Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(13):1936-1945. doi:10.7150/ijms.44330 This issue Cite
Research Paper
Inhibiting Caspase-12 Mediated Inflammasome Activation protects against Oxygen-Glucose Deprivation Injury in Primary Astrocytes
Lu Liu#, Manli Chen#, Kun Lin, Xuwu Xiang, Yueying Zheng ![]() , Shengmei Zhu
, Shengmei Zhu ![]()
Department of Anesthesiology, The First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, People's Republic of China.
#Co-first authors.
Received 2020-1-28; Accepted 2020-7-2; Published 2020-7-19
Abstract
Stroke is one of the leading causes of death worldwide. Accumulating evidence suggests that NLRP3 inflammasome activation plays an important role in ischemic stroke injury. However, the existence of the NLRP3 inflammasome in astrocytes remains controversial. In this study, we demonstrated the presence of the NLRP3 inflammasome in primary mouse astrocytes and investigated the role of caspase-12 in NLRP3 inflammasome activation and cell injury in an in vitro astrocyte oxygen-glucose deprivation (OGD) model. Astrocytes exposed to 2, 3, and 4 h of OGD exhibited increased cell injury and apoptosis, and the protein levels of caspase-12, cleaved caspase-3, NLRP3 inflammasome components, and IL-1β were also significantly elevated. Interestingly, pretreatment with the caspase-12-specific inhibitor Z-ATAD-FMK attenuated cell injury and apoptosis and decreased the levels of NLRP3, caspase-1, IL-1β, and cleaved caspase-3 in the OGD group. In conclusion, Z-ATAD-FMK protected astrocytes against OGD-induced cell death and inhibited NLPR3-inflammasome activation. Our results indicate that caspase-12 and its potential regulation of NLRP3 inflammasome activation might be a promising target for treatment of ischemic stroke.
Keywords: stroke, ischemia reperfusion injury, astrocytes, caspase-12, NLRP3-inflmmasome
Introduction
Ischemic stroke is one of the leading causes of death and serious long-term disability with prolonged physical, emotional, social, and financial consequences. A total of 113 million disability-adjusted life years (DALYs) are due to stroke; moreover, 10.3 million new strokes occur every year, of which 58% of DALYs and 67% of new cases are ischemic strokes [1]. There are several therapeutic options for ischemic stroke, including thrombolytic therapy, intravenous alteplase, intravascular thrombectomy, and neuroprotective drugs. However, the effectiveness of these treatments is limited in clinical practice due to narrow therapeutic windows [2]. Therefore, alternative effective medical interventions are urgently needed.
Astrocytes, as major glial cells in the central nervous system (CNS), play an important role in maintaining normal neuronal function, including regulation of cerebral blood flow, extracellular fluid, ion homeostasis, ion transportation, energy supply, synaptic function, and synaptic remodeling [3]. In ischemic stroke, accumulating evidence suggests that astrocytes play fundamental roles in the pathogenesis of ischemic neuronal death; however, the detailed mechanisms remain unclear. Importantly, endoplasmic reticulum stress (ERS) is particularly damaging to ischemic brains and was found to play a role in the pathophysiology of hypoxia/ischemia-induced neuronal death [4]. ERS can initiate the activation of caspase-12. Caspase-12 activation and processing have been detected following ischemic stroke, but the role of caspase-12 in apoptosis and inflammation is controversial. Experiments have shown that caspase-12 is responsible for further activation of caspase-3 and ERS-induced cell apoptosis in astrocytes following ischemic stroke [5-7]. Caspase-12 is classified in the group I family of caspases, which are characterized as inflammatory caspases [8, 9]. Caspase-12 has been shown to induce apoptosis in different cell and animal models; extensive cellular crosstalk may be involved [10-12]. However, there is limited evidence of other downstream mechanisms of caspase-12 in astrocytes subjected to ischemia/reperfusion (I/R) injury. Studies have focused on the relationship between caspase-12 and inflammation in an effort to understand the underlying mechanism. At present, it is believed that caspase-12 negatively modulates the innate inflammatory response as well as inflammasome activation and caspase-1 activity [8, 13]. However, Saleh et al. have challenged this opinion, as they failed to see increased caspase-1 activation in caspase-12-deficient mice in a recent study [14]. Moreover, irremediable ERS after I/R injury is associated with nucleotide-binding domain leucine-rich repeat containing receptor pyrin domain containing 3 (NLRP3) inflammasome activation [15]. The NLRP3 inflammasome has been reported to be involved in the pathophysiology of ischemic stroke [16]. Nevertheless, the existence of the NLRP3 inflammasome in primary mouse astrocytes remains controversial [17, 18].
To date, the relationship between caspase-12 and the NLRP3 inflammasome in ischemic stroke is unclear. The aims of this study were to investigate the existence of the NLRP3 inflammasome in primary mouse astrocytes, as well as the effect of caspase-12 on apoptosis, and to explore the molecular mechanisms of the caspase-12/NLRP3 inflammasome pathway.
Materials and Methods
Primary mouse astrocyte cultures
Primary astrocytes were separated from cerebral cortices of postnatal (0-1 day old) male and female C57BL/6 mice, as described previously [19, 20]. The astrocytes were cultured in complete medium composed of high-glucose Dulbecco's modified Eagle medium (DMEM; 4.5 g/l D-glucose, HyClone) supplemented with 10% fetal bovine serum (BI, #04-001-1A) and 1% penicillin/streptomycin. Cells were plated in T-75 flasks (Costar; Corning) at a density of three cortices per flask precoated with poly-D-lysine (10 µg/ml; Sigma-Aldrich). The cells were grown for at least 7 days at 37°C with 5% CO2, and the medium was changed every other day. After 8-10 days, confluent astrocytes were purified of microglia by shaking for 12-16 h at 250 rpm at 37°C on an orbital shaker followed by culture in medium containing 20 µM cytosine-1-β-D-arabinofuranosid (Sigma-Aldrich) for 2-3 days. Dissociated cells were then re-plated at a density of (0.03-0.05) × 106/cm2 in 6-well plates precoated with poly-D-lysine and then cu ltured at 37°C in a humidified incubator with 5% CO2. After 5-7 days, cultures at 90-95% confluence were used for subsequent experiments. Immunofluorescence (GFAP, 1:100, Cell Signaling Technology, #12389; IBA-1, 1:100, Novus, #NBP2-19019) and western blotting were used to test the purity of the astrocytes, which was found to be more than 95%.
Oxygen glucose deprivation and reperfusion
Oxygen-glucose deprivation (OGD) experiments were performed using a humidified incubator chamber at 37 °C with 95% nitrogen and 5% CO2, and the culture medium was replaced with DMEM without glucose. After 1-4 h of OGD, cultures were removed from the chamber and reperfused for 24 h with complete medium in a regular incubator. Control cell cultures were incubated in high-glucose DMEM under normoxia for the corresponding duration. Cell viability was measured using the lactate dehydrogenase (LDH) cytotoxicity assay.
Inhibition of caspase-12-mediated apoptosis using Z-ATAD-FMK
To verify if I/R-induced cell injury and apoptosis are mediated by caspase-12, primary mouse astrocytes were pretreated with the caspase-12 inhibitor Z-ATAD-FMK (ZAF; R&D; #FMK013). Briefly, primary mouse astrocytes were preincubated with 0, 0.05, 0.5, 5, or 10 μM ZAF 24 h before OGD. Then, the supernatant was replaced with normoxic medium, and the cells were incubated for another 30 min before OGD.
Lactate dehydrogenase cytotoxicity assay
The LDH detection kit is a colorimetric assay used to quantify cell death and cell lysis based on the release of LDH from the cytosol of damaged cells into the supernatant. The amount of dead or plasma membrane-damaged cells is reflected by the amount of LDH release in the culture supernatant. After 3 h of OGD and 24 h of reperfusion, the cell medium supernatant was collected and incubated with the reaction mixture provided in the LDH activity assay kit (Nanjing Jiancheng Bioengineering Institute). The absorbance was measured at a wavelength of 450 nm using a microplate reader (Molecular Devices SpectraMax i3x).
Immunofluorescence
Coverslips were placed in 6-well plates under normoxic conditions. The coverslips were washed three times with cold phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 min at room temperature, and washed three times with PBS. The coverslips were then incubated for 15 min with 0.1% Triton X-100 at room temperature followed by three washes with PBS. Then, 10% serum from the secondary antibody species was used to block the samples for 30 min at 37 °C followed by incubation with primary antibodies (GFAP, 1:200, Cell Signaling Technology, #12389 and #3670; IBA-1, 1:100, Novus, #NBP2-19019; NLRP3, 1:100, Cell Signaling Technology, #15101; caspase-1, 1:100, Novus, #NB-100-56565) overnight at 4 °C; cells were then washed three times with PBS. Then, fluorescent secondary antibodies (1:100, Proteintech, #SA00013-1, SA00013-2, SA00013-3, and SA00013-4) were added for 1 h. After three washes with PBS, 1 μg/ml DAPI (Cell Signaling Technology, #4083) solution was added to cover the cells for 5 min in the dark followed by three washes with PBS. Fluorescence images were captured using a confocal laser-scanning microscope (Nikon A1 Ti, 400× or 600× magnification).
Flow cytometry
Astrocyte apoptosis was measured by flow cytometry using a fluorescein isothiocyanate (FITC)-Annexin V/propidium iodide (PI) apoptosis detection kit (BD Biosciences, San Jose, CA, USA). Primary astrocytes cultured in 6-well plates were digested with 0.25% EDTA trypsin (Gibco). Cell precipitates were collected after centrifugation (1000 × g, 3 min) and washed with cold PBS. For Annexin V/PI analysis, cells were harvested and stained with 5 μl Annexin V and PI according to the manufacturer's instructions. The proportion of apoptotic cells was detected using a flow cytometer (BD Biosciences).
Western blotting analysis
Astrocytes were lysed with RIPA buffer at 4°C for 30 min. Lysates were centrifuged at 14,000 × g for 20 min at 4 °C. The supernatant was collected, and the protein concentration was measured using a BCA Protein Assay kit (Thermo Fisher Scientific). Each sample (30-40 μg) was loaded onto 12% gels, electrophoresed, and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA, #IPVH00010) at 4 °C. Non-fat milk (10%) was used to block the PVDF membranes for 1 h at room temperature. After blocking, the membranes were incubated with specific primary antibodies (caspase-12, 1:1000, Cell Signaling Technology, #2022; caspase-3, 1:1000, Abcam, #ab214430; NLRP3, 1:1000, Cell Signaling Technology, #15101; caspase-1, 1:1000, Novus, #NB-100-56565; IL-1β, 1:2000, Abcam, #ab9722; β-actin, 1:2000, Proteintech, #60008-1-Ig) overnight at 4 °C, followed by secondary antibodies (horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG) for 1 h at room temperature. Western blots were visualized using the Bio-Rad ChemiDocTM MP imaging system.
Statistics analysis
All experiments were performed at least in triplicate. The experimental data were analyzed by one-way ANOVA with post-hoc tests (LSD test and S-N-K test) using SPSS 23.0 software. Data are presented as means ± standard deviation (SD). The level of statistical significance was defined as P < 0.05.
Results
The NLRP3-inflammasome was expressed in primary mouse astrocytes
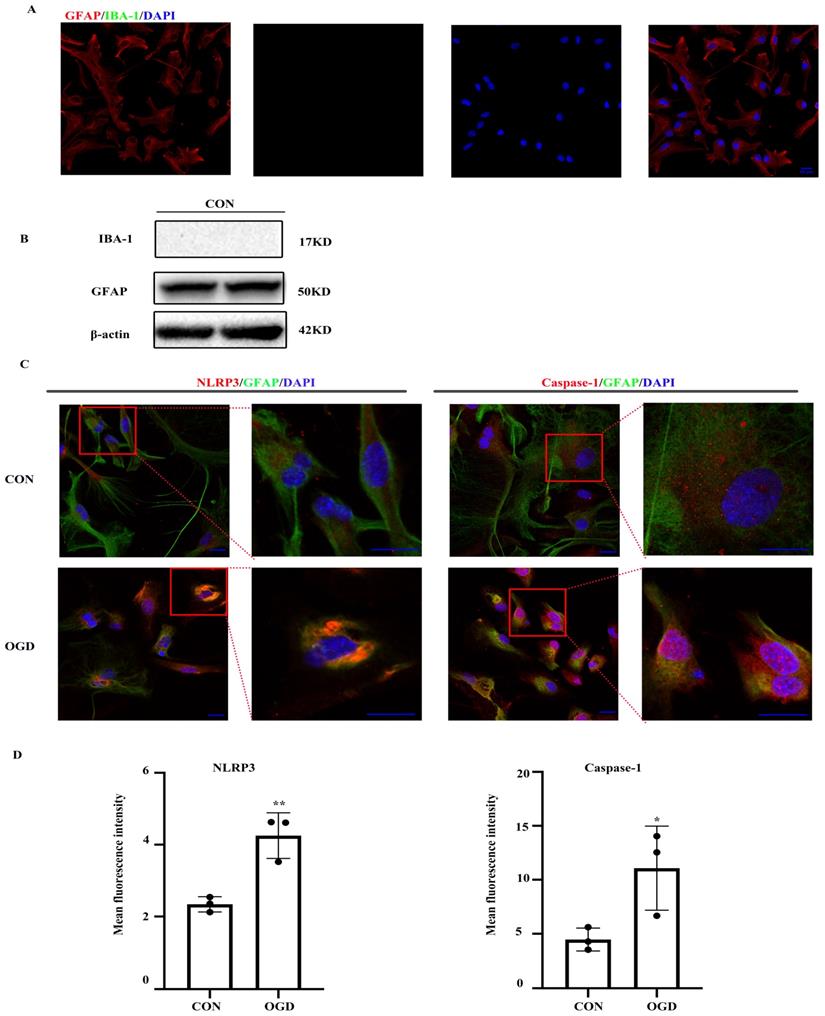
The NLRP3 inflammasome, which consists of NLRP3, the apoptosis-associated speck-like protein containing a caspase-activating recruitment domain adaptor, and procaspase-1, has been linked to a number of diseases including ischemic stroke. First, we ensured the purity of primary mouse astrocytes (> 95%) (Fig. 1A, B). We then determined the expression of NLRP3 inflammasome components by immunofluorescence. Both NLRP3 and caspase-1 expression was observed in primary astrocytes under normal and OGD conditions (Fig. 1C). As shown in Figure 1C and D, NLRP3 and caspase-1 expression was upregulated following OGD.
NLRP3-inflammasome was expressed in primary mouse astrocytes. NLRP3-inflammasome components were existed in primary mouse astrocytes. (A,B)The purity of primary mice astrocytes was detected by immunofluorescence and western blot (scale bar 20 µm, 400× magnification). (C) Immunofluorescence and was used to demonstrate the expression of NLRP3 and caspase-1 in primary mouse astrocytes in CON (control) group and OGD (3h hypoxia and 24h reperfusion) group (scale bar 20 µm, 600× or 1800x magnification). (D) Mean fluorescence intensity of NLRP3 and caspase-1 (comparisons to control group). Bar graphs were presented as means ± SD.*p<0.05, **p<0.01 (n=3).
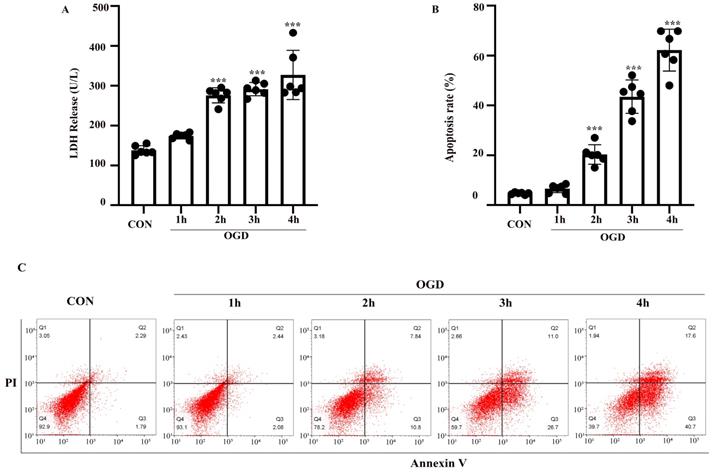
OGD induced cell injury and apoptosis in primary mouse astrocytes
The OGD model of astrocytes was established as described above. LDH and FITC-Annexin V/PI flow cytometry assays were performed to evaluate astrocyte injury and apoptosis. Astrocytes were exposed to 1-4 h of OGD and returned to normal conditions for 24 h. Compared with the control group, the rate of injury in astrocytes treated with OGD for 2-4 h was increased (P < 0.05) (Fig. 2A). Similarly, flow cytometry showed that apoptosis was increased after exposure to 2-4 h of OGD (Fig. 2B, C). The difference between the results shown in Fig. 2A and B might be due to the different properties of the two kits. The LDH release cytotoxicity assay measures membrane integrity, while flow cytometry was used to determine the rate of apoptosis. Therefore, astrocytes exposed to OGD for 3 h followed by 24 h of reoxygenation were used as the model for subsequent experiments.
OGD induced cell injury and apoptosis in primary mouse astrocytes. Astrocytes were exposed to OGD for 1 to 4 hours and then returned to normal culture condition for 24 hours. (A) Cell injury was detected by LDH release assay. (B) Cell apoptosis was detected by Annexin V- fluorescein isothiocyanate (FITC)/prodium iodide (PI) flow cytometry, and the apoptosis rate was presented as the percentage of apoptotic cells vs total cells. (C) Representative scatterplots of Annexin V-FITC/PI flow cytometry in each group. CON: control group; OGD: Oxygen Glucose Deprivation group. *P < 0.05, **P<0.01, ***P<0.001(all comparisons to control group). Bar graphs were presented as means ± SD. (n=6),
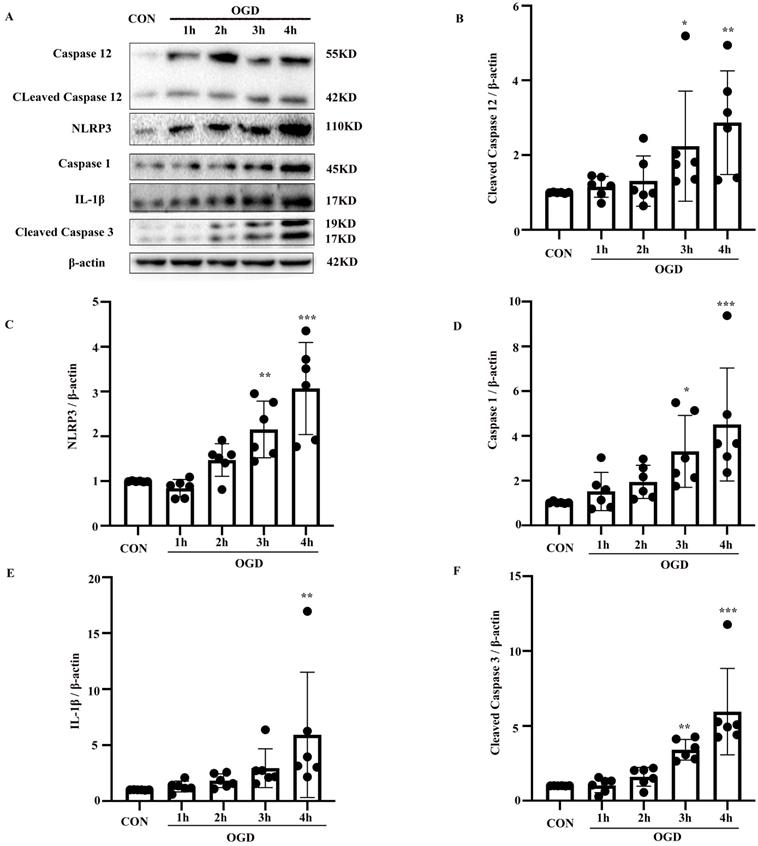
OGD increased the expression of NLRP3 inflammasome components, including NLRP3 and caspase-1, as well as caspase-12 and IL-1β expression in primary mouse astrocytes
To confirm the involvement of the NLRP3 inflammasome and caspase-12 in OGD-induced astrocyte apoptosis, NLRP3, caspase-1, IL-1β, and caspase-12, as well as the apoptotic biomarker cleaved caspase-3, were analyzed in primary mouse astrocytes subjected to OGD. Compared with the control group, the protein levels of cleaved caspase-3 and caspase-12 (P < 0.05) were elevated in the OGD model (P < 0.05) (Fig. 3A-F). IL-1β and NLRP3 inflammasome components including NLRP3 and caspase-1 were also increased in the OGD groups compared with the control group (P < 0.05). These results suggest that OGD increases caspase-12 and IL-1β expression and NLRP3 inflammasome activation.
OGD increased the expression of NLRP3 inflammasome components, including NLRP3 and caspase-1, as well as caspase-12 and IL-1β expression in primary mouse astrocytes. (A) Western blot analysis of NLRP3, caspase-1, IL-1β, caspase-12 and cleaved caspase-3 in astrocytes exposed to 1, 2, 3, 4 hours of OGD and then returned to normal condition for 24 hours. β-actin was used as a loading control. Protein levels of cleaved caspase-12 (B) NLRP3 (C), caspase-1 (D), IL-1β (E), and cleaved caspase-3 (F) were normalized to β-actin and quantified by Image Lab software. CON: control group; OGD: Oxygen Glucose Deprivation group. *P < 0.05, **P<0.01, ***P<0.001(all comparisons to control group). Bar graphs were presented as means ± SD (n=6).
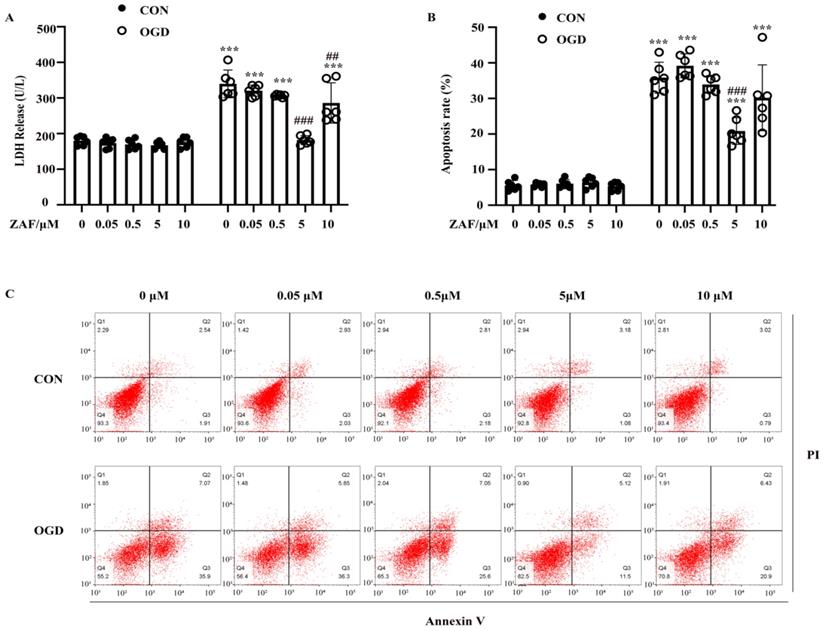
Inhibition of caspase-12 prevented OGD-induced cell injury in primary mouse astrocytes
To verify the role of caspase-12 in OGD-induced cell injury in primary mouse astrocytes, we pretreated astrocytes with 0.05, 0.5, 5, or 10 μM ZAF. As shown in Figure 4A and B, compared with the OGD (0 μM ZAF) group, cell injury and apoptosis were significantly reduced by ZAF in a dose-dependent manner (P < 0.05). Taken together, these data demonstrate that inhibition of caspase-12 prevented OGD-induced cell injury in primary mouse astrocytes.
Inhibition of caspase-12 prevented OGD-induced cell injury in primary mouse astrocytes. Astrocytes were exposed to 3 hours of OGD and then 24 hours of reperfusion with or without ZAF (0, 0.05, 0.5, 5, 10 µM). (A) Cell injury was detected by LDH release assay. (B) Cell apoptosis was detected by Annexin V- fluorescein isothiocyanate (FITC)/prodium iodide (PI) flow cytometry, and the apoptosis rate was presented as the percentage of apoptotic cells vs total cells. (C) Representative scatterplots of Annexin V-FITC/PI flow cytometry in each group. CON: control group; OGD: Oxygen Glucose Deprivation group. *P < 0.05, **P<0.01, ***P<0.001 (all comparisons to control (ZAF=0μM) group); # P < 0.05, ## P<0.01, ### P<0.001(all comparisons to OGD(ZAF=0μM) group). Bar graphs were presented as means ± SD (n=6).
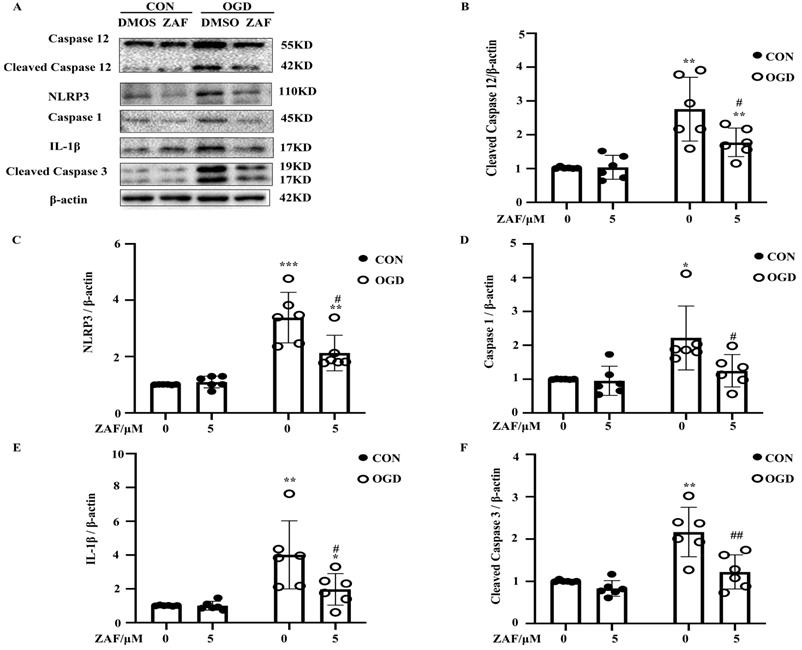
Inhibition of caspase-12 attenuated NLRP3-inflammasome activation in OGD in primary mouse astrocytes
As shown in Figure 5, activation of caspase-12 by OGD was significantly downregulated in primary mouse astrocytes when pre-incubated with ZAF (5 μM) (P < 0.05) (Fig. 5A, B). Cells pre-incubated with ZAF (5 μM) did not exhibit any significant changes in NLRP3 inflammasome components including NLRP3, caspase-1, and IL-1β as well as cleaved caspase-3 in the control group (P > 0.05) (Fig. 5). In addition, ZAF (5 μM) dramatically attenuated the upregulation of NLRP3, caspase-1, IL-1β, and cleaved caspase-3 in the OGD groups (Fig. 5A-F) (P < 0.05). These results indicate that inhibition of caspase-12 was essential for attenuating activation of the NLRP3 inflammasome in primary mouse astrocytes subjected to OGD.
Inhibition of caspase-12 attenuates NLRP3-inflammasome activation in OGD in primary mouse astrocytes. (A) Western blot analysis of NLRP3, caspase-1, IL-1β, caspase-12 and cleaved caspase-3 in astrocytes exposed to 3 hours of OGD followed by 24 hours reperfusion with or without ZAF (5 µM). β-actin was used as a loading control. Protein levels of cleaved caspase-12 (B) NLRP3 (C), caspase-1 (D), IL-1β (E), and cleaved caspase-3 (F) were normalized to β-actin and quantified by Image Lab software. CON: control group; OGD: Oxygen Glucose Deprivation group. *P < 0.05, **P<0.01, ***P<0.001 (all comparisons to control (ZAF=0 µM) group); # P < 0.05, ## P<0.01, ### P<0.001 (all comparisons to OGD (ZAF=0 µM) group). Bar graphs were presented as means ± SD (n=6).
Discussion
I/R injury elicits damage to neurons and non-neuronal cells. Reperfusion after ischemia triggers an inflammatory cascade, resulting in secondary brain injury after ischemia. Neurons are more vulnerable than astrocytes to I/R injury; thus, enhancing the survival of astrocytes provides neuroprotection against I/R injury [21]. Therefore, a better understanding of the potential mechanisms of astrocytes against I/R damage in stroke would help prevent further neuronal dysfunction. Astrocytes are the most abundant non-neuronal cells in the brain. Studies have suggested that astrocytes participate in regulation of innate immunity in the CNS. Astrocytes can affect the physiological status of neurons and may exacerbate neuronal damage during I/R injury and other types of brain damage [22-24] . The astrocytic response to stroke is complex and incompletely understood [25]. Following ischemic stroke, astrocytes perform both deleterious and beneficial functions. Considering the role of astrocytes in cerebral ischemic injury, targeting this cell type as a novel therapeutic strategy in ischemic stroke is worth exploring.
Evidence has demonstrated that ERS results in NLRP3 inflammasome activation [26], and in turn the NLRP3 inflammasome might also cause ERS, although the underlying mechanisms remain unclear [27] . Caspase-12 is predominantly localized in the ER and is especially activated by ERS [12, 28]. Caspase-12 has also been reported to activate the downstream molecule caspase-3, a traditional pro-apoptotic protein, in ERS-induced neuronal apoptosis [29]. In addition to its role in apoptosis, caspase-12 generally acts as a regulator of the inflammatory response, which has been attributed to its relationship with inflammasome complexes [30]. Caspase-12 is an inflammatory caspase that has been reported to negatively regulate inflammation [31, 32], although one study questioned its role in inflammasome activation [14].
Cleaved caspase-12 up-regulation has been observed after I/R injury but its roles in apoptosis are controversial [33, 34]. Our results indicate that the levels of cleaved caspase-12 and cleaved caspase-3 as well as rate of apoptosis were elevated following I/R injury in primary mouse astrocytes. To evaluate the role of caspase-12 in I/R injury, we evaluated the effects of ZAF. When astrocytes were pretreated with ZAF, both the proportion of apoptotic cells and level of cleaved caspase-3 were significantly lower compared with the untreated groups, providing strong evidence that caspase-12 plays an essential role in inducing injury in astrocytes after OGD.
The inflammatory response is important in the pathophysiology of ischemic stroke. Several potential molecules are implicated in cerebral ischemic inflammation, and essential functions of inflammasomes have been identified. Recent studies showed that various inflammasomes, containing NLRP1, NLRP2, NLRP3, NLRP10, NLR family card domain containing 4 (NLRC4), and absent in melanoma 2(AIM2) [35-39], are activated in ischemic stroke. Notably, evidence indicated that the NLRP2 inflammasome is also expressed in astrocytes [40]. The NLRP3 inflammasome in particular has been widely explored in brain ischemia because of its vital contributions to neuronal death and behavioral disorders following stroke [41]. Evidence has suggested that inhibition of NLRP3 activation attenuates infarction volumes and ameliorates stroke outcomes [42]. However, most of the current inflammasome studies of CNS disorders have focused on microglia. Gustin et al. suggested that the NLRP3 inflammasome is limited to mouse microglia but not astrocytes [17]. Nevertheless, increasing evidence supports the presence of the NLRP3 inflammasome in other brains cells, such as oligodendrocytes and astrocytes, in pathological conditions and some disease models [18, 43]. Primary astrocytes were purified of microglia in this study, and the existence of the NLRP3 inflammasome in primary mouse astrocytes was demonstrated (Fig. 1C, D). Interestingly, the expression of NLRP3, caspase-1, and IL-1β was attenuated by ZAF, which to our knowledge is the first report of the effect of ZAF on NLRP3 inflammasome activation. Inflammasomes are cytosolic protein complexes involved in the innate immune system. OGD treatment of primary mouse astrocytes stimulated NLRP3 inflammasome activation and thus upregulation of procaspase-1 and IL-1β expression, consistent with a previous study [44]. IL-1β is primarily an anti-inflammatory cytokine that contributes to I/R cerebral damage and CNS inflammatory responses [45]. The generation of IL-1β is mediated by inflammasomes and the protease caspase-1 [46]. As shown in the present study, ZAF could decrease the generation of IL-1β and thus alleviate astrocyte I/R injury.
Activation of caspase-12 is closely related to brain ischemia. Inhibition of ERS-induced caspase-12 activation potentially decreases cerebral damage in stroke and thereby damage-associated molecular pattern-induced NLRP3 inflammasome activation. However, there are several questions remain unanswered. First, the effect of ZAF in vivo will be addressed in future work. Second, the reciprocal effect of the NLRP3 inflammasome on caspase-12 is worth exploring. Finally, it would be interesting to investigate whether the pyroptosis pathway is involved in our model.
Conclusion
Taken together, we demonstrated the existence of the NLRP3 inflammasome in primary mouse astrocytes. ZAF inhibited caspase-12-mediated primary mouse astrocyte apoptosis following I/R injury, and ZAF may be involved in alleviation of NLRP3 inflammasome activation. This study suggests that caspase-12 and its potential regulation of NLRP3 inflammasome activation may be a promising target for treatment of ischemic stroke.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant NO. 81471171; 81771194) Shengmei Zhu; Zhejiang Medical Technology & Education (2017KY323) the Project of Experimental Animal Science and Technology Plan of Zhejiang Province (NO. 2018C37116) Yueying Zheng.
Abbreviations
OGD: oxygen-glucose deprivation; ZAF: caspase-12 specific inhibitor Z-ATAD-FMK; CNS: central nervous system; ERS: endoplasmic reticulum stress; I/R: ischemia/reperfusion; NLRP3: nucleotidebinding domain leucine-rich repeat containing receptors pyrin domain containing 3; LDH: Lactate dehydrogenase; PVDF: polyvinylidenedifluoride; ASC: apoptosis-associated speck-like protein containing a caspase-activating recruitment domain; NLRC4: NLR family card domain containing 4; AIM2: absent in melanoma 2; DAMPs: damage-associated molecular patterns.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Feigin VL, Krishnamurthi RV, Parmar P. et al. Update on the Global Burden of Ischemic and Hemorrhagic Stroke in 1990-2013: The GBD 2013 Study. Neuroepidemiology. 2015;45(3):161-76
2. Hankey GJ. Stroke. Lancet. 2017;389(10069):641-654
3. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7-35
4. Nakka VP, Prakash-Babu P, Vemuganti R. Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries. Mol Neurobiol. 2016;53(1):532-544
5. Shimoke K, Matsuki Y, Fukunaga K. et al. Appearance of nuclear-sorted caspase-12 fragments in cerebral cortical and hippocampal neurons in rats damaged by autologous blood clot embolic brain infarctions. Cell Mol Neurobiol. 2011;31(5):795-802
6. Zhong C-J, Chen M-M, Lu M. et al. Astrocyte-specific deletion of Kir6.1/K-ATP channel aggravates cerebral ischemia/reperfusion injury through endoplasmic reticulum stress in mice. Experimental neurology. 2019;311:225-233
7. Osada N, Kosuge Y, Ishige K. et al. Characterization of neuronal and astroglial responses to ER stress in the hippocampal CA1 area in mice following transient forebrain ischemia. Neurochemistry international. 2010;57(1):1-7
8. Garcia de la Cadena S, Massieu L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis. 2016;21(7):763-77
9. Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186-94
10. Zhang Q, Liu J, Chen S. et al. Caspase-12 is involved in stretch-induced apoptosis mediated endoplasmic reticulum stress. Apoptosis. 2016;21(4):432-42
11. Aoyama K, Burns DM, Suh SW. et al. Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. J Cereb Blood Flow Metab. 2005;25(3):358-70
12. Nakagawa T, Zhu H, Morishima N. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98-103
13. Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16(1):7-21
14. Vande Walle L, Jimenez Fernandez D, Demon D. et al. Does caspase-12 suppress inflammasome activation? Nature. 2016;534(7605):E1-4
15. Lerner AG, Upton JP, Praveen PV. et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16(2):250-64
16. Gao L, Dong Q, Song Z. et al. NLRP3 inflammasome: a promising target in ischemic stroke. Inflamm Res. 2017;66(1):17-24
17. Gustin A, Kirchmeyer M, Koncina E. et al. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS One. 2015;10(6):e0130624
18. Freeman L, Guo H, David CN. et al. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214(5):1351-1370
19. Borlongan CV, Yamamoto M, Takei N. et al. Glial cell survival is enhanced during melatonin-induced neuroprotection against cerebral ischemia. Faseb j. 2000;14(10):1307-17
20. Martin V, Sainz RM, Antolin I. et al. Several antioxidant pathways are involved in astrocyte protection by melatonin. J Pineal Res. 2002;33(4):204-12
21. Xia CF, Yin H, Borlongan CV. et al. Adrenomedullin gene delivery protects against cerebral ischemic injury by promoting astrocyte migration and survival. Hum Gene Ther. 2004;15(12):1243-54
22. Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Current molecular medicine. 2004;4(2):193-205
23. Ouyang Y-B, Voloboueva LA, Xu L-J. et al. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(16):4253-60
24. Xu L, Emery JF, Ouyang Y-B. et al. Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia. 2010;58(9):1042-9
25. Anderson MF, Blomstrand F, Blomstrand C. et al. Astrocytes and stroke: networking for survival? Neurochemical research. 2003;28(2):293-305
26. Ji T, Han Y, Yang W. et al. Endoplasmic reticulum stress and NLRP3 inflammasome: Crosstalk in cardiovascular and metabolic disorders. J Cell Physiol. 2019
27. Lebeaupin C, Proics E, de Bieville CH. et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015;6(9):e1879
28. Aoyama K, Burns DM, Suh SW. et al. Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2005;25(3):358-70
29. Hitomi J, Katayama T, Taniguchi M. et al. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci Lett. 2004;357(2):127-30
30. García de la Cadena S, Massieu L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis. 2016;21(7):763-77
31. Saleh M, Vaillancourt JP, Graham RK. et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429(6987):75-9
32. Saleh M, Mathison JC, Wolinski MK. et al. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature. 2006;440(7087):1064-8
33. Obeng EA, Boise LH. Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. The Journal of biological chemistry. 2005;280(33):29578-87
34. Di Sano F, Ferraro E, Tufi R. et al. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. The Journal of biological chemistry. 2006;281(5):2693-700
35. Fann DY, Lee SY, Manzanero S. et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013;4(9):e790
36. Cheon SY, Kim EJ, Kim SY. et al. Apoptosis Signal-regulating Kinase 1 Silencing on Astroglial Inflammasomes in an Experimental Model of Ischemic Stroke. Neuroscience. 2018;390:218-230
37. Li ZG, Shui SF, Han XW. et al. NLRP10 ablation protects against ischemia/reperfusion-associated brain injury by suppression of neuroinflammation. Exp Cell Res. 2020;389(2):111912
38. Denes A, Coutts G, Lénárt N. et al. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci U S A. 2015;112(13):4050-5
39. Zhang N, Zhang X, Liu X. et al. Chrysophanol inhibits NALP3 inflammasome activation and ameliorates cerebral ischemia/reperfusion in mice. Mediators Inflamm. 2014;2014:370530
40. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61(7):1113-21
41. Alishahi M, Farzaneh M, Ghaedrahmati F. et al. NLRP3 inflammasome in ischemic stroke: As possible therapeutic target. Int J Stroke. 2019;14(6):574-591
42. Yang F, Wang Z, Wei X. et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab. 2014;34(4):660-7
43. Johann S, Heitzer M, Kanagaratnam M. et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63(12):2260-73
44. Jian Z, Ding S, Deng H. et al. Probenecid protects against oxygen-glucose deprivation injury in primary astrocytes by regulating inflammasome activity. Brain research. 2016;1643:123-9
45. Savage CD, Lopez-Castejon G, Denes A. et al. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Front Immunol. 2012;3:288
46. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720-32
Author contact
![]() Corresponding authors: Shengmei Zhu, E-mail: smzhu20088edu.cn; Yueying Zheng, E-mail: 1507128edu.cn.
Corresponding authors: Shengmei Zhu, E-mail: smzhu20088edu.cn; Yueying Zheng, E-mail: 1507128edu.cn.