-1β
In the behavioral experiment completed, NMN treatment showed a neuroprotective effect on TBI. Previous studies showed the neuroinflammation, which is closely related to the activation of microglia and astrocyte activation, is one of the important mechanisms of TBI secondary injury [33-35]. Therefore, we further administrated whether NMN affected the activation of glial cells after TBI. The activated astrocytes maker GFAP and the microglia maker Iba-1 immunostaining showed that NMN treatment significantly reduced the TBI-induced activated astrocytes and microglia in the CA1 area of hippocampus, and the quantitative analysis of fluorescence intensity showed the same result (P < 0.05) (Fig. 3A-C).
NMN treatment alleviated microglial and astrocyte activation and inhibited inflammatory response in hippocampal CA1 at 8 days after TBI. A) Representative fluorescence images for staining of GFAP in CA1 region. Scale bar = 40 μm (n =3 per group). B) Representative fluorescence images for staining of IBA-1 in CA1 region. Scale bar = 40 μm (n =3 per group). C) quantitative analysis of GFAB and IBA-1 fluorescence intensity in the DG. D) RT-qPCR of IL-1β and TNF-α mRNA expression levels at 24h after TBI (n =3 per group). (*p < 0.05, **p < 0.01, and ***p < 0.001 TBI vs Sham group; #p < 0.05 and ##p < 0.01 TBI vs TBI +NMN group).
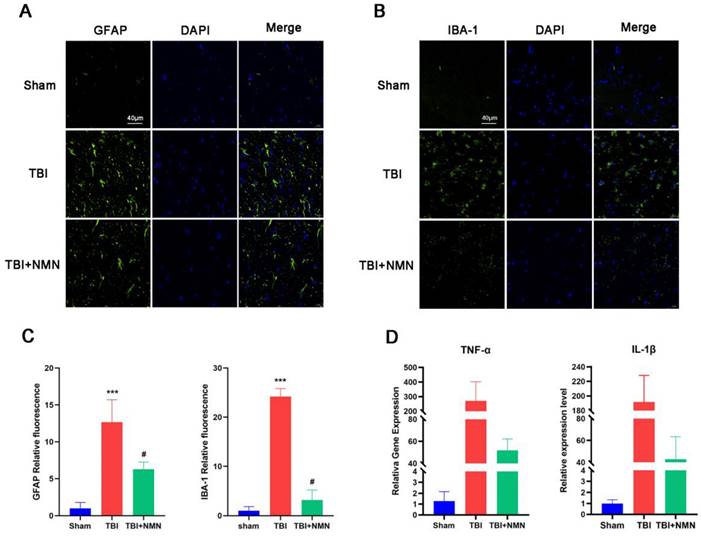
TNF-α and IL-1β as representative inflammatory factor in TBI, their expression level reflects the degree of inflammatory response. We detected the expressions of TNF-α and IL-1β by RT-qPCR. The finding illustrated that the expression of TNF-α and IL-1β genes in hippocampus of TBI+NMN group was significantly lower than TBI group (P < 0.05) (Fig. 3D). These results indicated NMN can relieve the inflammatory response after TBI.
NMN decreased neuronal apoptosis after TBI
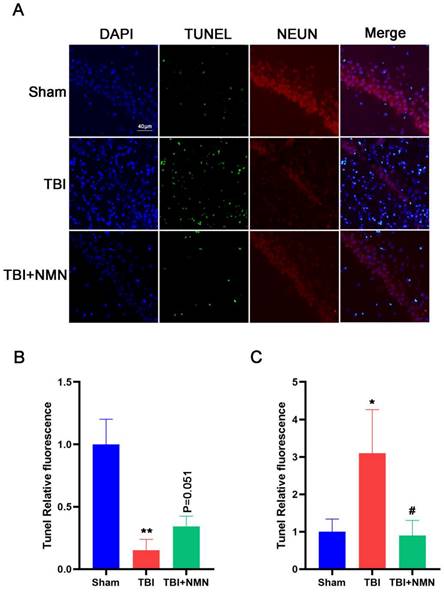
The improvement of neurological functions after TBI is not only related to the reduction of inflammation, but also related to the damage of neurons directly. Therefore, we next assessed neuronal apoptosis by TUNEL assay. Brain samples were taken for double immunofluorescence staining to detect the co-expression of TUNEL and NEUN in CA1 area (Fig. 4A). Consistent with the neuroinflammation result, the NEUN fluorescence intensity in hippocampus were decreased (P < 0.05) and the TUNEL fluorescence intensity were increased after TBI compared with the sham group (P < 0.05). And NMN administration significantly decreased the fluorescence intensity of TUNEL after TBI (P < 0.05). The fluorescence intensity of NEUN in TBI+NMN group also increased significantly compared with the TBI group (Fig. 4B, C). These results demonstrated that NMN treatment was able to reduce neuronal apoptosis.
NMN decreased neuronal apoptosis at 8 days after TBI. A) Representative images of NEUN, TUNEL, and DAPI costaining of brain sections. Scale bar: 40 nm (n =3 per group). B) Quantitative analysis of NEUN fluorescence intensity in CA1 region (n =3 per group). C) Quantitative analysis of TUNEL fluorescence intensity in CA1 region. (n =3 per group, *p < 0.05, **p < 0.01, and ***p < 0.001 TBI vs Sham group; #p < 0.05 and ##p < 0.01 TBI vs TBI +NMN group).
Hippocampal transcriptome analysis of three groups
To further probe the related mechanism of neuroprotective effect of NMN after TBI, transcriptome sequencing was performed on hippocampus of Sham, TBI, and TBI+NMN groups. The sequencing results showed that there were 1589 differentially expressed genes (DEGs) in TBI vs sham, including 1123 up-regulated and 466 down-regulated, and 157 up-regulated and 697 down-regulated were accessed due to the effect of NMN treatment (|log2FC| > 1 and p < 0.01) (Fig. 5A and Table S1). Of the 1589 DEGs genes that appeared after TBI, 792 were reversed in the TBI+NMN group, including 656 up-regulated and 136 down-regulated ones (Fig. 5B and Table S1). Hierarchical cluster analysis of 792 reversed genes shows that gene expression has been significantly reversed after TBI and treatment (Fig. 5D).
RNA-seq analysis on Sham, TBI, and TBI +NMN in rat 24h post-TBI (n=4 per group). A) Histogram of deferentially expressed genes (DEGs) between different groups. B) Venny analysis between DEGs in TBI vs Sham and DEGs in TBI+NMN vs TBI. C) Expression heatmap of 792 reversed genes. D and E) GO enrichment analysis and KEGG enrichment analysis based on the reversed genes.
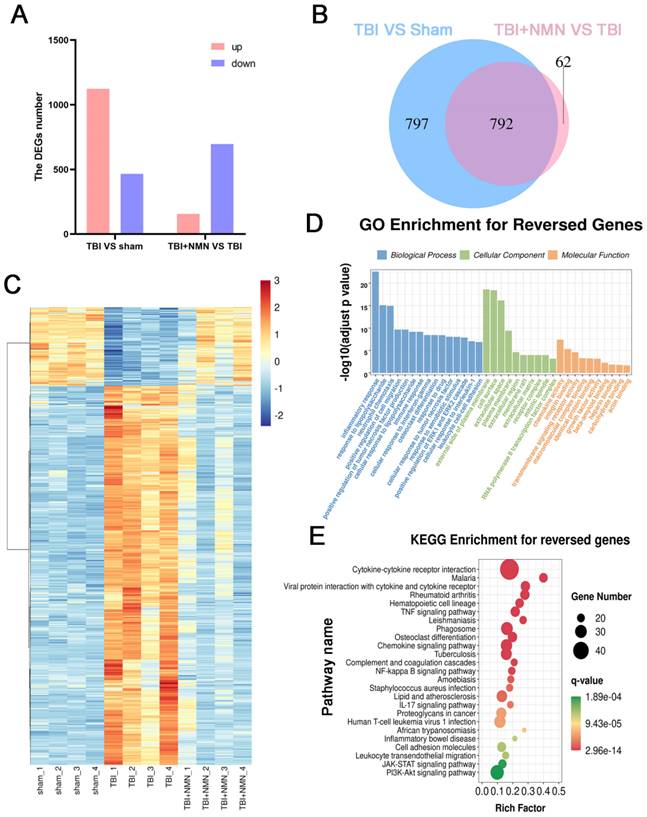
Gene ontology (GO) and KEGG enrichment analysis of these common differential genes were carried out to explore the function of them. The 792 reversed DEGs were enriched in inflammation related GO items, such as inflammatory response, positive regulation of cell migration, positive regulation of tumor necrosis factor production, immune response, cellular response to tumor necrosis factor, positive regulation of ERK1 and ERK2 cascade and cellular response to interleukin-1 (Fig. 5C and Table S2). They were also enriched in Cytokine-cytokine receptor interaction, TNF signaling pathway, Phagosome, Chemokine signaling pathway, NF-kappa B signaling pathway, Cell adhesion molecules, JAK-STAT signaling pathway, PI3K-Akt signaling pathway and some immunity-related pathway (Fig. 5E and Table S2). The above results suggested that NMN may exert neuroprotective effects via influencing inflammation and immunity related signaling pathways.
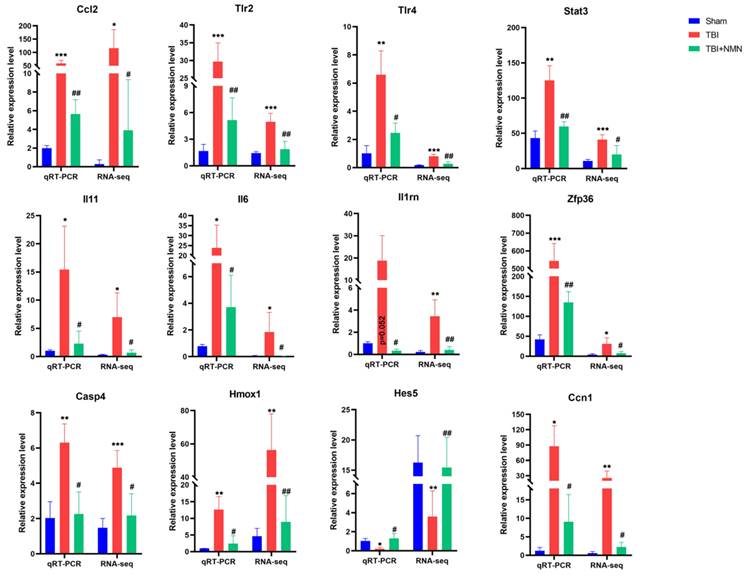
At last, in order to confirm the reliability of RNA sequencing results, a series of genes were selected to detect their expression levels using RT-qPCR. As showed in Fig. 6, 10 genes (Ccl2, Trl2, Trl4, Stat3, Il11, Il6, Il1rn, Zfp36, Casp4, Hmox1, and Ccn1) were up-regulated in TBI group compared to sham group, and were significantly down-regulated in TBI+NMN group compared to TBI group. Moreover, Hes5 was down-regulated in TBI group compared to sham group and reversed by NMN treatment. The sequencing results and PCR results were mutually verified.
RT-qPCR of Ccl2, Tlr2, Tlr4, Stat3, Il11, Il6, Il1rn, Zfp36, Casp4, Homx1, Hes5 and Ccn1 mRNA expression levels at 24h after TBI. (n = 6 per group, *p < 0.05, **p < 0.01, and ***p < 0.001 TBI vs Sham group; #p < 0.05 and ##p < 0.01 TBI vs TBI +NMN group).
Discussion
TBI is initially caused by mechanical damage, but its secondary damage is complex and diverse. Substantial evidence suggested that cellular cascade of inflammation participates in secondary brain injury [36-39]. However, short-term inflammation after TBI is protective, while long-term, intense inflammation can damage the brain [40]. And a number of studies have reported that NAD+ play critical roles in many biological processes including metabolism, inflammation, and stress and damage response resulting from its consuming enzymes [41, 42]. Our study demonstrated that the administration of NMN, a NAD+ intermediates, is capable of alleviating TBI-associated neurological impairment by mitigating neuronal inflammation. In the present study, we first examined the effect of NMN treatment on the pathological changes after TBI. We found that NMN reduced the death of nerve cells, maintained the structure of hippocampus, and alleviated the brain edema after TBI, which proved that NMN can effectively reduce brain damage caused by TBI. Previous studies have shown that the degree of neuronal death in the hippocampus due to contusion, inflammation, edema and other reasons affects the postoperative recovery of TBI [43, 44]. Thus, we further investigated the neurological function of NMN in TBI rats. We found that NMN-treated TBI rats substantially got lower mNSS scores and lower escape latency compared to TBI rats. Zhao's study reported that NAD+ ameliorate cognitive impairment in chronic cerebral hypoperfusion (CCH) models [45], which is consistent with our results. These findings proved that NMN can play a protective role in brain neurons after TBI.
Astrocytes and microglia which play crucial roles in the inflammatory response of the central nervous system can lead to neurodegenerative diseases [46]. According to our findings, decreased GFAP and Iba-1were observed under NMN administration. Pro-inflammatory response astrocytes induce proinflammatory factors (e.g., IL-1β, TNF-α) that are known to have deleterious functions [47]. Besides, microglia were reported have ability to reduce clearance effect, which leads to the aggravation of nerve cell damage [48]. Moreover, NAD+ treatment have been reported to reduce CCH-induced microglia and subsequent production of pro-inflammatory factors [45]. Similarly, TBI rats in our study exhibited activated TNF-α and IL-1β that were mitigated by NMN treatment. Additionally, Post-traumatic neuroinflammation have been reported to significantly contribute to the neuronal death observed after neurotrauma [49]. TUNEL assay in our study also demonstrated that NMN treatment decreased TBI-induced neuronal apoptosis. Given these results, we hypothesized that the improvement of neurological function in TBI rats may be result from the fact that NMN alleviated neuroinflammation.
To further investigated the related mechanism of NMN in TBI, we identified differentially expressed genes in TBI by RNA sequencing and explored genes and enrichment pathways for NMN reversal. We found 1589 genes were changed in TBI and 792 genes were reversed by NMN administration. And GO analysis showed that the different reversed genes were strongly related to inflammatory response, which was consistent to our experiment findings. Our data proved that MNM treatment reversed the elevated CCL2 which induced by TBI. Zhao et al. reported CCL2 which was derived from microglia, astrocytes and neurons can aggravate tissue damage by conducting secondary inflammatory response [50]. In addition, the reversal of proinflammatory cytokines such as IL-6, IL-11 and IL1rn also proved that NMN could reduce the level of inflammation after TBI. Moreover, we also found that Toll-like receptors TLR2 and TLR4 were also reduced by NMN treatment. Tu et al. found that inhibition of TLR2/4-NF-κB signaling reduced brain damage and protected nerves [51, 52]. Lin et al. also revealed that TLR2/4-NF-κB signaling has an inhibitory effect in the later stage of TBI [53]. Wan et al. reported NMN prevented DIC in rats by inhibiting NLRP3 inflammatory body activation [54]. These findings were in agreement with our results. Similarly, KEGG analysis also showed that the NF-kappa B signaling pathway, Jak-STAT signaling pathway, TNF signaling pathway were significantly enriched. Besides, present studies have revealed that oxidative and inflammation are indispensable in TBI [55, 56]. The increased NAD+ can preserve cellular respiration. reduce oxidative stress, and further alleviate inflammation [57].
Conclusion
In summary, this study provide evidence that NMN administration alleviated TBI-associated neurological impairment by anti-neuroinflammation bioactivities. Besides, the underlying molecular mechanisms of these beneficial effects may involve TLR2/4-NF-κB signaling. Our data might provide a novel therapeutic strategy for TBI. However, the precise mechanism on NMN still needs to be explored in further study.
Supplementary Material
Supplementary table 1.
Supplementary table 2.
Acknowledgements
Funding statement
This study was supported by the 2021 Medical Research Project from Health Bureau of Wuchang District of Wuhan, 2020 Annual Funding for Discipline Construction from Zhongnan Hospital of Wuhan University, and Discipline Cultivation Funding from Zhongnan Hospital of Wuhan University. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of Wuhan University (ZN2021192).
Author contributions
XZ, JC, HM and YZ designed the project; XZ and JC performed the experiments; XZ, JC, HM, JY collected and analyzed the data; XZ, JC, HM prepared the draft; HM and YZ revised the paper.
Data availability
The datasets showed in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.ncbi.nlm.nih.gov/bioproject/902029, PRJNA902029.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ghajar J. Traumatic brain injury. The Lancet. 2000;356:923-9
2. Callcut RA, Kornblith LZ, Conroy AS, Robles AJ, Meizoso JP, Namias N. et al. The why and how our trauma patients die: A prospective Multicenter Western Trauma Association study. J Trauma Acute Care Surg. 2019;86:864-70
3. Khellaf A, Khan DZ, Helmy A. Recent advances in traumatic brain injury. J Neurol. 2019;266:2878-89
4. Jiang J-Y, Gao G-Y, Feng J-F, Mao Q, Chen L-G, Yang X-F. et al. Traumatic brain injury in China. The Lancet Neurology. 2019;18:286-95
5. Jha S, Ghewade P. Management and Treatment of Traumatic Brain Injuries. Cureus. 2022;14:e30617
6. Abdelmalik PA, Draghic N, Ling GSF. Management of moderate and severe traumatic brain injury. Transfusion. 2019;59:1529-38
7. Ma X, Aravind A, Pfister BJ, Chandra N, Haorah J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol Neurobiol. 2019;56:5332-45
8. Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:171-91
9. Capizzi A, Woo J, Verduzco-Gutierrez M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med Clin North Am. 2020;104:213-38
10. Kane AW, Diaz DS, Moore C. Physical Therapy Management of Adults with Mild Traumatic Brain Injury. Semin Speech Lang. 2019;40:36-47
11. Maas AIR, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. The Lancet Neurology. 2008;7:728-41
12. Wang H, Zhou XM, Wu LY, Liu GJ, Xu WD, Zhang XS. et al. Aucubin alleviates oxidative stress and inflammation via Nrf2-mediated signaling activity in experimental traumatic brain injury. J Neuroinflammation. 2020;17:188
13. Chen X, Chen C, Fan S, Wu S, Yang F, Fang Z. et al. Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury. J Neuroinflammation. 2018;15:116
14. Okabe K, Yaku K, Tobe K, Nakagawa T. Implications of altered NAD metabolism in metabolic disorders. J Biomed Sci. 2019;26:34
15. Rich PR. The molecular machinery of Keilin's respiratory chain. Biochemical Society transactions. 2003;31:1095-105
16. Rajman L, Chwalek K, Sinclair DA. Therapeutic Potential of NAD-Boosting Molecules: The In vivo Evidence. Cell Metab. 2018;27:529-47
17. Yao Z, Yang W, Gao Z, Jia P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci Lett. 2017;647:133-40
18. Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28-42
19. Satchell MA, Zhang X, Kochanek PM, Dixon CE, Jenkins LW, Melick J. et al. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J Neurochem. 2003;85:697-708
20. Kiss T, Nyúl-Tóth Á, Balasubramanian P, Tarantini S, Ahire C, Yabluchanskiy A. et al. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. GeroScience. 2020;42:527-46
21. Zhao N, Xia J, Xu B. Physical exercise may exert its therapeutic influence on Alzheimer's disease through the reversal of mitochondrial dysfunction via SIRT1-FOXO1/3-PINK1-Parkin-mediated mitophagy. J Sport Health Sci. 2021;10:1-3
22. Chandrasekaran K, Choi J, Arvas MI, Salimian M, Singh S, Xu S. et al. Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. Int J Mol Sci. 2020 21
23. Sasaki Y, Zhu J, Shi Y, Gu W, Kobe B, Ve T. et al. Nicotinic acid mononucleotide is an allosteric SARM1 inhibitor promoting axonal protection. Exp Neurol. 2021;345:113842
24. Klimova N, Fearnow A, Long A, Kristian T. NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp Neurol. 2020;325:113144
25. Wang X, Hu X, Yang Y, Takata T, Sakurai T. Nicotinamide mononucleotide protects against β-amyloid oligomer-induced cognitive impairment and neuronal death. Brain research. 2016;1643:1-9
26. Siebold L, Obenaus A, Goyal R. Criteria to define mild, moderate, and severe traumatic brain injury in the mouse controlled cortical impact model. Exp Neurol. 2018;310:48-57
27. Li T, Zhang W, Hu E, Sun Z, Li P, Yu Z. et al. Integrated metabolomics and network pharmacology to reveal the mechanisms of hydroxysafflor yellow A against acute traumatic brain injury. Computational and structural biotechnology journal. 2021;19:1002-13
28. Park JH, Long A, Owens K, Kristian T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol Dis. 2016;95:102-10
29. Zhou Y, Tao T, Liu G, Gao X, Gao Y, Zhuang Z. et al. TRAF3 mediates neuronal apoptosis in early brain injury following subarachnoid hemorrhage via targeting TAK1-dependent MAPKs and NF-kappaB pathways. Cell Death Dis. 2021;12:10
30. Wu H, Zheng J, Xu S, Fang Y, Wu Y, Zeng J. et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflammation. 2021;18:2
31. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nature protocols. 2006;1:848-58
32. Lan X, Han X, Li Q, Li Q, Gao Y, Cheng T. et al. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav Immun. 2017;61:326-39
33. Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol. 2016 275 Pt 3: 305-15
34. <The contribution of astrocytes and microglia to traumatic brain injury.pdf>
35. Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. British journal of pharmacology. 2016;173:692-702
36. Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229-48
37. Schwartz M, Raposo C. Protective Autoimmunity: A Unifying Model for the Immune Network Involved in CNS Repair. Neuroscientist. 2014;20:343-58
38. Jacquens A, Needham EJ, Zanier ER, Degos V, Gressens P, Menon D. Neuro-Inflammation Modulation and Post-Traumatic Brain Injury Lesions: From Bench to Bed-Side. Int J Mol Sci. 2022 23
39. Wangler LM, Bray CE, Packer JM, Tapp ZM, Davis AC, O'Neil SM. et al. Amplified Gliosis and Interferon-Associated Inflammation in the Aging Brain Following Diffuse Traumatic Brain Injury. J Neurosci. 2022
40. Postolache TT, Wadhawan A, Can A, Lowry CA, Woodbury M, Makkar H. et al. Inflammation in Traumatic Brain Injury. J Alzheimers Dis. 2020;74:1-28
41. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464-71
42. Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y. et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016;24:795-806
43. Huie JR, Chou A, Torres-Espin A, Nielson JL, Yuh EL, Gardner RC. et al. FAIR Data Reuse in Traumatic Brain Injury: Exploring Inflammation and Age as Moderators of Recovery in the TRACK-TBI Pilot. Front Neurol. 2021;12:768735
44. Jamjoom AAB, Rhodes J, Andrews PJD, Grant SGN. The synapse in traumatic brain injury. Brain. 2021;144:18-31
45. Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang XJ, Wang H. et al. NAD(+) improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1alpha pathway. J Neuroinflammation. 2021;18:207
46. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9:42
47. Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity. 2017;46:957-67
48. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918-34
49. Badaut J, Ajao DO, Sorensen DW, Fukuda AM, Pellerin L. Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: signs of blood-brain barrier healing? Neuroscience. 2015;285:215-26
50. Zhao X, Liu S, Yang X, Liu Y, Liu G, Fan K. et al. Cathepsin C aggravates neuroinflammation via promoting production of CCL2 and CXCL2 in glial cells and neurons in a cryogenic brain lesion. Neurochem Int. 2021;148:105107
51. Tu XK, Yang WZ, Shi SS, Chen Y, Wang CH, Chen CM. et al. Baicalin inhibits TLR2/4 signaling pathway in rat brain following permanent cerebral ischemia. Inflammation. 2011;34:463-70
52. Tu XK, Yang WZ, Chen JP, Chen Y, Ouyang LQ, Xu YC. et al. Curcumin inhibits TLR2/4-NF-kappaB signaling pathway and attenuates brain damage in permanent focal cerebral ischemia in rats. Inflammation. 2014;37:1544-51
53. Lin SJ, Cao LX, Cheng SB, Dai QF, Lin JH, Pu L. et al. Effect of acupuncture on the TLR2/4-NF-kappaB signalling pathway in a rat model of traumatic brain injury. Acupunct Med. 2018;36:247-53
54. Wan Y, He B, Zhu D, Wang L, Huang R, Zhu J. et al. Nicotinamide mononucleotide attenuates doxorubicin-induced cardiotoxicity by reducing oxidative stress, inflammation and apoptosis in rats. Arch Biochem Biophys. 2021;712:109050
55. Wang J, Jiang C, Zhang K, Lan X, Chen X, Zang W. et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic Biol Med. 2019;131:345-55
56. Eastman CL, D'Ambrosio R, Ganesh T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology. 2020;172:107907
57. Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC. et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426-33
Author contact
![]() Corresponding authors: Haoli Ma and Yan Zhao, E-mail: mahaoliedu.cn; doctoryanzhaoedu.cn.
Corresponding authors: Haoli Ma and Yan Zhao, E-mail: mahaoliedu.cn; doctoryanzhaoedu.cn.