Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(10):1320-1327. doi:10.7150/ijms.33728 This issue Cite
Review
Distinct gene expression characteristics in epithelial cell-Porphyromonas gingivalis interactions by integrating transcriptome analyses
Dongmei Zhang1, Jingya Hou2, Yun Wu2, Yanqing Liu2, Rong Li2, Tong Xu2, Junchao Liu1, Yaping Pan1 ![]()
1. Department of Periodontics and Oral Biology, School of Stomatology, China Medical University, Shenyang 110002, China
2. Department of Periodontics, School of Stomatology, China Medical University, Shenyang 110002, China
Received 2019-2-1; Accepted 2019-8-24; Published 2019-9-7
Abstract
Porphyromonas gingivalis is a pivotal periodontal pathogen, and the epithelial cells serve as the first physical barrier to defend the host from bacterial attack. Within this host-bacteria interaction, P. gingivalis can modify the host immune reaction and adjust the gene expression, which is associated with periodontitis pathogenesis and developing strategies. Herein, a meta-analysis was made to get the differential gene expression profiles in epithelial cells with or without P. gingivalis infection. The network-based meta-analysis program for gene expression profiling was used. Both the gene ontology analysis and the pathway enrichment analysis of the differentially expressed genes were conducted. Our results determined that 290 genes were consistently up-regulated in P. gingivalis infected epithelial cells. 229 gene ontology biological process terms of up-regulated genes were discovered, including “negative regulation of apoptotic process” and “positive regulation of cell proliferation/migration/angiogenesis”. In addition to the well-known inflammatory signaling pathways, the pathway associated with a transcriptional misregulation in cancer has also been increased. Our findings indicated that P. gingivalis benefited from the survival of epithelial cells, and got its success as a colonizer in oral epithelium. The results also suggested that infection of P. gingivalis might contribute to oral cancer through chronic inflammation. Negative regulation of the apoptotic process and transcriptional misregulation in cancer pathway are important contributors to the cellular physiology changes during infection development, which have particular relevance to the pathogenesis and progressions of periodontitis, even to the occurrence of oral cancer.
Keywords: Differentially expressed gene, Epithelial cell, Meta-analysis, Microarray, Periodontitis, Porphyromonas gingivalis
Introduction
Periodontitis is a ubiquitous inflammatory disease and the primary cause of tooth extraction in adults resulted from infection of periodontopathic bacteria. The host manifests an inflammatory immune response to this bacterial infection. Porphyromonas gingivalis is an opportunistic pathogen and mainly colonized in periodontal tissues [1]. More and more evidence shows that it is also a pathogen of systemic diseases. P. gingivalis may modify gene expression, even host immune response by degrading host cell surface proteins or receptors [2, 3]. Within this host-bacteria balance, the gene expression is directly correlated with individual susceptibility and influences the pathogenesis of periodontitis and disease progression. Several studies have suggested that plenty of genes were regulated by a host-protective response in gingival tissue of periodontitis. These related genes are critical components of immunological response, biological behavior, and metabolic signal pathways. Recently Kebschull M et al. have employed microarray analyses to speculate the gene expression signatures in periodontitis [4-8]. It is proved that microarray could supply more insight into the etiology of periodontitis. The growing microarray data offer some valuable clue for analyzing the host immune response of periodontitis. However, the discrimination against critical genes and relevant pathways based on these reports were limited because of the sample size, differences in research design and reporting methods in the separate studies.
A meta-analysis deals with the transcriptome data by combining the individual microarray study, and counters the above-mentioned disadvantage [9]. In the current study, a meta-analysis consisting of three separate microarray datasets was made to distinguish the gene expression profile in the epithelial cells with or without P. gingivalis infection. The present study gives us a synthesized assessment of the gene expression signatures in the epithelial cells with P. gingivalis infection. The differentially expressed genes (DEGs), the gene ontology (GO) terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways concerned in the transcription signatures were identified [10-13]. Our analysis would offer new understanding in the comprehension of periodontitis pathobiology, more information on the design of future research, and more tactics to block the progression of periodontitis.
Materials and Methods
Gene expression array data collection
An electronic search was performed in Gene Expression Omnibus (GEO, NCBI) database. Microarray data of the gene expression with the keywords “Porphyromonas gingivalis” or “gingival epithelial cells” or “oral epithelial cells” were downloaded. Epithelial cells infected with P.gingivalis were considered as “case group”, while non-infected epithelial cells as “control group”. Sample-sourced datasets from epithelial cells infected with other bacteria were excluded. Three separate microarray datasets were enrolled with raw data. The details of these datasets were listed in Table 1.
Information on transcriptome datasets used in the present analysis
| No. | GEO accession | Sample source | Platform | Sample size Case Control | Reference |
|---|---|---|---|---|---|
| 1 | GSE 97539 | human immortalized oral epithelial cell | GPL22516, [OElncRNAs520855F] Affymetrix Human OElncRNAs520855F Array | 3 3 | [2] |
| 2 | GSE 12121 | gingival epithelial HIGK cell | GPL570,[HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 4 4 | [7] |
| 3 | GSE 9723 | gingival epithelial HIGK cell | GPL96, [HG-U133A] Affymetrix Human Genome U133A Array | 4 4 | [8] |
The basic data were obtained from the 3 individual studies, including GEO accession; sample source; platform; numbers of cases or controls in the array data. Two of the datasets were performed in Affymetrix Human Genome U133 Array. The third dataset was conducted in Affymetrix Human OElncRNAs520855F Array. This analysis contains totally 22 GEO transcriptome datasets for epithelial cells infected with P.gingivalis or non-infected control. Microarray data for epithelial cells with P.gingivalis infection (n = 11) and non-infected control (n = 11) from GEO transcriptome database (http://www.ncbi. nlm.nih.gov/geo/) were compared for their transcriptome profiles.
The research was authorized by the Ethics Committee of School of Stomatology, China Medical University (Shenyang). They determined the current analysis of the public data set did not require the consents of the patients.
Differential gene expression analysis
DEGs were identified in epithelial cells with P.gingivalis infection or not, the datasets extracted from the individual microarray assays were submitted to the network-based meta-analysis program for gene expression profiling (http://www.inmex.ca/INMEX/), starting with multiple gene expression tables for meta-analysis [13-16]. First, the gene ID or probe ID was converted to its identical Entrez ID using the gene/probe conversion tool in INMEX. Second, the data were enrolled, processed, annotated and the intensity measurement of every ID was log2 transformed and normalized to zero mean and unit variance. While performing differential expression analysis on individual data set, the False Discovery Rate (FDR) of Benjamini-Hochberg's was set 0.05 in order to adjust the cut-off P-value. Third, data integrity was assessed as described previously [17-18]. Here we used the batch effect correction option so as to minimize the potential batch effect [19]. Then the meta-analysis for DGEs was made exploiting the INMEX web-based computational tool. Statistical analysis was performed by the INMEX program. The combined P values were detected according to Fisher's method. A P value of less than 0.05 was considered as the criterion of statistically significant in our study. The genes selected were classified based on the grade products of the combined P-value. The statistics were downloaded and visual exploration was performed. A Venn diagram was made to compare the difference between the findings of the meta-analysis and the individual study (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Gene Ontology Biological Process terms and KEGG pathway analysis
Subsequently, we exploited a web-based tool (named Database for Annotation, Visualization, and Integrated Discovery) to conduct the GO Biological Process (GO_BP) terms and KEGG pathway analysis [9, 18, 20]. DAVID Version 6.8 was used here. In this database, genes were divided into different classes according to their biological processes or molecular functions. Using this GO analysis we compared the DEGs and distributed them into a functional systematization. Gene Entrez ID was uploaded and analyzed for its GO Biological Process Annotation with functional annotation chart after the identifier was selected. Pathway annotations of the DEGs were acquired from the KEGG database [12, 19]. Pathway categories with a Benjamini-corrected P-value < 0.05 were utilized to determine a critical analysis. Our figures listed the typical GO biological process terms, which were chosen from the functional annotation charts that were significantly enriched at the top. Representative KEGG pathways chosen from the most significantly enriched charts were shown in our figures.
Results
DEGs in infected and non-infected epithelial cells
First of all, we compared the genome-wide gene expressions of infected and non-infected epithelial cells across microarray datasets. There were 362 genes differentially expressed in the two groups of epithelial cells. Among the 362 DEGs, 290 genes were significantly up-regulated (Table S1), and 72 genes were significantly down-regulated (Table S2). The up-regulated DEGs consisted of PLK2, CYP24A1, TNFAIP3, PTGS2, EDN1, IL6, GADD45A, IL1B, etc. The top 50 up-regulated DEGs were shown in the heatmap (Fig. 1).
Heat map comparison of the differential gene expression in epithelial cells across the individual datasets with P. gingivalis infection or not. Each row means a differentially expressed gene (DEG), each column represents a different GSE data sets. The top 50 misregulated DEGs were included in the map.
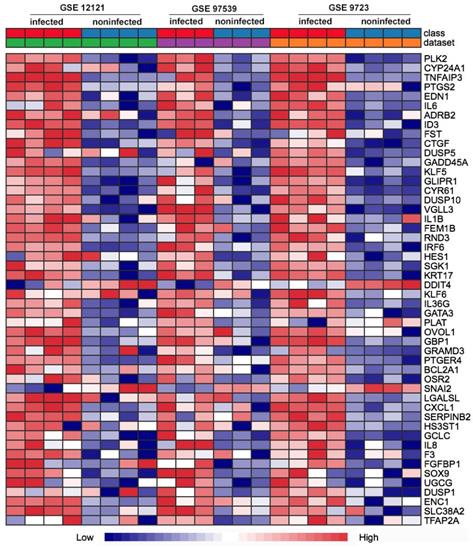
The most consistently up- and down-regulated DEGs of top 20 in epithelial cells, in the comparison of infected and non-infected epithelial cells, were shown in Table 2.
Top 20 up-regulated and down-regulated differentially expressed genes (DEGs) in P. gingivalis-infected epithelial cells compared to non-infected cells
| Entrez ID | Gene Symbol | Combined T stat | Combined P value | Entrez ID | Gene Symbol | Combined T stat | Combined P value |
|---|---|---|---|---|---|---|---|
| Up-regulated genes | Down-regulated genes | ||||||
| 10769 | PLK2 | -75.763 | <1.0E-10 | 54541 | DDIT4 | 39.654 | <1.0E-04 |
| 1591 | CYP24A1 | -59.072 | <1.0E-07 | 6591 | SNAI2 | 35.697 | <0.001 |
| 7128 | TNFAIP3 | -54.414 | <1.0E-06 | 7071 | KLF10 | 28.802 | <0.01 |
| 5743 | PTGS2 | -52.335 | <1.0E-06 | 26276 | VPS33B | 26.858 | <0.01 |
| 1906 | EDN1 | -52.685 | <1.0E-06 | 11067 | C10orf10 | 26.815 | <0.01 |
| 3569 | IL6 | -53.09 | <1.0E-06 | 6574 | SLC20A1 | 26.779 | <0.01 |
| 154 | ADRB2 | -51.163 | <1.0E-06 | 55844 | PPP2R2D | 26.31 | <0.01 |
| 3399 | ID3 | -50.168 | <1.0E-06 | 117247 | SLC16A10 | 26.115 | <0.01 |
| 10468 | FST | -49.696 | <1.0E-06 | 10224 | ZNF443 | 24.8 | <0.01 |
| 1490 | CTGF | -49.837 | <1.0E-06 | 4016 | LOXL1 | 24.512 | <0.01 |
| 1847 | DUSP5 | -49.125 | <1.0E-06 | 79759 | ZNF668 | 24.307 | <0.01 |
| 1647 | GADD45A | -48.243 | <1.0E-06 | 10561 | IFI44 | 24.232 | <0.01 |
| 688 | KLF5 | -47.733 | <1.0E-05 | 27034 | ACAD8 | 24.179 | <0.01 |
| 11010 | GLIPR1 | -47.307 | <1.0E-05 | 54014 | BRWD1 | 24.036 | <0.01 |
| 3491 | CYR61 | -46.897 | <1.0E-05 | 9778 | KIAA0232 | 23.467 | <0.01 |
| 11221 | DUSP10 | -46.685 | <1.0E-05 | 5154 | PDGFA | 23.45 | <0.01 |
| 389136 | VGLL3 | -46.265 | <1.0E-05 | 8772 | FADD | 23.374 | <0.01 |
| 3553 | IL1B | -44.656 | <1.0E-05 | 1390 | CREM | 23.33 | <0.01 |
| 10116 | FEM1B | -44.385 | <1.0E-05 | 211 | ALAS1 | 23.256 | <0.01 |
| 390 | RND3 | -43.556 | <1.0E-05 | 283871 | PGP | 23.211 | <0.01 |
Left columns: up-regulated DEGs, combined T stat < 0; Right columns: down-regulated DEGs, Combined T stat > 0
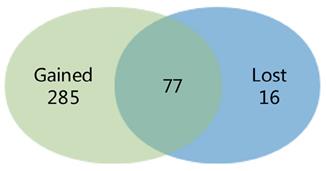
We compared the difference between the findings of the meta-analysis and the separate study, and a Venn diagram was made. 285 DEGs were distinguished in the meta-analysis only, which called gained DEGs. While only 16 DEGs were found in the separate study only, which called lost DEGs (Fig. 2).
Venn diagram showing the comparison of the results of the meta-analysis and the individual study. The overlying part represents differentially expressed genes (DEGs) determined in epithelial cells in the two investigations, gained DEGs indicate those charactered in the meta-analysis only and lost DEGs indicate those in the individual analysis only.
Functional classification and pathway assignment of DEG
GO analysis was carried out, and the DEGs were classified into different hierarchical categories based on the GO database. 229 GO Biological Process terms of up-regulated genes were discovered (Table S3). It has been shown that the significantly enriched GO terms for the DEGs included “negative regulation of apoptotic process”, “aging”, “positive regulation of cell proliferation”, “positive regulation of cell migration”, and “positive regulation of angiogenesis”. The top 20 enriched GO terms for up-regulated DEG in epithelial cells were shown in Fig. 3.
The gene ontology classification of the up-regulated genes in epithelial cells with P. gingivalis infection than those without infection. P and FDR < 0.05 were considered as the criterion of statistically significant in GO category investigation.
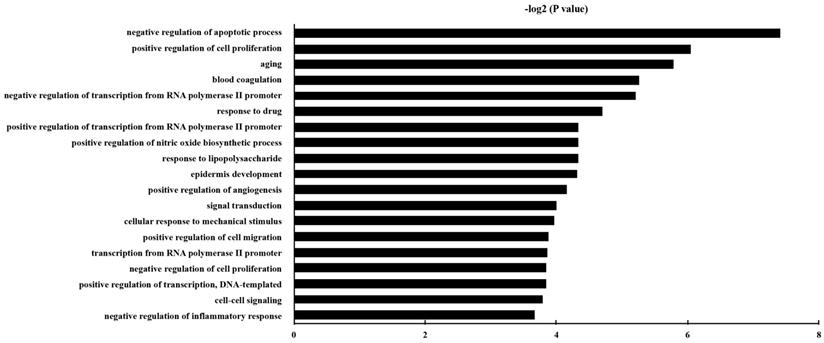
Here, we paid more attention to the first GO terms, namely negative regulation of apoptotic process. And the involved DEGs associated with up-regulated biological process categories of negative regulation of apoptotic process were listed in Table 3.
Top 10 of differentially expressed genes list associated with up-regulated gene ontology biological process categories of negative regulation of the apoptotic process
| Entrez ID | Gene Symbol | Gene Name |
|---|---|---|
| 597 | BCL2A1 | BCL2 related protein A1 |
| 596 | BCL2 | BCL2, apoptosis regulator |
| 580 | BARD1 | BRCA1 associated RING domain 1 |
| 8837 | CFLAR | CASP8 and FADD like apoptosis regulator |
| 2627 | GATA6 | GATA binding protein 6 |
| 55679 | LIMS2 | LIM zinc finger domain containing 2 |
| 1482 | NKX2-5 | NK2 homeobox 5 |
| 5292 | PIM1 | Pim-1 proto-oncogene, serine/threonine kinase |
| 6662 | SOX9 | SRY-box 9 |
| 301 | ANXA1 | annexin A1 |
KEGG pathway analysis was made in order to annotate the functional categories of DEGs as well. Here, 38 up-regulated pathways were found (Table S4). The 20 most significantly up-regulated pathways for DEGs were demonstrated in Fig. 4.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of the up-regulated genes in P. gingivalis-infected cells compared to the non-infected cells. P and FDR < 0.05 were considered as the criterion of statistically significant in the pathway analysis.
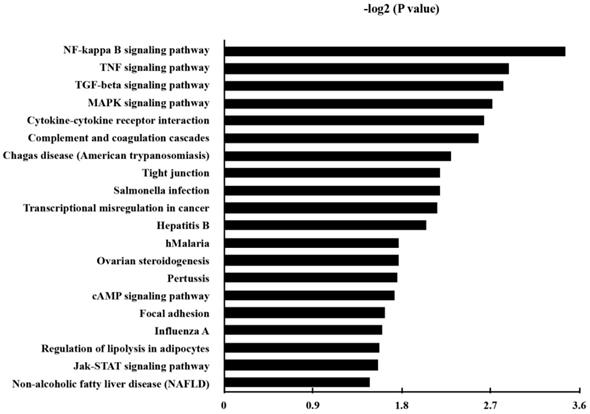
Besides the well-known inflammatory signaling pathways, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway, tumor necrosis factor (TNF) signaling pathway, mitogen-activated protein kinase (MAPK) signaling pathway, we found that the pathway associated with a transcriptional misregulation in cancer, Janus kinases/signal transducer and activator of transcription (JAK/Stat) pathway, and transforming growth factor-β (TGF-β) signaling pathway, had also been increased. Representatively, the involved DEGs of the KEGG pathways of transcriptional misregulation in cancer were listed in Table 4.
Top 10 of differentially expressed genes list associated with up-regulated Kyoto Encyclopedia of Genes and Genomes pathways of transcriptional misregulation in cancer
| Entrez ID | Gene Symbol | Gene Name |
|---|---|---|
| 597 | BCL2A1 | BCL2 related protein A1 |
| 3576 | CXCL8 | C-X-C motif chemokine ligand 8 |
| 1053 | CEBPE | CCAAT/enhancer binding protein epsilon |
| 4211 | MEIS1 | Meis homeobox 1 |
| 3569 | IL6 | interleukin 6 |
| 7403 | KDM6A | lysine demethylase 6A |
| 4353 | MPO | myeloperoxidase |
| 5327 | PLAT | plasminogen activator, tissue type |
| 5328 | PLAU | plasminogen activator, urokinase |
| 4609 | MYC | v-myc avian myelocytomatosis viral oncogene homolog |
Discussion
As a Gram-negative anaerobic bacterium, P. gingivalis is one of the most prominent pathogens in the etiology of chronic periodontitis. Epithelial cells are now considered to be the first immune barrier in the pathogenesis of periodontal disease confronted by the microorganism that colonizes or even intrudes mucosal surfaces. The intercommunication of P. gingivalis with epithelial cells involves several intricate signaling pathways [21-23]. Recognition of the pertinent genes and signal pathways concerned is pivotal, so we could comprehend the cellular and molecular changes during the periodontitis onset and disease progression. Considering P. gingivalis can not only invade the local periodontal tissue but also persist in other systems of the human being (for example esophagus tissue, respiratory and vascular), we should be more cautious in the active performance of P. gingivalis with epithelial cells.
Now we have obtained the transcriptomic profile with our meta-analysis using several public microarray data. Taking advantage of this enhanced statistical power, our results were more reliable [24-25]. We identified the DEGs, the corresponding Biological Process and pathways in P. gingivalis-infected epithelial cells based on comparisons to the GO [8,9] and KEGG [12-13] databases, respectively. The present results indicated the changes in gene expression feature modulated by some signal pathways were related to host-bacterial balance.
In our meta-analysis we showed several genes about negative regulation of the apoptotic process, including BCL2A1, BCL2, BARD1, CFLAR, were significantly up-regulated in epithelial cells infected with P. gingivalis compared to non-infected ones. It is conceived that P. gingivalis improves the survival of host cells by directly inhibiting different pro-apoptotic pathways [3, 26-28]. Inducing the durability of epithelial cells is crucial for the survival of P. gingivalis in these cells. It is reported that this resistance to apoptosis may be related to the BCL2 protein family, including BCL-2 and BAX [29]. This suppressed apoptosis during P. gingivalis infection was phosphatidylinositol 3 kinase/Protein Kinase B (PI3K/Akt) -dependent, furthermore, the PI3K signal pathway could regulate transcription or post-translational modification of certain BCL2 family member [3].
Our results verified that P. gingivalis infection has numerous anti-apoptotic effects on epithelial cells (such as activation of JAK/Stat pathways, NF-κB signal pathway, etc). The diversity of these multiple genes and pathways demonstrates that P. gingivalis may affect apoptotic pathways at different levels. Some of the up-regulated genes and pathways surround the mitochondria, which is a critical component of the intrinsic apoptosis process. The analysis of our study, along with others reports, showed that P. gingivalis suppressed the apoptosis of epithelial cell partially through the JAK/Stat signal pathway, which regulates the inherent mitochondria cell death mediated by the caspase-dependent apoptotic pathway.
At the meantime, P. gingivalis participates in the regulation of the extrinsic apoptotic pathway. Our KEGG pathway analysis demonstrated NF-κB signal pathway was up-regulated. It is reported that NF-κB, together with TNF and FasL, takes its role in the composition of the extrinsic apoptotic process [30]. NF-κB activation serves as a primary mechanism to protect cells against apoptotic stimulus such as TNF [31]. Current findings and early reports verify that the ability of P. gingivalis to benefit the survival of epithelial cells may assist its successful colonization in the oral mucosal epithelium.
Besides, the current analysis found that the transcriptional misregulation in cancer, another KEGG pathway, was up-regulated. Related genes expression, including C-X-C motif chemokine ligand 8 (CXCL8), CCAAT/enhancer binding protein epsilon (CEBPB), Meis homeobox 1 (MEIS1), interleukin 6 (IL-6), myeloperoxidase (MPO) and plasminogen activator urokinase (PLAU) were increased significantly.
IL6 is regarded as a tumor-promoting cytokine in diverse kinds of cancer including oral squamous cell carcinoma (OSCC) [32-35]. It is considered as one of the most promising markers for early diagnosis of tongue squamous cell carcinoma, which was the most usual kind of OSCC. Geng et al. revealed that both CEBPB and IL6 were up-regulated in late OSCC and CEBPB was activated as an upstream regulator of IL6. Moreover, CEBPB was recently identified as one of the master regulators in cancer biology [35,36].
Nowadays, more researches have reported a significant increase of CXCL8 and its mRNA in P. gingivalis-infected epithelial cells or oral squamous cells [37-39]. Ha's study found that infection by P. gingivalis stimulated the tumorigenic features and invasiveness of oral squamous cells, which was correlated with enhanced expression of CXCL8 [37]. PLAU, the serine protease, has been indicated in promoting pericellular proteolysis and oncogenic signal transduction in cancer cells. Yoshizawa implied that cases with PLAU/PLAU receptor/maspin expression should be considered as the high risk of poor prognosis of OSCC, which may lead to severe invasiveness, as well as the increased risk for cervical lymph node metastasis [40].
In addition, over-expression of MEIS1 in acute leukemia has been observed [41], which is related to the shorted latency and accelerated progression of acute leukemia [42]. The enhancer E9 mediates an auto-regulatory loop which contributes to the consistent expression of MEIS1 in leukemia. It was reported that MPO levels were dramatically enhanced in the bronchoalveolar lavage fluid and positively related to the carcinogenicity of inflammatory in lung cancer formation [43-44]. MPO is conceived as a critical mediator of inflammatory-promoted tumorigenesis, and its pro-tumor effects may take place in the window just after tumor onset in the lung [45].
So far, several studies have attempted to elucidate the role of P. gingivalis in contribution to oral cancer. Previous research by our group showed P. gingivalis enhanced the cell proliferation, migration, and invasive ability of human oral epithelial cells. Thus it promoted the cells tumorigenic properties [2]. Gonda et al. reported that chronic inflammation induced by bacterial infection drove carcinogenesis, involving induction, progression, invasion, and metastasis [33]. It is believed that the inflammatory microenvironment was an integral part of oral cancer [2,32-35,46,47].
Our KEGG pathway analysis further confirmed P. gingivalis played a role in promoting tumor transformation in some inflammatory conditions. Here pathways enrichment investigation showed that several pathways were enhanced significantly, including the NF-κB pathway, TNF pathway, TGF-beta pathway, MAPK signal pathway, and cytokine-cytokine receptor interaction. These inflammation-related signaling pathways were listed as the top 5 of our KEGG pathway analysis results. Here we believe that this chronic bacteria infection should be identified as probable effector which may result in oral cancer.
Generally speaking, the current meta-analysis using microarray datasets offered us an outlook about the gene expression characteristics in epithelial cells infected with P.gingivalis. The bioinformatics analyses of the up-regulated genes showed us that the GO terms of the negative regulation of apoptotic process, several inflammation-related signaling pathways, and the transcriptional misregulation in cancer were significantly enriched. It provided us some new sights into the alteration in cytopathogenicity that took place in the process of infection, which had particular relevance to periodontitis pathogenesis and developing strategies. Meanwhile, it provided more evidence to support the view that P.gingivalis might be related to the occurrence and development of OSCC.
Abbreviations
DEGs: differentially expressed genes; FDR: False Discovery Rate; GEO: Gene Expression Omnibus; GO: gene ontology; INMEX: integrative meta-analysis of expression data; KEGG: Kyoto Encyclopedia of Genes and Genomes; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; OSCC: oral squamous cell carcinoma; TGF-β: transforming growth factor-beta; TNF: tumor necrosis factor.
Supplementary Material
Supplementary table: All the up-regulated genes were listed in supplementary table S1; All the down-regulated genes were listed in supplementary table S2; GO Biological Process terms of up-regulated genes were listed in supplementary table S3; KEGG pathway of up-regulated genes were listed in supplementary table S4.
Acknowledgements
Funding Statement
This study was supported by grants from National Natural Science Foundation of China (81970943) and Provincial Natural Science Foundation of Liaoning, China (201800506).
Data Availability
The datasets used and/or analyzed during the current study are available in the Supplementary Materials.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jungnam L, Roberts JS, Atanasova KR. et al. Human Primary Epithelial Cells Acquire an Epithelial-Mesenchymal-Transition Phenotype during Long-Term Infection by the Oral Opportunistic Pathogen, Porphyromonas gingivalis. Front Cell Infect Microbiol. 2017;7:493. doi: 10.3389/fcimb.2017.00493
2. Geng F, Liu J, Guo Y. et al. Persistent Exposure to Porphyromonas gingivalis Promotes Proliferative and Invasion Capabilities, and Tumorigenic Properties of Human Immortalized Oral Epithelial Cells. Front Cell Infect Microbiol. 2017;7:57. doi: 10.3389/fcimb.2017.00057
3. Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008;154:2897-2903
4. Kebschull M, Demmer RT, Grün B. et al. Gingival Tissue Transcriptomes Identify Distinct Periodontitis Phenotypes. J Dent Res. 2014;93:459-468
5. Abe D, Kubota T, Morozumi T. et al. Altered gene expression in leukocyte transendothelial migration and cell communication pathways in periodontitis-affected gingival tissues. J Periodontal Res. 2011;46:345-353
6. Demmer RT, Behle JH, Wolf DL. et al. Transcriptomes in Healthy and Diseased Gingival Tissues. J Periodontol. 2008;79:2112-2124
7. Mans JJ, von Lackum K, Dorsey C. et al. The degree of microbiome complexity influences the epithelial response to infection. BMC Genomics. 2009;10:1-13
8. Handfield M, Mans JJ Zheng G. et al. Distinct transcriptional profiles characterize oral epithelium-microbiota interactions. Cell Microbiol. 2005;7:811-823
9. Zhang LL, Zhang ZN, Wu X. et al. Transcriptomic meta-analysis identifies gene expression characteristics in various samples of HIV-infected patients with nonprogressive disease. J Transl Med. 2017;15:91. doi: 10.1186/s12967-017-1294-5
10. Ashburner M, Ball CA, Blake JA. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25-29
11. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44-57
12. Du J, Yuan Z, Ma Z. et al. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol BioSyst. 2014;10:2441-2447
13. Xia J, Fjell CD, Mayer ML. et al. INMEX—a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 2013;41:63-70
14. Lee YH, Song GG. Meta-analysis of differentially expressed genes in ankylosing spondylitis. Genet Mol Res. 2015;14:5161-5170
15. Toro-Domínguez D, Carmona-Sáez P, Alarcón-Riquelme ME. Shared signatures between rheumatoid arthritis, systemic lupus erythematosus and Sjögren's syndrome uncovered through gene expression meta-analysis. Arthritis Res Ther. 2014;16:489
16. Santiago JA, Potashkin JA. Network-based metaanalysis identifies HNF4A and PTBP1 as longitudinally dynamic biomarkers for Parkinson's disease. Proc Natl Acad Sci USA. 2015;112:2257-2262
17. Bolstad BM, Irizarry RA, Astrand M. et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185-193
18. Sun Y, Sang Z, Jiang Q, Ding X. et al. Transcriptomic characterization of differential gene expression in oral squamous cell carcinoma: a meta-analysis of publicly available microarray data sets. Tumour Biol. 2016;37:15913-15924
19. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118-127
20. Barrett T, Troup DB, Wilhite SE. et al. NCBI GEO: archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011;39:1005-1010
21. Lamont R J, Jenkinson HF. Life Below the Gum Line: Pathogenic Mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244-1263
22. Nagihan Bostanci, Belibasakis GN. Doxycycline inhibits TREM-1 induction by Porphyromonas gingivalis. FEMS Immunol Med Microbiol. 2012;66:37-44
23. Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. 2012;27:409-419
24. Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat Rev Genet. 2013;14:89-99
25. Lee YH, Nath SK. Systemic lupus erythematosus susceptibility loci defined by genome scan meta-analysis. Hum Genet. 2005;118:434-443
26. Yilmaz O, Jungas T, Verbeke P. et al. Activation of the Phosphatidylinositol 3-Kinase/Akt Pathway Contributes to Survival of Primary Epithelial Cells Infected with the Periodontal Pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743-3751
27. Mao S, Park Y, Hasegawa Y. et al. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007;9:1997-2007
28. Whitmore SE, Lamont RJ. Oral bacteria and cancer. PLoS Pathog. 2014;10(3):e1003933. doi: 10.1371/journal.ppat.1003933
29. Nakhjiri S F, Park Y, Yilmaz Ö. et al. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol Lett. 2001;200:145-149
30. Beyaert R, Loo G V, Heyninck K. et al. Signaling to gene activation and cell death by tumor necrosis factor receptors and fas. Int Rev Cytol. 2002;214:225-272
31. Brozovic S, Sahoo R, Barve S. et al. Porphyromonas gingivalis enhances FasL expression via up-regulation of NFκB-mediated gene transcription and induces apoptotic cell death in human gingival epithelial cells. Microbiology. 2006;152:797-806
32. Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation and Cancer. Cell. 2010;140:883-899
33. Gonda TA, Tu S, Wang TC. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle. 2009;8:2005-2013
34. Hussein AA, Forouzanfar T, Bloemena E. et al. A review of the most promising biomarkers for early diagnosis and prognosis prediction of tongue squamous cell carcinoma. Br J Cancer. 2018;119:724-736
35. Geng F, Wang Q, Li C. et al. Identification of Potential Candidate Genes of Oral Cancer in Response to Chronic Infection With Porphyromonas gingivalis Using Bioinformatical Analyses. Front Oncol. 2019;9:91. doi: 10.3389/fonc.2019.00091
36. Califano A, Alvarez MJ. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer. 2017;17:116-130
37. Na NH, Woo BH, Kim DJ. et al. Prolonged and repetitive exposure to Porphyromonas gingivalis increases aggressiveness of oral cancer cells by promoting acquisition of cancer stem cell properties. Tumour Biol. 2015;36:9947-9960
38. Savitri IJ, Ouhara K, Fujita T. et al. Irsogladine maleate inhibits Porphyromonas gingivalis-mediated expression of toll-like receptor 2 and interleukin-8 in human gingival epithelial cells. J Periodontal Res. 2015;50:486-493
39. Kusumoto Y, Hirano H, Saitoh K. et al. Human gingival epithelial cells produce chemotactic factors interleukin-8 and monocyte chemoattractant protein-1 after stimulation with Porphyromonas gingivalis via toll-like receptor 2. J Periodontol. 2004;75:370-379
40. Yoshizawa K, Nozaki S, Kitahara H. et al. Expression of urokinase-type plasminogen activator/urokinase-type plasminogen, activator receptor and maspin in oral squamous cell carcinoma: Association with, mode of invasion and clinicopathological factors. Oncol Rep. 2011;26:1555-1560
41. Kumar AR, Li Q, Hudson WA. et al. A role for MEIS1 in MLL-fusion gene leukemia. Blood. 2009;113:1756-1758
42. Wang Q, Li Y, Dong J. et al. Regulation of MEIS1 by Distal Enhancer Elements in Acute Leukemia. Leukemia. 2014;28:138-146
43. Vaguliene N, Zemaitis M, Lavinskiene S. et al. Local and systemic neutrophilic inflammation in patients with lung cancer and chronic obstructive pulmonary disease. BMC Immunol. 2013;14:36. doi: 10.1186/1471-2172-14-36
44. Rymaszewski AL, Tate E, Yimbesalu JP. et al. The Role of Neutrophil Myeloperoxidase in Models of Lung Tumor Development. Cancers. 2014;6:1111-1127
45. Pertuska JM, Moosebrook DR, Jakab GJ. et al. Myeloperoxidase-enhanced formation of (+-)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene-DNA adducts in lung tissue in vitro: a role of pulmonary inflammation in the bioactivation of a procarcinogen. Carcinogenesis. 1992;13:1075-1081
46. Chang C, Geng F, Shi X. et al. The prevalence rate of periodontal pathogens and its association with oral squamous cell carcinoma. Appl Microbiol Biotechnol. 2019;103:1393-1404
47. Mantovani A, Allavena P, Sica A. et al. Cancer-related inflammation. Nature. 2008;454:436-444
Author contact
![]() Corresponding author: Prof. Yaping Pan, Department of Periodontics and Oral Biology, School of Stomatology, China Medical University, Heping Distrct, Nanjing North Street No.117, Shenyang 110002, China. Email: yppanedu.cn; Phone/Fax: 86-24-31927813
Corresponding author: Prof. Yaping Pan, Department of Periodontics and Oral Biology, School of Stomatology, China Medical University, Heping Distrct, Nanjing North Street No.117, Shenyang 110002, China. Email: yppanedu.cn; Phone/Fax: 86-24-31927813