Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(14):3197-3205. doi:10.7150/ijms.61944 This issue Cite
Research Paper
p53-dependent apoptosis is essential for the antitumor effect of paclitaxel response to DNA damage in papillary thyroid carcinoma
Wenshuang Wu1,2*, Tao Wei1*, ZhiHui Li1,2, Jingqiang Zhu1 ![]()
1. Department of Thyroid Surgery, West China Hospital, Sichuan University, Chengdu, China
2. Laboratory of Thyroid and Parathyroid Disease, Frontiers Science Center for Disease-related Molecular Network, West China Hospital, Sichuan University, Chengdu, China
* Contributed equally
Received 2021-4-23; Accepted 2021-7-1; Published 2021-7-11
Abstract

A functional p53 protein plays an important role in killing tumor cells. Previous studies showed that chemotherapeutic drug, paclitaxel (PTX), showed anti-tumor activity through inducing G2/M arrest and apoptosis by targeting microtubules in tumor cells. However, PTX was not sensitive to p53-inactivated papillary thyroid carcinoma (PTC) cells by inducing G2/M arrest only. Recombinant adenovirus-p53 (rAd-p53) was used to increase the level of p53, which significantly increased the sensitivity of PTC cells to PTX by inducing S arrest, G2/M arrest and apoptosis. To discuss the anti-tumor mechanism of rAd-p53 + PTX and found p53 activation was necessary for anti-tumor effect of PTX in PTC cells. There was high level of p53 in rAd-p53-treated PTC cells. rAd-p53 + PTX increased the level of p21, p-ATM and γ-H2AX and decreased the level of Cyclin D1/E1, suggesting p53 activated p21 which negatively regulated cyclins to induce S arrest response to DNA damage in PTC cells. rAd-p53 + PTX increased the levels of cleaved-PARP-1, cleaved -Caspase 3, and BAX and decreased the level of BCL-XL, suggesting p53 regulates the expression of BAX/BCL-XL to mediate DNA damage-induced apoptosis in PTC cells. Furthermore, rAd-p53 + PTX showed significant tumor inhibition in TPC-1 xenograft model, with an inhibitory rate of 79.39%. TUNEL assay showed rAd-p53 + PTX induced notable apoptosis in tumor tissues. rAd-p53 showed good sensitization of PTX in vitro and in vivo through inducing DNA damage induced-apoptosis indicated p53-dependent apoptosis was essential for the antitumor effect of PTX in PTC.
Keywords: PTC, PTX, p53, cell cycle arrest, apoptosis
Introduction
Papillary thyroid carcinoma (PTC) represents ~85% of all thyroid cancer cases [1]. In clinical treatment, the majority of patients with localized, well-differentiated PTC patients have a good prognosis under the traditional treatment approaches, including surgery followed by thyroid-stimulating hormone suppressive therapy with/without radioactive iodine therapy, with a 10-year survival rate of >90% [2]. However, some cases challenge the traditional therapy approaches. For example, patients with distant metastatic diseases that are not suitable for surgery, of which 50% of them lose the ability to take up radiation, hinder the prognosis significantly, demonstrating a 10-year survival rate of 42% [3]. The radioiodine refractory disease has a more dismal prognosis, with a 10-year survival rate of 10% [4]. Therefore, further basic research that focuses on pathology and pharmacology may be required for these PTC cases.
The TP53 gene (P53), which encodes the p53 protein, is one of the most common and important tumor suppressor genes that inhibits the growth and development of tumors [5]. Tumor cells always have instable genomic, which is associated with the accumulation of DNA damage. The main function of p53 is to repair damaged-DNA in order to prevent altered DNA being passed on to daughter cells. p53 is a transcriptional factor containing DNA binding and transcription activation domains [6]. Once DNA damage in cells occurs, DNA damage results in phosphorylation of the ATM protein. Activated-ATM phosphorylates Chk2, which activates the p53 pathway to negatively regulate cell cycle proteins [7,8]. p21, as the main targeted gene of p53 during cell cycle arrest, negatively regulates Cyclins to induce cell cycle arrest [9]. If DNA damage is too extensive to be repaired, p53 regulates cells to undergo programmed death, including apoptosis, senescence or necrosis [6]. In the DNA damage-induced apoptosis pathway, p53 initiates the expression of apoptosis-related genes, including BAX (apoptotic gene), BCL-2 and BCL-XL (anti-apoptotic genes), to regulate cell apoptosis [10,11]. Once the ratio of apoptotic gene/anti-apoptotic genes increases, it causes a change in the mitochondrial membrane permeability and leads to the release of cytochrome c from the mitochondrial to the cytoplasm [12-14]. Then, cytosolic cytochrome c activates Caspase 9, which activates Caspase 3 to induce apoptosis [14].
However, tumor cells are very smart. They have developed some ways to avoid the protection of p53. There are main two ways including mutating p53 gene and inhibiting p53's function by its antagonist to inactivate p53 in tumor cells. Studies have reported that p53 is one of the most frequently mutated genes in human cancer, and p53 mutations occurs in ~50% of tumor cells [15]. In the absence of p53 mutation in tumors, the function of wild type (wt)-p53 protein is frequently inhibited by its negative regulators, such as E3 ubiquitin-protein ligase Mdm2 (MDM2) and protein Mdm4 (MDMX) [16]. MDM2 directly binds to p53 and inhibits its transcriptional activity, causes ubiquitination and proteasomal degradation of p53, and exports p53 out of the nucleus [16,17]. MDMX is a homolog of MDM2, which is encoded by the MDM4 gene. MDMX also directly binds to the transactivation domain of p53 and inhibits its activity, but does not cause the degradation of p53 [16,18]. Inactivation of p53 contributes to some functions that are helpful for tumor cell growth, including resistance to apoptosis and chemotherapy or losing the ability to repair damaged DNA to induce genomic instability [19].
In the present study, the effect of chemotherapy on the treatment of PTC was investigated in vitro and it was found that PTC cells were not sensitive to the chemotherapeutic drug, paclitaxel (PTX). However, using recombinant adenovirus-p53 (rAd-p53), a recombinant replication-incompetent human serotype 5 adenovirus, in which the E1 region is replaced by a human wild type-p53 expression [20], could significantly increase the sensitivity of PTC cells to PTX. Thus, the present study illustrated the molecular mechanism of antitumor action of rAd-p53 combined with PTX in PTC cells.
Materials and methods
Materials
PTX, dimethyl sulfoxide (DMSO), XTT, propidium iodide (PI), Hoechst 33258, and the anti-β-actin antibody were purchased from Sigma-Aldrich (Merck KGaA); anti-p53, and anti-cleaved Caspase-3 antibodies were obtained from Abcam; anti-CDK4, anti-CDK6, anti-Cyclin D1, anti-Cyclin E1, anti-serine-protein kinase ATM (ATM), anti-Chk2, anti-phosphorylated Chk2 (p-Chk2), anti-phosphorylated H2A histone family member X (γ-H2AX), anti-H2AX, anti-cleaved poly(ADP-ribose)polymerase 1 (PARP-1), anti-cytochrome C, anti-BAX, anti-BCL-2 and anti-BCL-XL were obtained from Cell Signaling Technology, Inc.; and the adenoviral vector [recombinant human p53 adenovirus injection, rAd-p53 (Gendicine™)] was purchased from Shenzhen Sibiono GeneTech Co., Ltd.
Cell culture
The human PTC cell lines, TPC-1 and BCPAP, were obtained from The Cell Bank of Type Culture Collection of The Chinese Academy of Sciences. TPC-1 and BCPAP cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 50 U/ml penicillin and streptomycin. Human hepatocellular carcinoma cells (HepG2) were obtained from the American Type Culture Collection and cultured in DMEM supplemented with 10% FBS and 50 U/ml penicillin and streptomycin. The cells all had authenticated using STR profiling.
Cell proliferation assay
Cancer cells (1 × 104) were cultured in 96-well plates overnight, and then treated with different concentrations of PTX (0.625, 1.25, 2.5, 5, 10, and 20 μM) and/or rAd-p53 [1010 viral particles (VP)] for 48 h. XTT solution (20 μl of 3.3 mM) was added into each well and incubated at 37°C for 1-2 h. The absorbance was determined in each well at 450 nm with the SpectraMax M5 Microtiter Plate Luminometer (Molecular Devices, LLC). The proliferation of the treated cells was compared with that of the control cells, which were arbitrarily assigned 100% viability.
Flow cytometry
After treatment with different concentrations of PTX, the PTC and HepG2 cells were fixed in 70% ethanol for ≥24 h and stained with PI for 30 min. Then, after washing with 1X PBS, the stained cells were analyzed using a CytoFlex flow cytometer (Beckman Coulter, Inc.) to determine the cell cycle distribution. To determine apoptosis, control or PTX-treated cells were stained with Annexin V-FITC (green) and PI (red) for 30 min and analyzed with the flow cytometer. The percentage of apoptotic cells (Annexin V+/PI-) was determined using flow cytometry.
Western blotting
Cells from various treatment groups were lysed in lysis buffer (Beyotime Institute of Biotechnology) with 1 mM phenylmethylsulfonyl fluoride on ice for 30 min. Then, the cell lysates were centrifuged at 12,000 x g for 15 min and the protein lysate was quantified using the Bio-Rad DC™ Protein assay (Bio-Rad Laboratories, Inc.). An equal amount of proteins (30 μg) was separated via SDS-PAGE on 6%-12% gels at 80 mV for 2 h. The separated proteins were transferred onto a polyvinylidene difluoride membrane at 100 mV for 1 h. The blots were then blocked with 5% milk and incubated with primary antibodies overnight at 4oC. Next, following washing with TBS with Tween-20, the blots were incubated with secondary antibodies for 1 h at room temperature. The blots were developed with enhanced chemiluminescence (EMD Millipore) and the protein bands were imaged and semi-quantified with Image Lab Software (Bio-Rad Laboratories, Inc.).
Immunofluorescence
Cells were cultured on poly-L-lysine-coated glass coverslips and treated with different drugs for 24 h. The cells were fixed with 4% formaldehyde for 10 min, followed by incubation with the blocking buffer (1X PBS containing 3% bovine serum albumin, 2% goat serum, and 0.2% Triton X-100) for 1 h. Then, the fixed cells were incubated overnight with a primary antibody against γ-H2AX (1:100; cat. no. 9718; Cell Signaling Technology, Inc.) at 4oC. Next, they were incubated with Alexa Fluor-conjugated secondary antibody (Alexa Fluor® 568; 1:800; Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. Then, the cells were stained with the nuclear dye, Hoechst 33258. Images were obtained using an inverted fluorescence microscope.
In vivo PTC tumor xenograft
TPC-1 cells (5x106 in 100 μl saline) were subcutaneously injected into the right flanks of 6-week-old female BALB/cA-nu/nu mice (Beijing HFK Bioscience Co., Ltd.). When the tumor size reached 100 mm3, the mice were randomly divided into the following groups (n = 6/group): i) Vehicle control group, mice were intratumorally injected with 100 μl adenovirus vector/100 mm3 every 3 days; ii) rAd-p53 group, mice were intratumorally injected with 1010 VP rAd-p53/100 mm3 every 3 days; iii) PTX group, mice were intraperitoneally injected with 30 mg/kg PTX in 200 μl saline every 5 days; and iv) rAd-p53 + PTX group, mice were intratumorally injected with 1010 VP rAd-p53/100 mm3 every 3 days and intraperitoneally injected with 30 mg/kg PTX in 200 μl saline every 5 days. Tumor volume was measured every 3 days with a caliper as π/6 x length x width x width (mm3). The experiment was carried out over 30 days.
Histopathological analysis
Tumor tissues were fixed in 4% neutral-buffered formalin for 24 h and embedded in paraffin. Then, 3-5-μm thick sections were cut on a microtome. A TUNEL assay was performed with the In Situ Cell Death Detection kit (Roche Applied Science), according to the manufacturer's instructions, to determine the effect of different treatments on tumor cell apoptosis. Immunohistochemistry was performed by incubating the tumor sections with mouse anti-p53 primary antibody (1:50; cat. no. ab1101; Abcam), followed by HRP-conjugated secondary antibodies. Then, the slides were developed with DAB.
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM) and were analyzed by using the Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
PTC cells were resistant to PTX
Chemotherapy is a systemic therapy and is helpful for refractory solid tumors, and thus is a possible treatment for refractory PTC, including metastatic disease and radioiodine refractory diseases. However, studies have reported PTC resistance to chemotherapy. Therefore, the effect of chemotherapy on PTC was first determined in the present study.
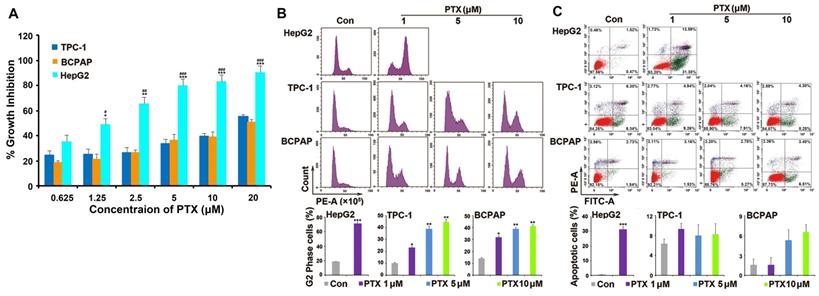
PTX is widely used as a broad-spectrum antitumor drug. Two PTC cell lines (TPC-1 and BCPAP) were chosen to investigate the sensitivity of PTC cells to PTX. One drug-sensitive tumor cell line (HepG2) was used as positive control cells. Compared with the response to PTX in HepG2 cells, TPC-1 and BCPAP cells were resistant to PTX. PTX inhibited the proliferation of HepG2 cells after treatment for 48 h, with a 50% inhibitory rate (IC50) of 1.25 μM. However, the concentration of PTX was 20 μM in TPC-1 and BCPAP cells (Fig. 1A).
PTC cells were resistant to PTX. (A) XTT assay analyzed the effect of PTX on the proliferation in tumor cells. PTC cells (TPC-1 and BCPAP) and HepG2 cells were treated with different concentrations (0.625, 1.25, 2.5, 5, 10, and 20 μM) of PTX. An XTT assay was performed after 48 h to determine the relative proliferation rates and the IC50 values were calculated for PTX in the three cell types. *P<0.05, **P<0.01 and ***P<0.001, TPC-1 compared with HepG2; #P<0.05, ##P<0.01 and ###P<0.001, BCPAP compared with HepG2. Flow cytometry analysis of (B) the cell cycle distributions and (C) apoptosis in TPC-1 and BCPAP cells treated with PTX. TPC-1 and BCPAP cells were treated with different concentrations of PTX (1, 5, and 10 μM), whereas HepG2 cells were treated with 1 μM PTX for 24 h and 48 h, respectively. Flow cytometry analysis of PI-stained cells was performed to determine the cell cycle distribution. Apoptosis was detected by flow cytometry staining with Annexin V-FITC (green) and PI (red). The percentage of apoptotic cells (Annexin V+/PI-) was determined using flow cytometry. *P<0.05, **P<0.01 and ***P<0.001, compared with control.
PTX did not induce apoptosis in PTC cells
Then, the underlying mechanism of drug resistance to PTX in PTC cells was investigated. As a microtubule polymerization enhancer, PTX performs antitumor activity by inducing G2/M phase accumulation and apoptosis in tumor cells [21]. Because HepG2 cells were much more sensitive than PTC cells to PTX, we found that 1 μM PTX induced significant G2/M phase accumulation after treatment for 24 h and apoptosis following treatment for 48 h in HepG2 cells. However, different concentrations (1, 5 and 10 μM) of PTX induced G2/M phase accumulation after 24 h of treatment (Fig. 1B), they could not induce apoptosis after treatment for 48 h in TPC-1 and BCPAP cells (Fig. 1C). Moreover, 1, 5 and 10 μM PTX induced G2/M phase accumulation at 48 h in the two PTC cells (Fig. S1). This suggested that the mechanism of PTC cells resistance to PTX was associated with the inability of PTX to induce apoptosis of PTC cells.
rAd-p53 increased the sensitivity of PTC cells to PTX
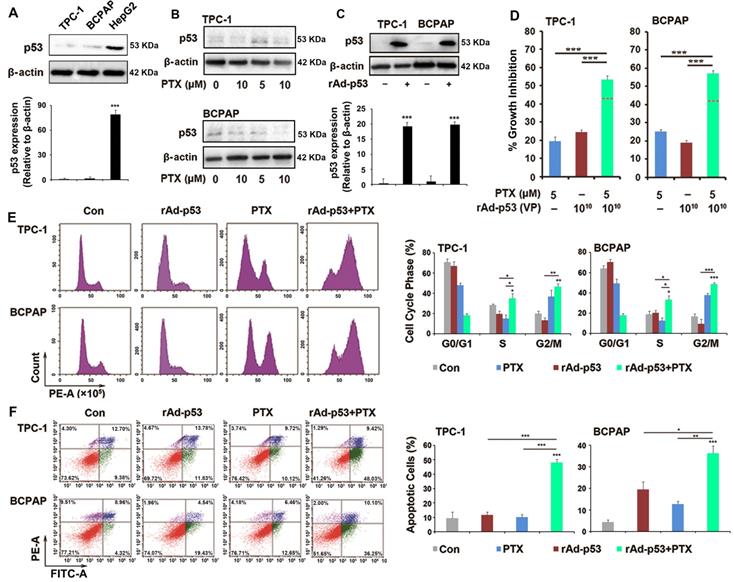
Previous studies have reported that the inactivation of p53 plays an important role in drug resistance and apoptosis in various tumors types [19,22]. When the status of the P53 gene was previously determined in TPC-1 and BCPAP cells, it was found that wt-p53 could be detected in TPC-1 cells and p53 mutant could be detected in BCPAP cells [23]. However, the western blotting results of the present study showed that p53 expression was low in TPC-1 and BCPAP cells (Fig. 2A) and PTX could not increase the expression of p53 in the two PTC cells (Fig. 2B). rAd-p53 was used to increase the expression of wt-p53 in tumor cells (Fig. 2C). Because 0.1% DMSO and adenovirus vector both had no interfering effect in PTC cells in preliminary experiment (Fig. S2), just adenovirus vector-treated group was used as control group in next studies. We had done the preliminary experiments to make sure that 1010 VP was the most appropriate concentration of rAd-p53 (Table S1). 1010 VP rAd-p53 markedly increased the sensitivity of PTC cells to PTX (Fig. 2D). 1010 VP rAd-p53 + 5 μM PTX led to >50% inhibition of cell proliferation (54.02 ± 0.43% and 57.72 ± 0.38%) of TPC-1 and BCPAP cells, while 5 μM PTX treatment alone caused ~20% inhibition of cell proliferation in the two PTC cell lines (Fig. 2D). 1010 VP rAd-p53 combined with 5 μM PTX or 10 μM PTX showed sensitization in TPC-1 and BCPAP cells (the predicted additivity is shown with the dotted line in Fig. 2D).
rAd-p53 increased the sensitivity of PTC cells to PTX. (A-C) Western blotting was performed to analyze the expression of p53 protein in PTC cells. ***P<0.001. (D) rAd-p53 enhanced the antitumor activity of PTX in TPC-1 and BCPAP cells. TPC-1 and BCPAP cells were treated with rAd-p53, PTX and rAd-p53 + PTX for 48 h. The dotted line denotes the predicted additivity [(eA + eB) - (eA x eB)] (eA, the inhibitory rate of A; eB, the inhibitory rate of B). ***P<0.001, compared with control. Flow cytometry was performed to determine (E) cell cycle distributions (cells stained with PI) and (F) the percentage of apoptotic cells (cells stained with Annexin V/PI) in TPC-1 and BCPAP cells. Cells were treated with vehicle, 1010 VP rAd-p53, 5 μM PTX or 1010 VP rAd-p53 + 5 μM PTX for 24 h or 48 h. *P<0.05, **P<0.01 and ***P<0.001, compared with control.
Combined rAd-p53 and PTX treatment induced cell cycle arrest and apoptosis
Because 5 and 10 μM of PTX combined with 1010VP rAd-p53 revealed no significant difference (Table S2), we therefore chose the small dose (5 μM) of PTX to combine with rAd-p53 in current study. Cell cycle analysis showed that rAd-p53 + PTX not only increased the number of cells in G2/M phase accumulation, but also induced S phase accumulation in TPC-1and BCPAP cells (Fig. 2E). PTC cells treated with rAd-p53 + PTX had a higher percentage of S phase cells (TPC-1 cells, 35.22 ± 4.52% and BCPAP cells, 33.24 ± 4.12%) than those treated with rAd-p53 alone (19.67 ± 2.97% and 20.21 ± 2.61%; P<0.05) and PTX alone (14.99 ± 3.58% and 12.58 ± 3.12%; P<0.05) (Fig. 2E). Furthermore, it was observed that rAd-p53 + PTX induced obvious apoptosis in TPC-1 and BCPAP cells. The number of apoptotic cells was 48.03 ± 2.45% for TPC-1 and 36.25 ± 3.39% for BCPAP cells, which was higher than the control (9.38 ± 4.48% and 4.32 ± 1.09%; P < 0.001), rAd-p53-treated alone (11.83 ± 1.99% and 19.43 ± 3.62%; P<0.05), and PTX-treated (10.12 ± 1.78% and 12.66 ± 1.39%; P<0.01) groups (Fig. 2F).
Combined rAd-p53 and PTX treatment induced S phase accumulation and apoptosis in a p53-dependent pathway response to DNA damage
Then, the signaling pathways that induce the S phase cell cycle arrest and apoptosis-induced by rAd-p53 + PTX in PTC cells were analyzed. Cyclins and Cyclin-dependent kinase (CDK) are two main types of proteins which involved in regulation of cell cycle progression. CDK4 and -6, which along with their Cyclins (Cyclin D1 and Cyclin E1 belong to the G1 cyclins), regulate the passage of cells through the G1 phase and their entry into the S phase [24,25]. Western blotting showed that rAd-p53 + PTX decreased the expression levels of cyclin D1 and -E1 protein (Fig. 3A), but had no effect on the expression of CDK 4 and -6 proteins (Fig. S3) in the PTC cells. As a main transcriptional regulator, p53 could modulates the cell cycle progression through regulating p21 expression. p21 inhibits cyclin E1-CDK2 and cyclin D1-CDK4,6 complexes and induces cells G1 and S phases accumulation [26-28]. p21 protein expression was notably increased in the rAd-p53 + PTX-treated TPC-1 and BCPAP cells (Fig. 3B). These results demonstrated that rAd-p53 + PTX treatment induced S phase accumulation in PTC cells by activating the p53/p21 pathway, which resulted in decreased expression levels of cyclin D1 and E1.
rAd-p53 + PTX induced S arrest in a p53-dependent pathway in response to DNA damage in PTC cells. (A-C) Representative western blotting images show the expression of (A) S phase cell cycle-related proteins, (B) p21 and (C) DNA damage-related proteins in TPC-1 and BCPAP cells. PTC cells were treated with vehicle, 1010 VP rAd-p53, 5 μM PTX, or 1010 VP rAd-p53 + 5 μM PTX for 24 h. *P<0.05; **P<0.01 and ***P<0.001, compared with control. (D) Representative immunofluorescence images (magnification, x200) show γ-H2AX-stained TPC-1 and BCPAP cells. PTC cells were treated with vehicle, 1010 VP rAd-p53, 5 μM PTX, or 1010 VP rAd-p53 + 5 μM PTX for 24 h. Nuclei were stained with Hoechst 33258.
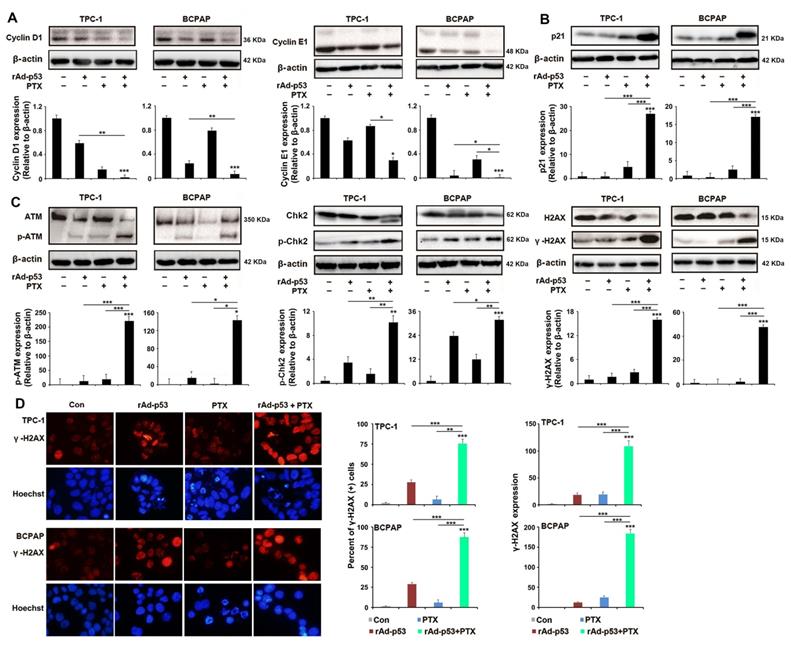
In most mammalian cells, cell cycle arrest is always induced by DNA damage in response to cellular stress [25]. Therefore, the status of the DNA damage response in PTC cells was analyzed in the present study. Western blotting analysis showed that rAd-p53 + PTX increased the expression levels of phosphorylated-ATM, phosphorylated-Chk2 and γ-H2AX proteins in PTC cells (Fig. 3C). Immunofluorescence analysis confirmed the highest level of γ-H2AX in the rAd-p53 + PTX-treated PTC cells (Fig. 3D). This suggested that rAd-p53 + PTX activated the DNA damage signaling pathway in PTC cells.
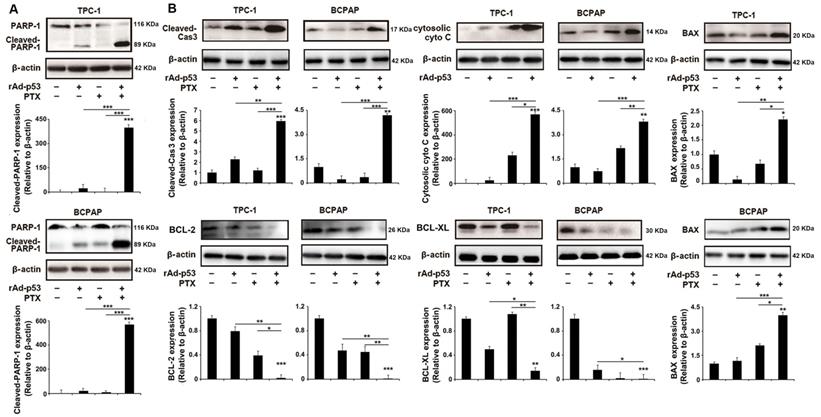
When DNA damage is irreparable, mammalian cells undergo programmed cell death via the apoptotic pathway [29]. PARP-1, as a critical DNA damage repair protein, is cleaved by cleaved Caspase 3-during DNA damage-induced apoptosis [11,30]. As shown in Fig. 4A, the level of cleaved PARP-1 was notably increased in rAd-p53 + PTX-treated PTC cells, suggesting that rAd-p53 + PTX promoted DNA damage-induced apoptosis in PTC cells. Western blotting analysis showed that rAd-p53 + PTX did increase the level of cleaved Caspase 3 in PTC cells (Fig. 4B). As one of the critical determinants of cell survival in response to DNA damage, p53 activates the expression of downstream genes such as BCL-2 family proteins to activate mitochondrial apoptotic pathway [14,31]. BCL-2 and BCL-XL are apoptotic inhibitors that decreases the accumulation of cytosolic cytochrome C, which activates caspase 3 in DNA damage-induced apoptosis [12]. In the present study the expression of these apoptosis-related proteins was measured in PTC cells. The results showed that rAd-p53 + PTX increased the levels of cytosolic cytochrome C and BAX proteins and decreased the levels of BCL-2 and BCL-XL proteins in PTC cells (Fig. 4B).
rAd-p53 + PTX induced DNA damage-induced apoptosis in PTC cells. Western blotting was performed to analyze the levels of (A) cleaved PARP-1 and (B) apoptotic pathway-related proteins in TPC-1 and BCPAP cells. PTC cells were treated with vehicle, 1010 VP rAd-p53, 5 μM PTX, or 1010 VP rAd-p53 + 5 μM PTX for 48 h. *P<0.05, **P<0.01 and ***P<0.001, compared with control.
Combined rAd-p53 and PTX treatment inhibited tumor growth of TPC-1 xenograft tumors in nude mice
A xenograft mouse model was generated to further investigate the inhibitory effect of rAd-p53 + PTX on tumor growth in PTC. The results showed that rAd-p53 + PTX significantly inhibited tumor growth in a TPC-1 xenograft model. The tumor volume in the rAd-p53 + PTX treatment group was the lowest (265.91 ± 9.24 mm3) of all treatment groups [compared with vehicle control group (1257.27 ± 134.29 mm3), P<0.001; compared with rAd-p53-treated group (779.16 ± 123.51 mm3), P = 0.005; compared with PTX-treated group (1026.63 ± 122.29 mm3), P<0.001] (Fig. 5A). The tumor inhibitory rates of the rAd-p53, PTX, and rAd-p53 + PTX treatment groups relative to the vehicle control group were 40.30, 17.45, and 79.39%, respectively (Fig. 5B and C).
Wt-p53 overexpression enhanced antitumor activity of PTX in the TPC-1 xenograft mouse model. (A) TPC-1 xenograft tumor volume curves for mice in the control, rAd-p53, PTX, and rAd-p53 + PTX groups (n = 6/group). (B) Bar chart showing the tumor weights in the control, rAd-p53, PTX, and rAd-p53 + PTX groups (n = 6/group). **P<0.01 and ***P<0.001. (C) Bar chart showing the inhibitory rate of tumor development in the rAd-p53, PTX and rAd-p53 + PTX groups mice relative to the control group. The dotted line denotes predicted additivity [(eA + eB) - (eA x eB)]. (D) Representative images (magnification, x100) of hematoxylin & eosin (H&E)- and TUNEL-stained tumor sections showing tumor necrosis and apoptosis. Immunohistochemical staining (magnification, x100) of p53 proteins in tumor sections belonging to the four groups. **P<0.01 and ***P<0.001, compared with control.
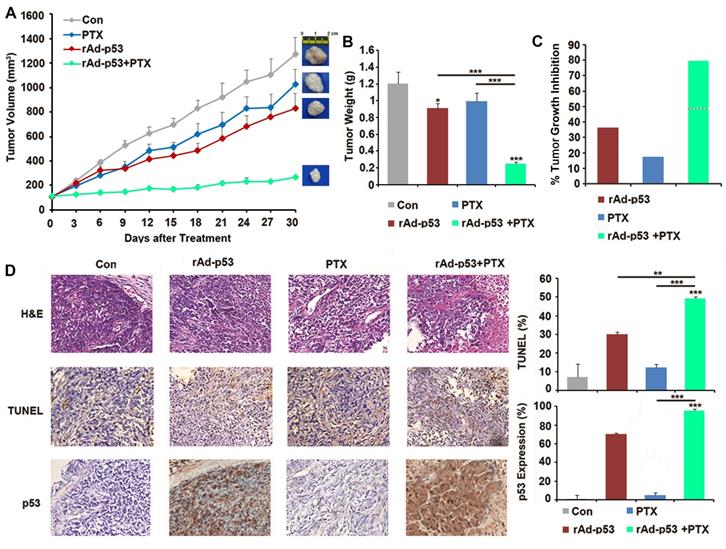
The results of the TUNEL assay demonstrated that rAd-p53 + PTX treatment induced apoptosis in the xenograft tumors (Fig. 5D). Immunohistochemical analysis revealed that there was no expression of p53 protein in the control group, low expression in PTX-treated tumors, and high expression in rAd-p53-treated and rAd-p53 + PTX-treated tumors (Fig. 5D).
The results of in vivo experiment indicated that PTX treatment alone could not inhibit tumor growth, but rAd-p53 + PTX significantly inhibited tumor growth in PTC, which indicated that p53 activation was necessary for the antitumor activity of PTX in PTC.
Discussion
Refractory types of PTC, including radioiodine refractory diseases, have poor prognosis rates due to the limitation of traditional treatment approaches [2,3]. Chemotherapy could be a potential strategy for these cases. However, various questions still need answering, for example what is the nature of PTC tumors? Are there any drug-sensitive pathways in PTC tumors? Thus, studying the pathology and pharmacology of PTC tumors is of great significance.
Apoptotic pathways are important for cellular activities and functions in tumor cells, and resistance to apoptosis may be a contributing factor in the development of chemoresistance in tumor cells [32]. A previous study reported that chemotherapeutic drugs that target apoptotic pathways would make tumor cells sensitive to chemotherapy and radiotherapy [33]. In the present study, the flow cytometry results showed that PTX only induced G2/M phase arrest, but not apoptosis in PTC cells. Under this condition, 10 μM PTX could not inhibit proliferation in 50% of PTC cells. However, treatment with rAd-p53 + 5 μM PTX induced obvious apoptosis in PTC cells, while they notably inhibited cells proliferation, with an inhibitory rate of >50%. rAd-p53 also significantly increased the antitumor activity of PTX in vivo by inducing tumor apoptosis. The results confirmed that resistance to apoptosis mediated PTC cells resistance to PTX.
Next, which signaling pathway or regulators were inactivated in the PTX-induced apoptotic pathway in PTC cells? Studies have reported that inactivation of p53 might contribute to resistance to cancer drugs and inhibit apoptosis of tumor cells [19]. In the present study, the status of the P53 gene was found to be the wt-p53 in TPC-1 cells and p53 mutant in BCPAP cells. Compared with in p53-inactivated PTC cells whereby PTX induced G2/M arrest by targeting the microtubule polymerization, PTX induced obvious S arrest and apoptosis in p53-activated PTC cells (Fig. 2E,F), suggesting that activation of p53 played an important role in the mechanism of antitumor action of PTX in PTC, and PTX induced S phase arrest and apoptosis in a p53-dependent pathway in PTC cells. In PTC cells, treatment with rAd-p53 increased the expression of wt-p53 protein. rAd-p53 + PTX activated p21 to negatively regulate Cyclin D1/E1 to induce S phase arrest in response to DNA damage within 24 h and regulated the expression of BAX, BCL-2 and BCL-XL to mediate the DNA damage-induced apoptosis after 48 h of treatment.
In summary, the results in the present study showed that p53-dependent pathway was necessary for the inhibition of PTX in PTC cells. The result that rAd-p53 + PTX significantly inhibited PTC tumor in vitro and in vivo through p53-dependent apoptosis response to DNA damage suggested activation of p53 had a great meaning for the clinical management and treatment of PTC and rAd-p53 combined with chemotherapy, which might be used in the treatment of advanced-stage PTC patients with tumors unsuitable for surgery.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
This project was supported by the Foundation of National Natural Science Foundation of China (81702977), the Sichuan Science and Technology Program (2021YFS0230 and 2021YFS0228), the Sichuan University West China Hospital Postdoctoral Science Foundation (2018HXBH016), the Foundation of Sichuan University (2018SCUH0067), the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZY2017309).
Author Contributions
WW and TW contributed equally to this work. JZ conceived and designed the experiments. WW, TW, and ZL performed the majority of the experiments and analyzed the data. WW, TW and JZ drafted the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Fagin JA, Wells SA Jr. Biologic and clinical perspectives on thyroid cancer. N Engl J Med. 2016;375:1054-1067
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30
4. Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP. et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91(8):2892-2899
5. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359-370
6. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413-431
7. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16(1):20-33
8. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499-506
9. Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL. et al. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013;3(5):1339-1345
10. Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32:3789-3797
11. Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM. et al. Apoptosis-inducing factor mediates poly (ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103:18314-18319
12. Kharbanda S. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:6939-6942
13. Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21(1):92-101
14. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43:95-118
15. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89-102
16. Sanz G, Singh M, Peuget S, Selivanova G. Inhibition of p53 inhibitors: progress, challenges and perspectives. J Mol Cell Biol. 2019;11(7):586-599
17. Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res. 2013;19:34-41
18. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83-96
19. Yamamoto S, Iwakuma T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers (Basel). 2018 11(1)pii: E4
20. Li Y, Li B, Li CJ. et al. Key points of basic theories and clinical practice in rAd-p53 (Gendicine™) gene therapy for solid malignant tumors. Expert Opin Biol Ther. 2015;15:437-454 2015
21. Yang CH, Horwitz SB. Taxol®: The First Microtubule Stabilizing Agent. Int J Mol Sci. 2017 18(8) pii:E1733
22. Ye S, Shen J, Choy E, Yang C, Mankin H, Hornicek F, et al p53 overexpression increases chemosensitivity in multidrug-resistant osteosarcoma cell lines. Cancer Chemother Pharmaco. 2016;l77(2):349-356.
23. Saiselet M, Floor S, Tarabichi M, Dom G, Hébrant A, van Staveren WC. et al. Thyroid cancer cell lines: an overview. Front Endocrinol (Lausanne). 2012;3:133
24. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nature reviews Clinical oncology. 2016;13:417-30
25. Monteiro LS, Diniz-Freitas M, Warnakulasuriya S, Garcia-Caballero T, Forteza-Vila J, Fraga M. Prognostic Significance of Cyclins A2, B1, D1, and E1 and CCND1 Numerical Aberrations in Oral Squamous Cell Carcinomas. Anal Cell Pathol (Amst). 2018;2018:7253510
26. Samar Al Bitar, Hala Gali-Muhtasib. The Role of the Cyclin Dependent Kinase Inhibitor p21 cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers (Basel). 2019;11(10):1475
27. Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518-528
28. Georgakilas AG, Martin OA, Bonner WM. p21: A Two-Faced Genome Guardian. Trends Mol Med. 2017;23:310-319
29. Strzyz P. Cell thriving despite DNA damage. Nat Rev Mol Cell Bio. 2016;17(7):396
30. Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodeling. Nat Rev Mol Cell Biol. 2017;18(10):610-621
31. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071-1078
32. Blagosklonny MV. Prospective strategies to enforce selectively cell death in cancer cells. Oncogene. 2004;23:2967-2975 2004
33. Wilson TR, Johnston PG, Longley DB. Anti-Apoptotic Mechanisms of Drug Resistance in Cancer. Curr Cancer Drug Target. 2009;9:307-319
Author contact
![]() Corresponding author: Dr Jingqiang Zhu, Department of Thyroid Surgery, West China Hospital, Sichuan University, Chengdu, 610041, China. E-mail: zhujingqiangedu.cn.
Corresponding author: Dr Jingqiang Zhu, Department of Thyroid Surgery, West China Hospital, Sichuan University, Chengdu, 610041, China. E-mail: zhujingqiangedu.cn.