Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(10):1338-1348. doi:10.7150/ijms.38219 This issue Cite
Research Paper
Investigating Novel Genes Potentially Involved in Endometrial Adenocarcinoma using Next-Generation Sequencing and Bioinformatic Approaches
Feng-Hsiang Tang1,2,3,#, Wei-An Chang1,4,5,#, Eing-Mei Tsai2,6, Ming-Ju Tsai1,4,5,7, ![]() , Po-Lin Kuo1,8,
, Po-Lin Kuo1,8, ![]()
1. Graduate Institute of Clinical Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
2. Department of Obstetrics and Gynecology, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 807, Taiwan
3. Department of Obstetrics and Gynecology, Kaohsiung Municipal Ta-Tung Hospital, Kaohsiung Medical University, Kaohsiung 807, Taiwan
4. Division of Pulmonary and Critical Care Medicine, Kaohsiung Medical University Hospital, Kaohsiung 807, Taiwan
5. Department of Internal Medicine, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
6. Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
7. Department of Respiratory Therapy, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
8. Institute of Medical Science and Technology, National Sun Yat-Sen University, Kaohsiung 804, Taiwan
#Contributed equally
Received 2019-7-6; Accepted 2019-8-22; Published 2019-9-7
Abstract
Endometrial cancer is one of the most common cancers in women worldwide, affecting more than 300,000 women annually. Dysregulated gene expression, especially those mediated by microRNAs, play important role in the development and progression of cancer. This study aimed to investigate differentially expressed genes in endometrial adenocarcinoma using next generation sequencing (NGS) and bioinformatics. The gene expression profiles and microRNA profiles of endometrial adenocarcinoma (cancer part) and normal endometrial tissue (non-cancer part) were assessed with NGS. We identified 56 significantly dysregulated genes, including 47 upregulated and 9 downregulated genes, in endometrial adenocarcinoma. Most of these genes were associated with defense response, response to stimulus, and immune system process, and further pathway analysis showed that human papillomavirus infection was the most significant pathway in endometrial adenocarcinoma. In addition, these genes were also associated with decreased cell death and survival as well as increased cellular movement. The analyses using Human Protein Atlas, identified 6 genes (PEG10, CLDN1, ASS1, WNT7A, GLDC, and RSAD2) significantly associated with poorer prognosis and 3 genes (SFN, PIGR, and CDKN1A) significantly associated with better prognosis. Combining with the data of microRNA profiles using microRNA target predicting tools, two significantly dysregulated microRNA-mediated gene expression changes in endometrial adenocarcinoma were identified: downregulated hsa-miR-127-5p with upregulated CSTB and upregulated hsa-miR-218-5p with downregulated HPGD. These findings may contribute important new insights into possible novel diagnostic or therapeutic strategies for endometrial adenocarcinoma.
Keywords: endometrial cancer, papillomavirus, next generation sequencing, bioinformatics, miR-127-5p, miR-218-5p, CSTB, HPGD
Introduction
Cancers of the corpus uteri, primarily from the endometrium, rank as the sixth most common neoplasm in women worldwide. The incidence increased from 290,000 in 2008 to over 380,000 in 2018 (1). Estrogen exposure, either endogenous or exogenous, is a major risk factor of endometrial cancer, while endometrial cancer is generally divided into two distinct types, type I (estrogen-related) and type II (non-estrogen-related) (2). As mentioned in a large review, strong evidence suggested that three factors were associated with endometrial cancer: increased body mass index and increased waist‐to‐hip ratio were associated with increased risk, while increased parity reduced the risk of disease (3). The genetic mechanism underlying the pathogenesis of endometrial cancer is not fully understood. In type I endometrial cancer, which account for nearly 80% of endometrial cancer, PTEN mutation, hMLH1 methylation, and hMSH6 mutation are important in atypical hyperplastic change of normal endometrium. Mutations in PTEN, KRAS, and CTNNB1 are associated with malignant change from atypical endometrial hyperplasia to low-grade endometrioid cancer, while P53 mutation plays an important role in advancing low-grade cancer into high-grade one (4). In type II endometrial cancer, mutations in P53 and HER2/neu are associated with non-endometrioid malignant transformation from normal or atrophic endometrium (4).
Traditionally, patients suffered from endometrial cancer have a favorable treatment outcome if diagnosed in the early stage. The overall five-year survival rate of endometrial cancer is 81%, but is only 17% if distal metastasis occurs (5). The three-year overall survival rate is 96.2% for women without recurrence; however, it is 73.4% for women with vaginal vault recurrence, loco-regional nodal recurrence, or local central pelvic recurrence, and is only 38.1% for those with distal metastases and/or peritoneal carcinomatosis (6). This might be result from the absence of a perfect treatment modality for advanced or recurrent disease currently.
The development of next-generation sequencing (NGS) technologies provides the capability to rapidly sequence exomes, transcriptomes, and genomes at relatively low cost. The application of this technology to catalog the mutational landscapes of tumor exomes, transcriptomes, and genomes has remarkably accelerated the progress in basic and clinical cancer researches (7), making precision medicine possible (8). Individual cancer patients can therefore receive personalized care with the most suitable drugs at the appropriate dose and at the right time (8).
As microRNAs have the ability to repress the expression of protein-coding genes, they might contribute to the pathogenesis of various diseases including cancer (9-13). Functional studies have shown that microRNA dysregulation plays important role in the development and progression of various cancers (9). Some microRNAs may act as either tumor suppressors (miR onco-suppressors) or tumor enhancers (onco-miRs), and anti-cancer treatment with microRNA mimics or molecules targeted at miRNAs are under development. With increasing knowledge of the microRNA-mediated changes in cancer cells, we will have better opportunity to develop a better microRNA-based anti-cancer treatment.
Through identifying novel gene expression signature and microRNA-gene interactions in endometrial adenocarcinoma, we may provide new perspectives for the development of novel diagnostic methods, prognostic predicting tools, and therapeutic strategies of endometrial adenocarcinoma. Therefore, in this study, we would like to identify the differentially expressed gene and the potential regulatory mechanisms through microRNAs in endometrial adenocarcinoma with systematic bioinformatics analysis.
Materials and methods
Study design
The flowchart of study design is illustrated in Figure 1. The cancer part and non-cancer part (normal endometrial tissue) were taken from the surgical specimen of a 53-year-old woman with stage Ia endometrial adenocarcinoma cancer after informed consent was obtained. This pair of tissues was sent for NGS to assess the expression profiles of mRNAs and microRNAs. Using bioinformatic tools, including Search Tool for the Retrieval of Interacting Genes (STRING), the Database for Annotation, Visualization and Integrated Discovery (DAVID), and Ingenuity® Pathway Analysis (IPA), the altered functions and pathways related to the dysregulated genes in endometrial cancer were investigated. In addition, the potential targets of the significantly dysregulated microRNAs were predicted with miRmap, TargetScan, and miRDB, and the potential microRNA-mRNA interactions in endometrial cancer were identified.
Flow chart of the study. Abbreviation: STRING, Search Tool for the Retrieval of Interacting Genes; DAVID, Database for Annotation, Visualization and Integrated Discovery; KEGG, Kyoto Encyclopedia of Genes and Genomes; IPA, Ingenuity® Pathway Analysis.
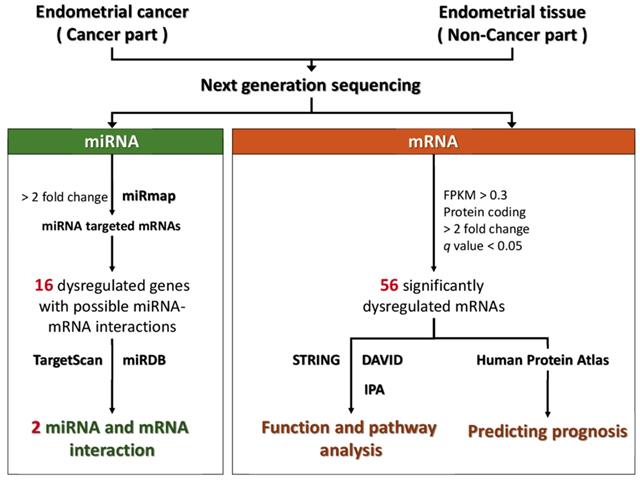
NGS for microRNA and mRNA expression profiles
The expression profiles of microRNAs and mRNAs were examined using NGS as in our previous studies (10, 11, 13-16). In brief, total RNA was extracted with Trizol® Reagent (Invitrogen, USA) as per the instruction manual. The purified RNAs were quantified at O.D.260nm with a ND-1000 spectrophotometer (Nanodrop Technology, Wilmington, DE, USA) and qualitatively assessed with Bioanalyzer 2100 and RNA 6000 LabChip kit (both from Agilent Technology, Santa Clara, CA, USA). Library preparation and sequencing were performed in Welgene Biotechnology Company (Taipei, Taiwan).
For transcriptome sequencing, the Agilent's SureSelect Strand Specific RNA Library Preparation Kit was used to construct the libraries, followed by AMPure XP Beads size selection. The sequence was directly determined using Illumina's sequencing-by-synthesis (SBS) technology. Sequencing data (FASTQ files) were generated by Welgene's pipeline based on Illumina's base-calling program bcl2fastq v2.2.0. After adaptor clipping and sequence quality trimming with Trimmomatics (Ver. 0.36) (17), alignment of the qualified reads were performed using HISAT2 (18, 19), which is a fast and sensitive alignment program for mapping NGS reads to genomes based on hierarchical graph FM index. The genes with low expression levels (< 0.3 fragment per kilobase of transcript per million mapped reads [FPKM]) in any group were excluded. The p values were calculated by Cuffdiff with non-grouped samples using the "blind mode”, in which all samples were treated as replicates of a single global "condition" and used to build a model for statistical test (20, 21). The q values were the p values adjusted with false discovery rate using the method by Benjamini and Hochberg (22). Genes with q-value < 0.05 (i.e., -log10(q value) > 1.3) and > 2-fold changes were considered significantly differentially expressed.
For small RNA sequencing, samples were prepared using Illumina sample preparation kit as per the TruSeq Small RNA Sample Preparation Guide. The 3' and 5' adaptors were ligated to the RNA, and then reverse transcription and PCR amplification were performed. The cDNA constructs were size-fractionated and purified using a 6% polyacrylamide gel electrophoresis and the bands corresponding to the 18-40 nucleotide RNA fragments (140-155 nucleotide in length with both adapters) were extracted. After sequencing on an Illumina (San Diego, CA, USA) instrument (75 bp single-end reads), the data was processed with the Illumina software. After trimming and filtering out low-quality data with Trimmomatics (17) and clipping the 3' adapter sequence and discarding reads shorter than 18 nucleotides with miRDeep2 (23), the qualified reads were aligned to the human genome from University of California, Santa Cruz (UCSC). Because microRNAs usually map to few genomic locations, only reads mapped perfectly to the genome ≤5 times were taken. MiRDeep2 is useful for estimating the expression levels of known microRNAs, as well as identifying novel microRNAs. The microRNAs with low levels (<1 normalized read per million (rpm)) in both groups were excluded. The microRNAs with >2 fold change are considered significantly changed.
Analyses using microRNA target predicting databases
miRmap (http://mirmap.ezlab.org/) is an open-source software library which can provide comprehensive prediction of microRNA targets (24). The putative target genes could be identified by calculating the complementary ability of microRNA-mRNA interactions. The prediction results provide a list of putative target genes with miRmap scores, which are predictive reference values representing the repression strength of the microRNAs on a target mRNA. In this study, the criteria for selection of putative microRNA targets were miRmap score ≥ 97.0.
TargetScan (http://www.targetscan.org) is an online database predicting the target of microRNA by searching for the presence of conserved 8mer, 7mer, and 6mer sites matching the seed region of each microRNA (25). The results of predictions are ranked by the predicted efficacy of targeting or by their probability of conserved targeting (25). TargetScan could provide a valuable resource for investigating the role of microRNAs in gene-regulatory networks.
miRDB (http://mirdb.org) provides web-based microRNA-target prediction and functional annotations in five species, including human, mouse, rat, dog, and chicken (26, 27). In miRDB, all targets were predicted by MirTarget, which was developed by analyzing microRNA-target interactions from high-throughput sequencing experiments.
Analysis using STRING
The functional interactions between expressed proteins in cells are very important and complicated. STRING database (https://string-db.org/) has collected and integrated this information, by consolidating known and predicted protein-protein association data of various organisms (28). The protein-protein interactions, including direct (physical) and indirect (functional) interactions, collected in STRING are derived from five main sources, including conserved co-expressions, high-throughput lab experiment, genomic context predictions, automated text-mining, and previous knowledge in database. In this study, the significantly dysregulated genes were input into STRING for protein-protein interaction network analysis. The minimum required interaction score was set to the medium confidence (score = 0.400). In addition, STRING also provides information of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway.
Analysis using DAVID
DAVID (https://david.ncifcrf.gov/) is a powerful tool for functional classification of genes (29). It integrates gene ontology, biological process, and KEGG pathway. In DAVID database, a list of interesting genes can be classified into clusters of related biological functions, signaling pathways, or diseases by calculating the similarity of global annotation profiles with an agglomeration algorithm method. An Expression Analysis Systematic Explorer (EASE) score is a modified Fisher's exact p value in DAVID database which represents how specifically the genes are involved in a category. In this study, we selected EASE score = 0.1 as the default and defined pathways with a q value (p value adjusted with false discovery rate using the method by Benjamini, et al.) <0.05 as significant.
Analysis using IPA
IPA (Ingenuity systems, Redwood City, CA, USA) is a database software containing large database with detailed and structured findings reviewed by experts, which was derived from thousands of biological, chemical and medical researches (30). IPA enables rapid searching, analysis, integration, and recognition of data from gene and single nucleotide polymorphism (SNP) arrays, RNA and small RNA sequencing, proteomics and many other biological experiments (30). Deeper understanding and identification of related signaling pathways, upstream regulators, molecular interactions, disease process, and candidate biomarkers are also available (30). In this study, we used IPA to assess the diseases and functions associated with the significantly dysregulated genes in endometrial adenocarcinoma. The disease and function with a p value < 0.05 was considered significant.
Analysis using the Human Protein Atlas
The Human Protein Atlas is a Swedish-based program initiated in 2003 with the aim to map all the human proteins in cells, tissues, and organs using integration of various omics technologies, including antibody-based imaging, mass spectrometry-based proteomics, transcriptomics, and systems biology (31). All data collected in it is open-access to allow scientists, either in academia or industry, to freely access the data for exploration of the human proteome. The Human Protein Atlas consists of three separate parts, each focusing on a particular aspect of the genome-wide analysis of the human proteins, including the Tissue Atlas showing the distribution of the proteins across all major tissues and organs in the human body, the Cell Atlas showing the subcellular localization of proteins in single cells, and the Pathology Atlas showing the impact of protein levels for survival of cancer patients. This program has already contributed to several thousands of publications in the field of human biology and disease and it is selected by the organization ELIXIR (http://www.elixir-europe.org) as a European core resource due to its fundamental importance for a wider life science community. In this study, the prognosis-predicting values of the significantly dysregulated genes in endometrial cancer were assessed with the Human Protein Atlas. Based on the FPKM value of each gene, the patients were classified into two groups, low-expression and high-expression, to compare their prognoses. The prognosis (survival) of each group of patients was examined and compared with Kaplan-Meier survival estimators and log-rank tests. To determine the best cut-off FPKM values for grouping the patients, all FPKM values from the 20th to 80th percentiles were used to group the patients, and the cut-off FPKM value that yielded the lowest log-rank p value was selected.
Results
Differential gene expressions in endometrial cancer
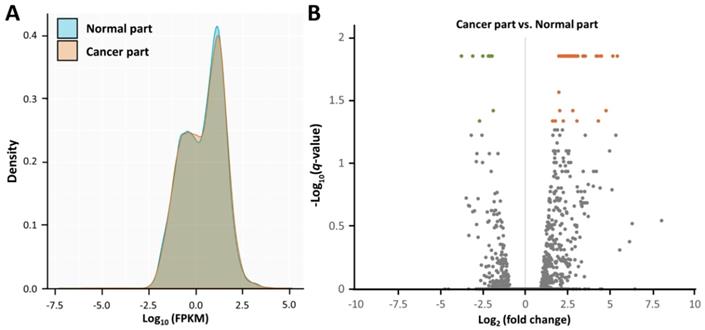
Using NGS, the gene expression profiles of the cancer part and non-cancer part of the surgical specimen from the patient with endometrial adenocarcinoma were assessed (Figure 2A). As shown in the volcano plots (Figure 2B), significantly dysregulated genes in endometrial adenocarcinoma (cancer part vs. non-cancer part) (those with -log10(q value) > 1.3 and fold change > 2) were identified (Table 1), including 47 upregulated and 9 downregulated genes.
Differentially expressed genes in endometrial adenocarcinoma (cancer part versus non-cancer part).
| Official gene symbol | FPKM | Ratio (C/N) | Log2(ratio) | p value | q value* | |
|---|---|---|---|---|---|---|
| Cancer (C) part | Non-cancer (N) part | |||||
| DKK4 | 169.43 | 3.95 | 42.84 | 5.42 | <0.0001 | 0.0141 |
| RXFP1 | 97.99 | 2.73 | 35.86 | 5.16 | <0.0001 | 0.0141 |
| LY6D | 41.57 | 1.54 | 27.05 | 4.76 | 0.0002 | 0.0384 |
| DPP4 | 165.45 | 7.38 | 22.41 | 4.49 | <0.0001 | 0.0141 |
| CST1 | 122.75 | 5.56 | 22.07 | 4.46 | <0.0001 | 0.0141 |
| BMP2 | 18.22 | 0.92 | 19.80 | 4.31 | 0.0002 | 0.0462 |
| PTGES | 51.92 | 2.67 | 19.41 | 4.28 | <0.0001 | 0.0141 |
| MUC13 | 28.01 | 1.55 | 18.08 | 4.18 | <0.0001 | 0.0141 |
| VNN1 | 36.31 | 3.14 | 11.56 | 3.53 | <0.0001 | 0.0141 |
| TFAP2A | 8.60 | 0.79 | 10.89 | 3.45 | <0.0001 | 0.0141 |
| MACROD2 | 39.50 | 3.77 | 10.47 | 3.39 | <0.0001 | 0.0141 |
| SFN | 145.86 | 17.02 | 8.57 | 3.10 | <0.0001 | 0.0141 |
| GPRC5A | 61.80 | 7.33 | 8.43 | 3.08 | <0.0001 | 0.0141 |
| PCSK5 | 9.55 | 1.15 | 8.29 | 3.05 | 0.0002 | 0.0462 |
| PEG10 | 21.40 | 2.71 | 7.89 | 2.98 | <0.0001 | 0.0141 |
| GLDC | 16.03 | 2.07 | 7.74 | 2.95 | <0.0001 | 0.0141 |
| ISG15 | 316.92 | 42.11 | 7.53 | 2.91 | <0.0001 | 0.0141 |
| ITGA3 | 76.72 | 10.39 | 7.39 | 2.88 | <0.0001 | 0.0141 |
| LAMC2 | 97.12 | 13.89 | 6.99 | 2.81 | <0.0001 | 0.0141 |
| BATF2 | 37.82 | 5.46 | 6.93 | 2.79 | 0.0002 | 0.0384 |
| PIGR | 47.74 | 7.12 | 6.70 | 2.74 | <0.0001 | 0.0141 |
| CLDN1 | 93.32 | 14.02 | 6.66 | 2.74 | <0.0001 | 0.0141 |
| RHOF | 45.21 | 6.79 | 6.65 | 2.73 | <0.0001 | 0.0141 |
| SEMA6A | 9.58 | 1.44 | 6.65 | 2.73 | <0.0001 | 0.0141 |
| IFI6 | 488.10 | 74.30 | 6.57 | 2.72 | <0.0001 | 0.0141 |
| APOL1 | 259.11 | 40.36 | 6.42 | 2.68 | <0.0001 | 0.0141 |
| LCN2 | 165.99 | 27.42 | 6.05 | 2.60 | <0.0001 | 0.0141 |
| B3GNT3 | 26.10 | 4.40 | 5.94 | 2.57 | <0.0001 | 0.0141 |
| GPX3 | 77.86 | 13.69 | 5.69 | 2.51 | <0.0001 | 0.0141 |
| ASS1 | 88.14 | 15.91 | 5.54 | 2.47 | <0.0001 | 0.0141 |
| SPP1 | 1680.24 | 304.79 | 5.51 | 2.46 | <0.0001 | 0.0141 |
| F3 | 151.88 | 27.62 | 5.50 | 2.46 | <0.0001 | 0.0141 |
| RSAD2 | 97.04 | 17.65 | 5.50 | 2.46 | <0.0001 | 0.0141 |
| PLAC8 | 80.96 | 14.73 | 5.49 | 2.46 | <0.0001 | 0.0141 |
| TGFA | 27.11 | 5.13 | 5.28 | 2.40 | <0.0001 | 0.0141 |
| CSTB | 152.48 | 30.76 | 4.96 | 2.31 | <0.0001 | 0.0141 |
| WNT7A | 57.35 | 11.73 | 4.89 | 2.29 | <0.0001 | 0.0141 |
| USP18 | 33.36 | 7.11 | 4.69 | 2.23 | 0.0002 | 0.0462 |
| MX1 | 73.00 | 15.58 | 4.69 | 2.23 | <0.0001 | 0.0141 |
| GDA | 99.40 | 22.50 | 4.42 | 2.14 | <0.0001 | 0.0141 |
| GDF15 | 71.56 | 16.45 | 4.35 | 2.12 | <0.0001 | 0.0141 |
| IFI44 | 277.77 | 67.96 | 4.09 | 2.03 | 0.0002 | 0.0384 |
| BST2 | 302.60 | 75.82 | 3.99 | 2.00 | <0.0001 | 0.0141 |
| PTGS1 | 44.54 | 11.28 | 3.95 | 1.98 | 0.0001 | 0.0274 |
| ATP11A | 24.91 | 6.32 | 3.94 | 1.98 | <0.0001 | 0.0141 |
| CDKN1A | 106.16 | 31.41 | 3.38 | 1.76 | 0.0002 | 0.0462 |
| MET | 78.65 | 25.94 | 3.03 | 1.60 | 0.0002 | 0.0462 |
| CXCL12 | 20.82 | 77.22 | 0.27 | -1.89 | 0.0002 | 0.0384 |
| MGP | 195.18 | 766.67 | 0.25 | -1.97 | <0.0001 | 0.0141 |
| SPARCL1 | 71.61 | 295.44 | 0.24 | -2.04 | <0.0001 | 0.0141 |
| TIMP3 | 7.22 | 30.83 | 0.23 | -2.09 | <0.0001 | 0.0141 |
| HPGD | 175.32 | 789.89 | 0.22 | -2.17 | <0.0001 | 0.0141 |
| LMOD1 | 2.55 | 14.59 | 0.17 | -2.52 | <0.0001 | 0.0141 |
| PDLIM3 | 3.18 | 20.66 | 0.15 | -2.70 | 0.0002 | 0.0462 |
| CNN1 | 2.98 | 25.60 | 0.12 | -3.10 | <0.0001 | 0.0141 |
| DES | 4.10 | 55.87 | 0.07 | -3.77 | <0.0001 | 0.0141 |
* p values adjusted with false discovery rate.
Abbreviation: FPKM, fragments per kilobase of transcript per million mapped reads. The genes highlighted with underlines were those significantly associated with poorer (in red color) or better (in blue color) prognosis, as shown in Figure 5.
Overview of the gene expression profiles in endometrial adenocarcinoma. (A) The density plot illustrates smoothed frequency distribution of the fragments per kilobase of transcript per million mapped reads (FPKM) among the cancer part and non-cancer part. (B) The volcano plot of differential gene expression patterns of the cancer part vs. non-cancer part. Significantly dysregulated genes in endometrial adenocarcinoma (cancer part vs. non-cancer part) (those with -log10(q-value) > 1.3 and fold change > 2) were shown in green (downregulated) or orange (upregulated).
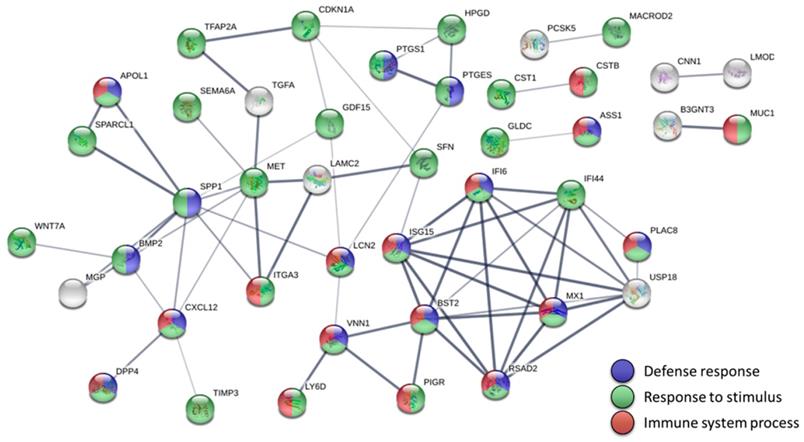
Using STRING to investigate the protein-protein interactions of the significantly dysregulated genes in endometrial adenocarcinoma, we built a highly interactive protein-protein interaction (PPI) network of 56 nodes and 67 edges (enrichment p value < 1.0 x 10-16) (Figure 3). Most genes in the PPI network were associated with three biological pathways, including defense response (19 genes), response to stimulus (44 genes), and immune system process (21 genes). Furthermore, the KEGG pathway analysis indicated that human papillomavirus (HPV) infection might be the most significant pathway involved in endometrial adenocarcinoma (q value = 0.0038) (Table 2).
KEGG pathway analysis of the significantly dysregulated genes in endometrial adenocarcinoma using the STRING database.
| Term description | Observed gene count | q value* | Matching proteins in the network |
|---|---|---|---|
| Human papillomavirus infection | 7 | 0.0038 | CDKN1A, ISG15, ITGA3, LAMC2, MX1, SPP1, WNT7A |
| Pathways in cancer | 8 | 0.0058 | BMP2, CDKN1A, CXCL12, ITGA3, LAMC2, MET, TGFA, WNT7A |
| PI3K-Akt signaling pathway | 6 | 0.0176 | CDKN1A, ITGA3, LAMC2, MET, SPP1, TGFA |
| Arachidonic acid metabolism | 3 | 0.0213 | GPX3, PTGES, PTGS1 |
| Renal cell carcinoma | 3 | 0.0213 | CDKN1A, MET, TGFA |
| Basal cell carcinoma | 3 | 0.0213 | BMP2, CDKN1A, WNT7A |
| Hepatocellular carcinoma | 4 | 0.0213 | CDKN1A, MET, TGFA, WNT7A |
| ECM-receptor interaction | 3 | 0.0234 | ITGA3, LAMC2, SPP1 |
| Focal adhesion | 4 | 0.0291 | ITGA3, LAMC2, MET, SPP1 |
| Proteoglycans in cancer | 4 | 0.0291 | CDKN1A, MET, TIMP3, WNT7A |
| Small cell lung cancer | 3 | 0.0291 | CDKN1A, ITGA3, LAMC2 |
* p values adjusted with false discovery rate.
The genes highlighted with underlines were those significantly associated with poorer (in red color) or better (in blue color) prognosis, as shown in Figure 5.
Abbreviation: KEGG, Kyoto Encyclopedia of Genes and Genomes; STRING, Search Tool for the Retrieval of Interacting Genes.
Protein-protein interaction network analysis of the dysregulated genes in endometrial adenocarcinoma. The 56 significantly dysregulated genes (47 upregulated and 9 downregulated) were input into the Search Tool for the Retrieval of Interacting Genes (STRING) database for protein-protein interaction (PPI) network analysis. The minimum required interaction score was set to the medium confidence (score = 0.400). Nodes represent proteins and edges represent protein-protein associations. Nodes without edges are not displayed. This analysis obtained a highly interactive PPI network of 56 nodes and 67 edges, with PPI enrichment p value of < 1.0 х 10-16. Most genes in the PPI network were associated with three biological pathways, including defense response (19 genes, shown in blue), response to stimulus (44 genes, shown in green), and immune system process (21 genes, shown in red).
We then used DAVID to analyze the biological processes, cellular components, and molecular functions associated with the 56 significantly dysregulated genes in endometrial adenocarcinoma (Table 3). The significant biological processes included response to virus (6 genes) and type I interferon signaling pathway (5 genes). The significant cellular components included extracellular space (19 genes), extracellular exosome (25 genes), and cell surface (9 genes). The only significant molecular functions associated with the 56 significantly dysregulated genes was protease binging (7 genes).
The significant biological processes (BP), cellular components (CC), and molecular function (MF) of the significantly dysregulated genes in endometrial adenocarcinoma shown in DAVID.a
| Category | Term | Gene count | Genes | q value* |
|---|---|---|---|---|
| BP | Response to virus | 6 | LCN2, BST2, RSAD2, IFI44, MX1, CXCL12 | 0.016 |
| BP | Type I interferon signaling pathway | 5 | ISG15, BST2, RSAD2, MX1, IFI6 | 0.015 |
| CC | Extracellular space | 19 | BMP2, SPARCL1, CST1, SFN, PIGR, TIMP3, CXCL12, LCN2, APOL1, F3, GPX3, CSTB, TGFA, LAMC2, GDF15, WNT7A, PCSK5, MUC13, SPP1 | <0.001 |
| CC | Extracellular exosome | 25 | GDA, ASS1, BST2, SPARCL1, PTGS1, MGP, ITGA3, SFN, PIGR, GPRC5A, TIMP3, CXCL12, LCN2, DES, F3, GPX3, CSTB, VNN1, GDF15, HPGD, RHOF, WNT7A, MUC13, DPP4, SPP1 | <0.001 |
| CC | Cell surface | 9 | BMP2, LY6D, BST2, F3, MET, TGFA, ITGA3, WNT7A, DPP4 | 0.006 |
| MF | Protease binding | 7 | LCN2, F3, CSTB, CST1, ITGA3, TIMP3, DPP4 | <0.001 |
a q value < 0.05 was considered significant. * p value adjusted with false discovery rate. The genes highlighted with underlines were those significantly associated with poorer (in red color) or better (in blue color) prognosis, as shown in Figure 5.
Abbreviation: DAVID, Database for Annotation, Visualization and Integrated Discovery.
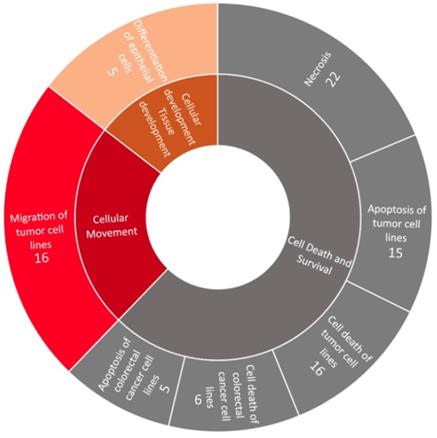
Using IPA, the associated diseases and functions of the 56 significantly dysregulated genes in endometrial adenocarcinoma were investigated (Figure 4). The diseases and functions significantly associated with these dysregulated genes belonged to three categories, including cell death and survival (downregulated), cellular movement (upregulated), and cellular development and tissue development (upregulated).
Disease and function analysis of the significantly dysregulated genes in endometrial adenocarcinoma. Using Ingenuity® Pathway Analysis, the associated diseases and functions of the significantly dysregulated genes in endometrial adenocarcinoma were analyzed. Significant diseases and functions (those with a p value < 0.05) are shown in the outer circle, while their categories are shown in the inner circle. The numbers show the counts of involved genes. The area of each disease and function reflects its significant level, based on their -log10(p-value). Downregulated diseases and functions are shown in gray color, while upregulated ones are shown in orange and red colors.
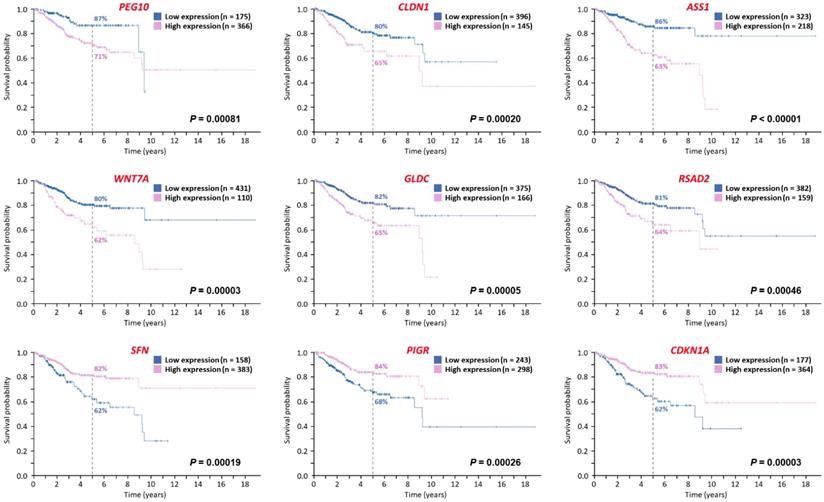
The possible genes associated with prognosis of endometrial cancer
Using the Human Protein Atlas, the prognosis-predicting values of the 56 significantly dysregulated genes in endometrial cancer were assessed (Figure 5). Totally, the information of 541 patients with endometrial cancer (Table 4) was obtained for analyses. The median follow-up time of this cohort was 2.5 years. Among these 56 significantly dysregulated genes, 6 genes were significantly associated with poorer prognosis (PEG10, CLDN1, ASS1, WNT7A, GLDC, and RSAD2) and 3 genes were significantly associated with better prognosis (SFN, PIGR, and CDKN1A).
Baseline demographics of the endometrial cancer patients obtained from Human Protein Atlas.
| Variable | Data |
|---|---|
| Number | 541 |
| Age (year) - mean (± standard deviation) † | 64 (±11) |
| Race - n (%) | |
| American Indian or Alaska native | 4 (1%) |
| Asian | 20 (4%) |
| African American | 106 (20%) |
| Native Hawaiian or other Pacific islander | 9 (2%) |
| White | 370 (68%) |
| Unknown | 32 (6%) |
† Ages of 3 subjects were unavailable.
Possible genes associated with the prognosis of endometrial cancer as predicted by the Human Protein Atlas. The prognosis-predicting values of the 56 significantly dysregulated genes in endometrial cancer were assessed using the Human Protein Atlas. The 12 Kaplan-Meier curves (with log-rank p values) show the genes significantly associated with prognosis, including 6 genes associated with poorer prognosis and 3 genes associated with better prognosis.
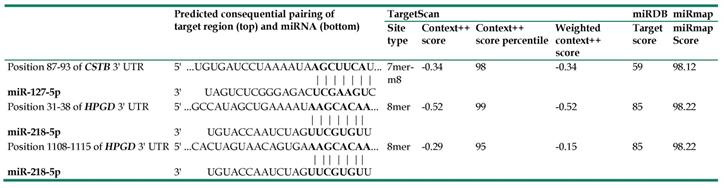
Potential dysregulated microRNA-mRNA interactions in endometrial cancer
Using NGS, 227 significantly dysregulated microRNAs (>2 fold change, including 186 upregulated and 41 downregulated microRNAs) were identified. We predicted the potential targets of these microRNAs with miRmap database, selecting the targets in the list of the 56 significantly dysregulated genes in endometrial cancer and the microRNA-mRNA interactions with miRmap score ≥ 97.0, and found 34 possible microRNA-mRNA interactions (including 12 interactions between a downregulated microRNA and an upregulated mRNA and 22 interactions between an upregulated microRNA and a downregulated mRNA) involving 16 mRNAs (9 upregulated and 7 downregulated mRNAs) (Figure 1). Further investigation using TargetScan and miRDB databases showed that only two microRNA-mRNA interactions, downregulated hsa-miR-127-5p with upregulated CSTB and upregulated hsa-miR-218-5p with downregulated HPGD, were validated in both TargetScan and miRDB databases (Table 5).
Potential miRNA-mRNA interactions involved in endometrial adenocarcinoma, validated with TargetScan, miRDB, and miRmap.
Discussion
In the current study, significantly dysregulated genes, especially those mediated by dysregulated microRNAs, in endometrial adenocarcinoma were investigated comprehensively using an approach with NGS and bioinformatics. We found 56 significantly dysregulated genes, including 47 upregulated and 9 downregulated genes. Most of these genes were associated with defense response, response to stimulus, and immune system process, suggesting the important association between endometrial adenocarcinoma and immune response. Interestingly, the KEGG pathway analysis in STRING showed that HPV infection was the most significant pathway in endometrial adenocarcinoma, suggesting the possible role of HPV in the carcinogenesis of endometrial cancer. Further analyses using DAVID also implied that endometrial adenocarcinoma might be associated with virus infection. On the other hand, the analyses using IPA found that these 56 genes were associated with decreased cell death and survival as well as increased cellular movement, which were common behaviors of malignant cells. The analyses using Human Protein Atlas, identified 6 genes significantly associated with poorer prognosis and 3 genes significantly associated with better prognosis. Combining with the data of microRNA profiles using microRNA target predicting tools, two significantly dysregulated microRNA-mediated gene expression changes in endometrial adenocarcinoma were found: downregulated hsa-miR-127-5p with upregulated CSTB and upregulated hsa-miR-218-5p with downregulated HPGD.
It has been debated whether endometrial cancer is associated HPV, although a strong association between HPV and cervical cancer is well-known. The close anatomical proximity to the cervix has led researchers to study whether HPV has a role in the carcinogenesis of endometrial cancer. However, a systematic review and meta-analysis revealed a pooled prevalence of HPV DNA in endometrial cancer tissue of 10.0% (95% confidence interval: 5.2-16.2%) with large between-study heterogeneity related to the HPV DNA detection methods, and concluded that HPV had a limited or no role in the carcinogenesis of endometrial cancer (2). Nevertheless, the absence of HPV DNA in the endometrial cancer tissue cannot totally exclude the possible contribution of HPV in the pathogenesis of endometrial cancer. In our study, however, the significantly dysregulated genes in endometrial adenocarcinoma was associated with immune responses, and the KEGG pathway analysis showed that HPV infection was the most significant pathway, suggesting the possible role of HPV in the carcinogenesis of endometrial cancer. Further large-scale study is needed to elucidate the association between HPV and endometrial cancer.
Aberrant expression of various microRNAs in endometrial cancer has been reported (32). Significantly higher serum levels of miR-186, miR-222, and miR-223 and significantly lower serum level of miR-204 were noted in patients of endometrial carcinoma than in the matched control subjects (33). Some studies also demonstrated the possibility of using extracellular vesicles isolated from the peritoneal lavage as biomarkers of endometrial cancer (34). In our study, we found significantly downregulated has-miR-127-5p and upregulated has-miR-218-5p in endometrial adenocarcinoma. In line with our findings, Dong et al. have also demonstrated downregulation of miR-127 in endometrial cancer using microRNA microarray profiling (35) and Delangle et al. have shown upregulation of miR-218 in endometrial cancer tissue using quantitative reverse transcription polymerase chain reaction (32, 36).
The CSTB gene encodes cystatin B in humans, which may interact with cathepsin B (37). Cysteine cathepsins are highly upregulated in many cancers (38). Being in various locations (being secreted, cell-surface, and intracellular space), they involve in many proteolytic pathways that may contribute to the progression of cancers (38). In human epithelial ovarian tumors, cystatin B was a progression marker, which was associated with the transforming growth factor β (TGF-β) signaling pathway (39). In a previous study enrolling 27 patients of endometrial cancer, increased expression of cathepsin B was found as a predictor of more aggressive cancer behavior over time, suggesting its potential as being a tumor marker of unfavorable outcome (40). In the present study, significantly increased CSTB expression was noted in the cancer tissue, which might be related to the early stage of the cancer in our patient. Further study should be taken to elucidate the role of CSTB.
The HPGD gene encodes 15-hydroxyprostaglandin dehydrogenase, an important enzyme responsible for inactivating prostaglandins and associated eicosanoids via reducing the 15S-hydroxyl group, which have been demonstrated as an important factor of disease-associated pain and inflammation in patients with endometriosis (41). Decreased HPGD expression is associated with abnormal prostaglandin metabolism in endometriosis (42). Generally speaking, 15-hydroxyprostaglandin dehydrogenase is considered a tumor suppressor (43). It has been reported that HPGD is associated with various types of cancer, such as bladder cancer, gastrointestinal cancer, breast cancer, and cervical cancer (44-47). In a recent study, decreased inactivation of prostaglandins E2 and F2α (PGE2 and PGF2α) via 15-hydroxyprostaglandin dehydrogenase was noted in type II endometrial cancer, while low HPGD expression was associated with worse progression-free survival and overall survival (48). In line with the previous studies, we found decreased HPGD expression in endometrial adenocarcinoma tissue, which might be suppressed by upregulated miR-218-5p.
A major limitation of this study should be specified. This study was conducted mainly using the NGS data of a pair of tissue from a patient. Since it was mainly a single subject study, the findings might not be applied to other patients. However, our study provided insights to understand the pathogenic mechanisms of endometrial adenocarcinoma. After further validation, the potential targets identified in our study might provide scientific basis for developing novel diagnostic and treatment modalities for endometrial cancer.
In summary, we identified 56 significantly dysregulated genes in endometrial adenocarcinoma. These genes were involved in defense response, response to stimulus, and immune system process, as well as the pathway associated with HPV infection. In these genes, 6 genes were associated with poorer prognosis and 3 genes were associated with better prognosis. We further found two significantly dysregulated microRNA-mediated gene expression alterations in endometrial adenocarcinoma: downregulated hsa-miR-127-5p with upregulated CSTB and upregulated hsa-miR-218-5p with downregulated HPGD. These findings may contribute important new insights into possible novel diagnostic or therapeutic strategies for endometrial cancer.
Acknowledgements
The authors thank the staff of the Center for Research Resources and Development of Kaohsiung Medical University for their support.
Funding
This study was supported in part by research grants from the Ministry of Science and Technology (MOST 107-2320-B-037-011-MY3; MOST-108-2314-B-037-097-MY3), Kaohsiung Medical University Hospital Research Foundation (KMUHS10701; KMUHS10712; KMUH107-7R14), and the Kaohsiung Medical University (KMU-DK108003; KMU-Q108005).
Availability of data and materials
The data used and analyzed in this study are available from the corresponding author on reasonable request.
Authors Contributions
FHT, EMT, and PLK conceived the study. FHT, WAC, and MJT analyzed and interpreted the data. FHT, WAC, MJT, and PLK prepared the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board in Kaohsiung Medical University Hospital (KMUHIRB-F(II)-20180036), and the patient provided informed consent to participate this study.
Patient consent for publication
The patient provided informed consent to participate this study before the operation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. World Cancer Research Fund International. Endometrial cancer statistics. 2019.
2. Olesen TB, Svahn MF, Faber MT. et al. Prevalence of Human Papillomavirus in endometrial cancer: a systematic review and meta-analysis. Gynecol Oncol. 2014;134:206-215
3. Raglan O, Kalliala I, Markozannes G. et al. Risk factors for endometrial cancer: An umbrella review of the literature. Int J Cancer. 2018
4. Banno K, Yanokura M, Iida M, Masuda K, Aoki D. Carcinogenic mechanisms of endometrial cancer: involvement of genetics and epigenetics. J Obstet Gynaecol Res. 2014;40:1957-1967
5. Surveillance Epidemiology, End Results Program. National Cancer Institute: Cancer Stat Facts: Uterine Cancer. 2018.
6. Ouldamer L, Bendifallah S, Body G. et al. Predicting poor prognosis recurrence in women with endometrial cancer: a nomogram developed by the FRANCOGYN study group. Br J Cancer. 2016;115:1296-1303
7. Le Gallo M. LF, Bell D.W.: Next-Generation Sequencing. Hedrick Ellenson L. (eds) Molecular Genetics of Endometrial Carcinoma. Advances in Experimental Medicine and Biology, vol 943. Springer, Cham. 2017
8. Deng X, Nakamura Y. Cancer Precision Medicine: From Cancer Screening to Drug Selection and Personalized Immunotherapy. Trends Pharmacol Sci. 2017;38:15-24
9. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-222
10. Tsai MJ, Tsai YC, Chang WA. et al. Deducting MicroRNA-Mediated Changes Common in Bronchial Epithelial Cells of Asthma and Chronic Obstructive Pulmonary Disease-A Next-Generation Sequencing-Guided Bioinformatic Approach. Int J Mol Sci. 2019:20
11. Sheu CC, Tsai MJ, Chen FW. et al. Identification of novel genetic regulations associated with airway epithelial homeostasis using next-generation sequencing data and bioinformatics approaches. Oncotarget. 2017;8:82674-82688
12. Chang WA, Tsai MJ, Jian SF, Sheu CC, Kuo PL. Systematic analysis of transcriptomic profiles of COPD airway epithelium using next-generation sequencing and bioinformatics. Int J Chron Obstruct Pulmon Dis. 2018;13:2387-2398
13. Tsai MJ, Chang WA, Jian SF, Chang KF, Sheu CC, Kuo PL. Possible mechanisms mediating apoptosis of bronchial epithelial cells in chronic obstructive pulmonary disease - A next-generation sequencing approach. Pathol Res Pract. 2018;214:1489-1496
14. Tsai MJ, Chang WA, Liao SH, Chang KF, Sheu CC, Kuo PL. The Effects of Epigallocatechin Gallate (EGCG) on Pulmonary Fibroblasts of Idiopathic Pulmonary Fibrosis (IPF)-A Next-Generation Sequencing and Bioinformatic Approach. Int J Mol Sci. 2019:20
15. Lee WH, Tsai MJ, Chang WA. et al. Deduction of novel genes potentially involved in hypoxic AC16 human cardiomyocytes using next-generation sequencing and bioinformatics approaches. Int J Mol Med. 2018;42:2489-2502
16. Sheu CC, Chang WA, Tsai MJ, Liao SH, Chong IW, Kuo PL. Bioinformatic analysis of nextgeneration sequencing data to identify dysregulated genes in fibroblasts of idiopathic pulmonary fibrosis. Int J Mol Med. 2019
17. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-2120
18. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357-360
19. Siren J, Valimaki N, Makinen V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans Comput Biol Bioinform. 2014;11:375-388
20. Galipon J, Ishii R, Suzuki Y, Tomita M, Ui-Tei K. Differential Binding of Three Major Human ADAR Isoforms to Coding and Long Non-Coding Transcripts. Genes (Basel). 2017:8
21. Trapnell C, Roberts A, Goff L. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562-578
22. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:289-300
23. Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37-52
24. Vejnar CE, Zdobnov EM. MiRmap: comprehensive prediction of microRNA target repression strength. Nucleic Acids Res 40: 11673-11683. 2012
25. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015:4
26. Wang X. Improving microRNA target prediction by modeling with unambiguously identified microRNA-target pairs from CLIP-ligation studies. Bioinformatics. 2016;32:1316-1322
27. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146-152
28. Szklarczyk D, Morris JH, Cook H. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362-D368
29. Huang DW, Sherman BT, Tan Q. et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183
30. Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics. 2010;4:353-360
31. Ponten F, Jirstrom K, Uhlen M. The Human Protein Atlas-a tool for pathology. J Pathol. 2008;216:387-393
32. Delangle R, De Foucher T, Larsen AK. et al. The Use of microRNAs in the Management of Endometrial Cancer: A Meta-Analysis. Cancers (Basel). 2019:11
33. Montagnana M, Benati M, Danese E. et al. Aberrant MicroRNA Expression in Patients With Endometrial Cancer. Int J Gynecol Cancer. 2017;27:459-466
34. Roman-Canal B, Moiola CP, Gatius S. et al. EV-Associated miRNAs from Peritoneal Lavage are a Source of Biomarkers in Endometrial Cancer. Cancers (Basel). 2019:11
35. Dong P, Ihira K, Xiong Y. et al. Reactivation of epigenetically silenced miR-124 reverses the epithelial-to-mesenchymal transition and inhibits invasion in endometrial cancer cells via the direct repression of IQGAP1 expression. Oncotarget. 2016;7:20260-20270
36. Jurcevic S, Klinga-Levan K, Olsson B, Ejeskar K. Verification of microRNA expression in human endometrial adenocarcinoma. BMC Cancer. 2016;16:261
37. Pol E, Bjork I. Role of the single cysteine residue, Cys 3, of human and bovine cystatin B (stefin B) in the inhibition of cysteine proteinases. Protein Sci. 2001;10:1729-1738
38. Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764-775
39. Wang X, Gui L, Zhang Y, Zhang J, Shi J, Xu G. Cystatin B is a progression marker of human epithelial ovarian tumors mediated by the TGF-beta signaling pathway. Int J Oncol. 2014;44:1099-1106
40. Devetzi M, Scorilas A, Tsiambas E. et al. Cathepsin B protein levels in endometrial cancer: Potential value as a tumour biomarker. Gynecol Oncol. 2009;112:531-536
41. Lousse JC, Defrere S, Colette S, Van Langendonckt A, Donnez J. Expression of eicosanoid biosynthetic and catabolic enzymes in peritoneal endometriosis. Hum Reprod. 2010;25:734-741
42. Cobellis L, Razzi S, De Simone S. et al. The treatment with a COX-2 specific inhibitor is effective in the management of pain related to endometriosis. Eur J Obstet Gynecol Reprod Biol. 2004;116:100-102
43. Frank B, Hoeft B, Hoffmeister M. et al. Association of hydroxyprostaglandin dehydrogenase 15-(NAD) (HPGD) variants and colorectal cancer risk. Carcinogenesis. 2011;32:190-196
44. Celis JE, Ostergaard M, Basse B. et al. Loss of adipocyte-type fatty acid binding protein and other protein biomarkers is associated with progression of human bladder transitional cell carcinomas. Cancer Res. 1996;56:4782-4790
45. Yan M, Rerko RM, Platzer P. et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci USA. 2004;101:17468-17473
46. Lehtinen L, Vainio P, Wikman H. et al. 15-Hydroxyprostaglandin dehydrogenase associates with poor prognosis in breast cancer, induces epithelial-mesenchymal transition, and promotes cell migration in cultured breast cancer cells. J Pathol. 2012;226:674-686
47. Yao S, Xu J, Zhao K. et al. Down-regulation of HPGD by miR-146b-3p promotes cervical cancer cell proliferation, migration and anchorage-independent growth through activation of STAT3 and AKT pathways. Cell Death Dis. 2018;9:1055
48. Cummings M, Massey KA, Mappa G. et al. Integrated eicosanoid lipidomics and gene expression reveal decreased prostaglandin catabolism and increased 5-lipoxygenase expression in aggressive subtypes of endometrial cancer. J Pathol. 2019;247:21-34
Author contact
![]() Corresponding authors: Dr. Ming-Ju Tsai, School of Medicine, College of Medicine, Kaohsiung Medical University, No. 100, Shih-Chuan 1st Road, Kaohsiung 807, Taiwan. E-mail: SiegfriedTsaicom. Professor Po-Lin Kuo, Graduate Institute of Clinical Medicine, College of Medicine, Kaohsiung Medical University, No. 100, Shih-Chuan 1st Road, Kaohsiung 807, Taiwan. E-mail: kuopolinnet.tw
Corresponding authors: Dr. Ming-Ju Tsai, School of Medicine, College of Medicine, Kaohsiung Medical University, No. 100, Shih-Chuan 1st Road, Kaohsiung 807, Taiwan. E-mail: SiegfriedTsaicom. Professor Po-Lin Kuo, Graduate Institute of Clinical Medicine, College of Medicine, Kaohsiung Medical University, No. 100, Shih-Chuan 1st Road, Kaohsiung 807, Taiwan. E-mail: kuopolinnet.tw