Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(7):1032-1041. doi:10.7150/ijms.34365 This issue Cite
Research Paper
Anti-Tumor Activity and Pharmacokinetics of AP25-Fc Fusion Protein
Dening Pei1,2,#, Jialiang Hu3,#, Chunming Rao2, Pengcheng Yu3, Hanmei Xu3, ![]() , Junzhi Wang1,2,
, Junzhi Wang1,2, ![]()
1. Department of Biochemistry and Molecular Biology, The State Key Laboratory of Cancer Biology, The Fourth Military Medical University, Xi'an 710032, China
2. Key Laboratory of the Ministry of Health for Research on Quality and Standardization of Biotech Products, National Institutes for Food and Drug Control, Beijing 100050, China
3. The Engineering Research Center of Peptide Drug Discovery and Development, China Pharmaceutical University, Nanjing 211198, China
#These authors contributed equally to this work.
Received 2019-2-24; Accepted 2019-5-21; Published 2019-6-10
Abstract
AP25 is an anti-tumor peptide with a high affinity for integrins. It exerts its anti-tumor activity by inhibiting angiogenesis and by directly inhibiting the growth of tumor cells. Its half-life time in vivo is only about 50 minutes, which limits its clinical application. In order to prolong the half-life time of AP25 while preserving its anti-tumor activity, several fusion proteins of AP25 and IgG4 Fc were designed and expressed; their anti-tumor activity and pharmacokinetics properties were evaluated. Firstly, four AP25-Fc fusion protein sequences were designed, and the corresponding proteins were expressed and purified. Based on the results of HUVEC migration inhibition assay, HUVEC and tumor cell proliferation inhibition assay and yields of expression by HEK293 cells, the fusion protein designated PSG4R was selected for further evaluation. The anti-tumor effect of PSG4R was then evaluated in vivo on HCT-116 nude mice xenograft model. And the pharmacokinetics properties of PSG4R were investigated in rats. The results showed that PSG4R could inhibit the growth of xenografts of human colon cancer cell line HCT-116 in nude mice by intravenous administration of 40 mg/kg once every two days. The half-life time of PSG4R was 56.270 ± 15.398 h. This study showed that the construction of AP25-Fc fusion protein could significantly prolong the half-life of AP25 while retaining its anti-tumor activity, which provides a new direction for new drug development of AP25.
Keywords: AP25-Fc, fusion protein, anti-angiogenesis, anti-tumor, half-life
Introduction
AP25 is a 25-amino-acid anti-tumor peptide with high affinity for integrin αvβ3 and α5β1 on the surface of vascular endothelial cells. It is a fusion polypeptide of an integrin ligand ACDCRGDCFCG (RGD-4C) peptide and an endostatin fragment ES-2 of 50-60 amino acids. It contains two disulfide bonds arranged as Cys2-Cys10 and Cys4-Cys8 [1]. In vivo experiments proved that it could effectively inhibit human breast cancer and colon cancer development with an effect equivalent to the chemical drug docetaxel [2,3]. The research on the mechanism of action of AP25 showed that it could inhibit angiogenesis by inhibiting the proliferation, migration and tubular formation of vascular endothelial cells, thus exerting its anti-tumor activity and directly inhibits the growth of tumor cells [4].
However, as a small molecular polypeptide with a molecular weight of 2.53 kDa, the half-life time of AP25 is only about 50 minutes, which greatly limits its clinical application [5]. At present, chemical modification and the fusion with a large protein are the most often used method to prolong the half-life of proteins or polypeptides. The most common techniques are Fc fusion proteins, serum albumin fusion proteins, and transferrin fusion proteins [7,8]. Fc-fusion proteins refer to the fusion of a functional protein with the Fc fragment of an antibody by the genetic engineering technology. This fusion strategy not only retains the biological activity of functional proteins, but also extends the half-life time, improves the stability, and facilitates the expression, purification and detection of the fusion proteins [9,10]. More than a dozen Fc-fusion protein drugs have been commercialized, with annual sales of more than 10 billion US dollars. In this study, four AP25-Fc fusion proteins with different sequences and linkers were designed and expressed. The fusion protein with high anti-tumor activity was screened out through in vitro experiments, and then the anti-tumor activity and pharmacokinetic parameters were evaluated in vivo, which presents a reference for its future development.
Materials and Methods
Materials
SPF-grade BALB/c male nude mice were obtained from Changzhou Cavens Laboratory Animal Co., Ltd; SPF-grade SD rats were from Qinglongshan Animal Breeding Farm, Nanjing. HUVEC, HCT-116 and HeLa cells were obtained from the American Type Culture Collection; HEK293 cell was from Shanghai Jin'an Co., Ltd. PcDNA3.1 (-) vector was purchased from Promega Company, USA. XD-202 fluorescent inverted biomicroscope was purchased from Nanjing Jiangnan Yongxin Optical Co., Ltd. Microplate reader was purchased from MD Company, USA. PCR amplifier was from ThermoFisher Company, USA. Nucleic acid protein quantifier was purchased from Thermo Company, USA; and Protein A beads (product number: AA0131) were purchased from Boglung (Shanghai) Biotechnology Co., Ltd.
AP25-Fc fusion proteins design, expression, and purification
Four fusion proteins were designed by fusing AP25 (ACDCRGDCFCGGGGIVRRADRAAVP) with the hIgG4 Fc fragment in different order using flexible linker (GGGGSGGGSGGGGGGS) or helical linker (AEAAAKEAAAKEAAAKEAAAKA). The domain arragements of the four fusion proteins were presented in Figure 1.
Schematic representation of AP25-Fc fusion protein domain arrangements. The fusion proteins displayed in panel A-D were named PPDN1, PPRT2, PRKN3 and PSG4R, respectively.

The DNA sequences of signal peptide-AP25-linker and hIgG4 FC fragment were separately amplified by PCR, then the two sequences were spliced to obtain the DNA sequence of signal peptide-AP25-linker-hIgG4 Fc. The target plasmids were obtained by a seamless cloning of the AP25-linker-hIgG4 Fc sequence into pcDNA3.1 (-) vector. The target plasmid was transformed into E. coli and sequenced after amplification. The correct plasmid was then transiently transfected into HEK293 cells and cultured in a shaking bed of 37 ˚C, 8% CO2 and 120 rpm for 5 days. The supernatants of cell culture medium, cell lysate supernatants and cell lysate precipitate were collected, respectively. The target protein was detected by SDS-PAGE and Western blot.
The supernatant of HEK293 cells was loaded into Protein A affinity chromatography column (1 ml) and the column was pre-equilibrated with 10 column volumes of equilibrium buffer (20 mM Tris, 500 mM NaCl, pH 8.0). The flow rate was adjusted to 2 mg/mL. The column was then washed with 5 column volumes of equilibrium buffer and eluted with 0.1 M Glycine (pH 3.0). The desired peaks were collected, dialyzed against PBS (pH 7.4) and preserved for further experiments.
HUVEC migration assay
Transwell upper chambers were coated with a thin film of serum-free matrigel and placed into 24-well plate. HUVEC cells were cultured in endothelial cell medium (ECM) supplemented with 5% fetal bovine serum (FBS) and 1% endothelial cell growth supplement (ECGS) at 37 ˚C and 5% CO2. After trypsinization, HUVEC cells were diluted to 1 × 108 cells/L with serum-free ECM and 100 µL of cells suspension was added to each chamber. The chambers were then supplied with 100 µL sample solution which includes the fusion protein (0.1, 0.2, 0.4, 0.8 or 1.6 µmol/L), AP25 (0.2, 0.4 or 0.8 µmol/L), the positive control Avastin (0.17 µmol/L), PBS solvent control, respectively. 100 µL serum-free ECM was used as a negative control. The migration of HUVEC cells was stimulated by adding 0.6 mL ECM containing 5% FBS and 1% ECGS into 24-well plate. The 24-well plate was incubated at 5% CO2 and 37 ˚C for 24 hours. Then the lower surfaces of the membranes were fixed at room temperature with absolute ethanol for 30 minutes, stained with 0.1% crystal violet for 10 minutes and observed under a microscopic. Four fields were randomly selected and photographed for cell counting. Five pictures were selected and the migrating cells were calculated by Photoshop software. Migration inhibition rate (MI) was calculated according to the formula MI (%) = 1- (Ntest/Ncontrol) × 100%, where N test was the number of cell migration in the test group and N control was the number of cell migration in the solvent control group.
HUVEC and tumor cell proliferation assay
HUVEC cells were cultured in ECM medium containing 5% FBS and 1% ECGS. HeLa cells and HCT-116 cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% new born calf serum (NBS). The cells were digested by trypsin and diluted into 2 × 107 cells/L. 100 µL cell suspension was inoculated into each well of a 96-well plate. After cell adherence, 100 µL of AP25-Fc fusion protein (0.4, 0.8, 1.6, 3.2, 6.3, 12.7, 25.4, 50.8 or 101.5 µmol/L) was added. Paclitaxel (111.7 µmol/L) and Endostatin (2.3 µmol/L) were, respectively, added as positive controls while serum-free DMEM medium was added into the negative control group well. 96-well plate was cultured in 37 ˚C, 5% CO2 for 48 hours, then 20 µL/well thiazolyl blue tetrazolium bromide (MTT) was added. After incubation for 4 hours, the medium was discarded, and 150 µL/well dimethyl sulfoxide (DMSO) was added. The absorbance was measured at 570 nm using microplate reader, and the half maximal inhibitory concentration (IC50) of each sample was calculated with 630 nm as the reference wavelength.
In vivo bioactivity evaluation of the fusion protein AP25-Fc
HCT-116 cells were subcutaneously injected in the right flank of 70 nude mice under sterile conditions at a density of 5 × 106 cells per mouse, and the diameter of transplanted tumors was measured by a vernier caliper every day. Sixty nude mice with good growth condition and good homogeneity of tumor size were selected when the tumors grew to about 80-100 mm3. They were randomly divided into 9 groups with 6 mice in each group, and the number of animals in the negative control group was doubled. The mice in each group were intravenously administrated with drugs at 0.2 mL/20 g body weight. After drug treatment for 21 days, the mice were observed for 2 days. The control group was given the same volume of saline. The treatment strategy was shown in Table 1. The diameter of the tumors and the weight of nude mice were measured every other day. On the 23rd day, the mice were executed and the tumor tissues were surgically removed. The tumors were weighed and fixed with 10% neutral formalin. The formula for calculating tumor volume (TV) is: TV = 1/2 × a × b2, where a and b represent length and width of the tumor, respectively. The relative tumor volume (RTV) is RTV = TVt/TV0, in which TV0 is measured on d0 and TVt is measured at indicated time. Relative tumor proliferation rate was indicated as T/C (%). T/C (%) = TRTV/CRTV × 100. TRTV was the treatment group RTV and CRTV was the control group RTV. The inhibition rate (%) = (average tumor weight in treatment group/average tumor weight in control group) × 100. The study (Code: YKDX20180123) was approved by the Ethic Committee of Nanjing OGpharma Co., Ltd on 29 Jan, 2018.
Experimental strategy for in vivo tumor inhibitory effect of the fusion protein.
| Group | Administration frequency | Dosage |
|---|---|---|
| Negative control | every day | 0.2 ml/20g |
| Avastin | day 1, 8 and 15 | 5 mg/kg |
| AP25 | every day | 10 mg/kg |
| Fusion protein (10 mg/kg) | once every five days | 10 mg/kg |
| Fusion protein (10 mg/kg) | once every three days | 10 mg/kg |
| Fusion protein (10 mg/kg) | once every two days | 10 mg/kg |
| Fusion protein (40 mg/kg) | once every five days | 40 mg/kg |
| Fusion protein (40 mg/kg) | once every three days | 40 mg/kg |
| Fusion protein (40 mg/kg) | once every two days | 40 mg/kg |
AP25-Fc fusion protein pharmacokinetic study
Six SD rats, half male and half female, were injected intravenously with the fusion protein at a dose of 5 mg/kg. 100 to 200 µL orbital blood samples were taken at 5 min before drug administration or 0 min, 5 min, 10 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 12 h, 18 h, 24 h, 36 h, 48 h, 72 h, 96 h, 120 h, 144 h, 168 h, 192 h, 216 h after drug administration. The blood was centrifuged, and the supernatants were collected and preserved at - 80 ˚C.
Plate was coated with 3200 ng/mL of the antigen diluted in the coating solution (carbonate buffer) and kept overnight at 4 ˚C. On the second day, the solution was removed, and the wells were washed, dried and blocked in a water bath at 37 ˚C for 1.5-2 hours. After washing, blood samples were added to the wells with 4 repeats for each condition. At the same time the antibody was diluted at a ratio of 1:32,000 and added to the plate. The reaction was conducted at 37 ˚C water bath for 2-2.5 h. Then the plate was washed, dried and 1:2000 diluted HRP enzyme labeled goat anti-mouse IgG was added to the wells. After incubation of the plate at 37 ˚C for 1.5 h, the plate was washed, dried and TMB substrate was added and incubated in dark for 20-30 minutes. Finally, the absorbance at 450 nm was measured using a microplate reader. To establish the standard curve, the fusion protein standard sample was diluted into concentrations (C) of 100-12800 ng/mL with blank SD rat plasma and PBS solution; the samples were preceded with the above method. The standard curve was made, and the OD (450 nm) value of each concentration gradient standard sample was recorded as B, and the OD (450 nm) value of sample without the fusion protein was recorded as B0. Logit-log linear regression fitting is performed by enzyme-linked immunosorbent assay (ELISA) Calc software, and the equation is y = a + bx, where p = B/B0, q = 1-p, y = ln(p/q), x = lg(C). The concentration of AP25-Fc fusion protein in blood samples was calculated according to the standard curve equation.
Results
Design, expression and purification of AP25-Fc fusion proteins
Four AP25-Fc fusion protein sequences were designed according to the linker sequence and the arrangement of AP25 and Fc segments. They were named PPDN1, PPRT2, PRKN3 and PSG4R, respectively. The domain arragements of the four fusion proteins were presented in Figure 1.
The target plasmid was transiently transfected into HEK293 cells. The supernatant of cell culture, cell lysate supernatant and cell lysate precipitate were collected, respectively, and the target protein was detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot (Figure 2A, B). The results showed that the four fusion proteins are mainly secreted into the supernatant of cell culture medium. Therefore, target protein was further purified from the supernatant of cell culture medium.
SDS-PAGE and Western blot analysis of the fusion protein with PPDN1 as an example. (A) SDS-PAGE analysis of HEK293 cells culture products. (B) Western blot analysis of HEK293 cells culture products. (C) SDS-PAGE analysis of the final product after Protein A purification. Sample 1: molecular weight marker; Sample 2: supernatant of cell culture; Sample 3: supernatant of cell lysate; Sample 4: precipitate of cell lysate; Sample 5: supernatant of cell culture; Sample 6: supernatant of cell lysate; Sample 7: precipitate of cell lysate; Sample 8: molecular weight marker; Sample 9: PPDN1 (reduced); Sample 10: PPDN1 (non-reduced).

Cell culture supernatant was purified by Protein A affinity column. The desired peaks were collected, dialyzed against PBS (pH 7.4) buffer and analyzed with reduced and non-reduced SDS-PAGE to detect the target protein (Figure 2C). The molecular weight of PPDN1 shown on the gel is between 30-40 kD which is higher than the molecular weight of 29.1kD calculated based on the theoretical amino acid sequence. This may be due to the glycosylation of the target protein. The yields of PPDN1, PPRT2, PRKN3 and PSG4R were 15, 20, 10 and 30 mg/L, respectively. The signal peptide for PPDN1 is the human IL-22 signal peptide. The signal peptide for PPRT2 is signal peptide for Mesencephalic astrocyte-derived neurotrophic factor. The signal peptide for PRKN3 is the signal peptide for human IgG2 heavy chain. The signal peptide for PSG4R is the signal peptide for Gaussia luciferase. Four signal peptides were used for comparison of their protein secretion efficiency.
HUVEC migration assay
As shown in Figure 3, AP25, PPDN1, PPRT2, PRKN3 and PSG4R significantly inhibited HUVEC migration with an inhibition rate of 53.0% ± 5.5%, 53.0% ± 4.5%, 22.0% ± 5.3%, 52.4% ± 6.8% and 64.4% ± 2.5%, respectively. The effect was higher for fusion protein PSG4R, PPDN1 and PRKN3, compared with the solvent control group. Typical images of the migrating cells under each treatment condition were shown in Supplementary Figure S1 to S5.
HUVEC migration inhibition assay. A, B, C, D, E refer to HUVEC migration inhibition rates by AP25, PPDN1, PPRT2, PRKN3 and PSG4R at different concentrations. PBS was used as a negative control and Avastin as a positive control. Data were expressed as Mean ± SD. *P<0.05 and **P<0.01, compared with the control group.

Cell proliferation assay
The results of the proliferation inhibition assay showed that AP25, fusion protein PPDN1, PPRT2, PRKN 3 and PSG4R could inhibit the proliferation of HUVEC, HCT116 and HeLa cells in a dose-dependent manner. As shown in Figure 4, the IC50 for AP25, PPDN1, PPRT2, PRKN 3 and PSG4R on HUVEC proliferation were 1.60 ± 0.15, 3.42 ± 0.44, 3.98 ± 0.63, 2.36 ± 0.27 and 1.82 ± 0.25 μmol/L, respectively. For inhibition of HCT116 cell proliferation, the IC50 values for AP25, PPDN1, PPRT2, PRKN 3 and PSG4R were 1.79 ± 0.31, 3.19 ± 0.41, 3.91 ± 0.49, 2.45 ± 0.23 and 2.02 ± 0.28 μmol/L, respectively. For inhibition of HeLa cell proliferation, the IC50 values for AP25, PPDN1, PPRT2, PRKN 3 and PSG4R were 4.27 ± 0.51, 6.92 ± 0.96, 7.84 ± 0.78, 5.86 ± 0.78 and 5.16 ± 0.81 μmol/L, respectively. Among the four fusion proteins, PSG4R and PRKN3 showed a relatively higher inhibitory activity in HUVEC proliferation inhibition whereas their inhibitory activities in cancer cell proliferation were similar with the other fusion proteins. From Figure 4, the IC50 values of AP25 and fusion proteins for HUVEC proliferation inhibition were similar to those for HCT116 cell proliferation inhibition and lower than those for HeLa cell proliferation inhibition. Therefore, HUVEC proliferation is more sensitive to these integrin antagonist proteins than some tumor cell lines. From Figure 3, AP25 and fusion protein PSG4R, PPDN1 and PRKN3 inhibited HUVEC migration with IC50 values below 1 μmol/L. As endothelial cell migration and proliferation are important processes for tumor angiogenesis, the data in Figure 3 and Figure 4 showed that PSG4R and PRKN3 have relatively better in vitro anti-angiogenic activity. As the preparation yield of PSG4R is the highest among the four fusion proteins, this fusion protein was prepared in a relatively large amount for in vivo bioactivity evaluation.
Cell proliferation inhibition assay. A, B and C were dose-response curves of HUVECs, HCT116 cells and HeLa cells under the treatment of AP25, PPDN1, PPRT2, PRKN3 and PSG4R at different concentrations.

In vivo bioactivity evaluation
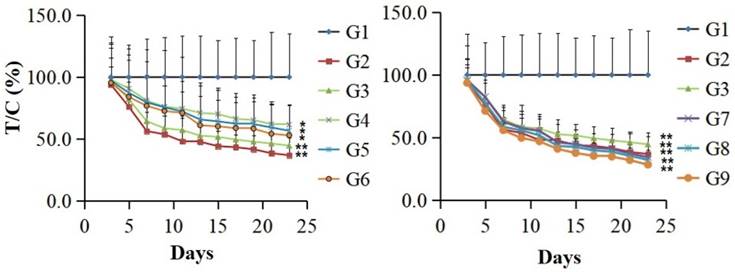
The in vivo anti-tumor activity of the fusion protein PSG4R was evaluated in a xenograft of colon cancer cell line HCT-116 in nude mice and was presented in Figure 5 and Figure 6. The results showed that for the mice that were administrated with Avastin (5mg/kg) on day 1, 8 and 15, they showed a T/C (%) of 36.9 ± 14.0% based on tumor volume and an inhibition rate of 61.2 ± 4.2% based on tumor weight on day 23. For the mice that were daily administrated with 10 mg/kg AP25, they showed a T/C (%) of 44.6 ± 9.5% and an inhibition rate of 52.4 ± 3.9% on day 23. Mice administrated with 10 mg/kg PSG4R with an interval of 5, 3 and 2 days showed a T/C (%) of 61.6 ± 16.4%, 56.7 ± 20.4% and 52.9 ± 24.2%, respectively and the corresponding inhibition rates were 37.8 ± 4.0%, 41.4 ± 4.5% and 46.3 ± 4.3%. Mice administrated with 40 mg/kg PSG4R with an interval of 5, 3 and 2 days showed a T/C (%) of 34.4 ± 5.9%, 32.3 ± 5.8% and 28.4 ± 6.2% respectively and the corresponding inhibition rates were 43.1 ± 1.7%, 47.7 ± 4.6% and 53.6 ± 2.8%. There was no significant difference in body weight of the mice between each group and the control group, and no obvious adverse reactions were observed. Administration of PSG4R (40 mg/kg) once every 2 days had a significant inhibition rate of HCT-116 growth in the xenograft tumor model in nude mice, which was lower than that of Avastin but higher than that of AP25.
Inhibition of the fusion protein PSG4R in the growth of colon cancer cell line HCT-116 on a xenograft model in nude mice. HCT-116 cells were subcutaneously injected in the right flank of nude mice. There were 6 mice in each drug treatment group and 12 mice in the negative control group. The mice in each group were intravenously administrated with drugs. Drug treatment strategy was shown in Table 1. The diameter of the tumors and the weight of nude mice were measured every other day. The relative tumor volume (RTV) is RTV=TVt/TV0, in which TV0 is measured on d0 and TVt is measured at indicated time. Relative tumor proliferation rate was indicated as T/C (%). T/C (%) = TRTV/CRTV × 100. TRTV was the treatment group RTV and CRTV was the control group RTV. G1, negative control; G2, 5 mg/kg Avastin on day 1, 8 and 15; G3, 10 mg/kg AP25 every day; G4, 10 mg/kg Fusion protein every five days; G5, 10 mg/kg Fusion protein every three days; G6, 10 mg/kg Fusion protein every two days; G7, 40 mg/kg Fusion protein every five days; G8, 40 mg/kg Fusion protein every three days; G9, 40 mg/kg Fusion protein every two days (*p<0.05, **p<0.01).
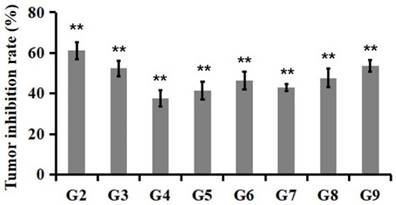
Tumor inhibition rate at the 23rd day after PSG4R administration. Animal grouping and drug treatment strategy were the same as in Figure 4. Mice were treated with drugs for 21 days. On the 23rd day, the mice were executed and the tumors removed. Tumor inhibition rates were calculated based on the weight of tumor tissues. The inhibition rate (%) = (average tumor weight in treatment group/average tumor weight in control group) × 100. G2, 5 mg/kg Avastin on day 1, 8 and 15; G3, 10 mg/kg AP25 every day; G4, 10 mg/kg Fusion protein every five days; G5, 10 mg/kg Fusion protein every three days; G6, 10 mg/kg Fusion protein every two days; G7, 40 mg/kg Fusion protein every five days; G8, 40 mg/kg Fusion protein every three days; G9, 40 mg/kg Fusion protein every two days (*p<0.05, **p<0.01).
The detection of pharmacokinetic parameters
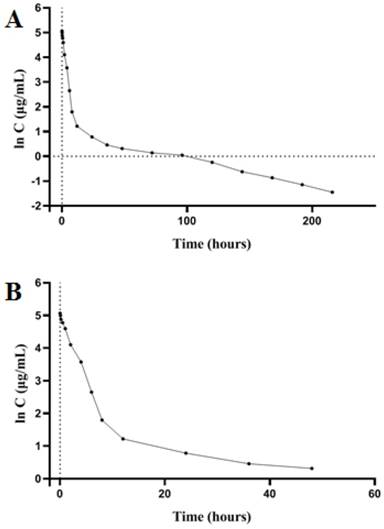
SD rats were intravenously injected with PSG4R and the pharmacokinetic procedure of PSG4R in rats was according to a one-compartment model. Pharmacokinetic parameters were calculated with DAS 1.0. The peak concentration (Cmax) was 158.429 ± 32.453 ug/mL. The elimination half-life (t1/2β) was 56.270 ± 15.398 h and the mean residence time (MRT) after administration was 37.441 ± 13.349 h. The clearance rate (CL) was 0.010 ± 0.009 L/h/kg, and the apparent distribution volume (Vd) was 2.769 ± 3.502 L/kg. The blood concentration-time curve is shown in Figure 7 and the pharmacokinetic parameters are shown in Table 2. From the results of pharmacokinetic study, the elimination half-life of PSG4R (56 hours) was much longer than AP25 (around 50 minutes).
Parmacokinetic parameters of PSG4R following a single i.v. administration to rats at a dose of 5 mg/kg.
| Parameter | Rats | Mean | SD | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| Ke (h-1) | 0.017 | 0.022 | 0.009 | 0.013 | 0.010 | 0.010 | 0.014 | 0.005 |
| t1/2β (h) | 41.878 | 32.171 | 76.810 | 53.874 | 66.540 | 66.348 | 56.270 | 15.398 |
| Vd (L/kg) | 1.871 | 10.472 | 0.767 | 2.243 | 0.575 | 0.683 | 2.769 | 3.502 |
| Tmax (h) | 0.167 | 0.083 | 0.083 | 0.083 | 0.083 | 0.083 | 0.097 | 0.031 |
| MRT 0-216 (h) | 11.775 | 16.987 | 25.197 | 31.715 | 26.679 | 25.428 | 22.963 | 6.613 |
| MRT 0-∞(h) | 14.820 | 40.597 | 33.344 | 50.610 | 54.954 | 30.323 | 37.441 | 13.349 |
| CL (L/h/kg) | 0.031 | 0.007 | 0.007 | 0.005 | 0.006 | 0.007 | 0.010 | 0.009 |
| Cmax (μg/mL) | 172.673 | 128.376 | 127.389 | 125.928 | 193.342 | 202.863 | 158.429 | 32.453 |
Plasma concentration-time curve of PSG4R after a single i.v. administration to rats at a dose of 5 mg/kg. (A) Plasma concentration-time curve of PSG4R within 216 hours after i.v. administration of PSG4R (y axis represents log scale while x axis is linear). Six rats were administrated with PSG4R. 100 µL blood sample was collected at each indicated time point. After centrifugation, the supernatant was collected for detection with an indirect competitive ELISA method. For pharmacokinetic parameters, please refer to Table 2. For convenience of observation, plasma concentration-time curve of PSG4R within 48 hours after i.v. administration of PSG4R was shown in panel B (y axis represents log scale while x axis is linear).
Discussion
Malignant tumors are the primary cause of deaths in human. Traditional chemical drugs have brought great pain to cancer patients because of their non-specific cytotoxicity and drug resistance development. In 1972, Folkman proposed the theory of tumor angiogenesis [11,12]. Since then many researches about the treatment of tumors by inhibiting tumor angiogenesis have been reported. At present, several anti-angiogenetic drugs have been commercialized and have achieved a good therapeutic effect, including Avastin [13,14], Abesip [15,16], Sunitinib [17] and endostatin [18,19].
Integrin blockers are an important class of anti-angiogenesis and anti-tumor drugs. Integrin is a transmembrane receptor that mediates the connection between cells and their external environment (e.g. extracellular matrix), which is mediated by the binding of the ligand with the RGD sequence. Integrins are the physiological "bridges" between extracellular matrix and cytoskeleton. They play an important role in cell adhesion and can also transmit extracellular signals to cells [20-22]. As regulators of tumor micro vascularization, integrins play an important role in the process of tumor invasion and metastasis. Many integrin molecules can influence angiogenesis (e.g. αvβ3, α5β1, α2β1, α1β1, etc.). Among them, αvβ3 is particularly important. It participates in activation and migration of endothelial cells, mediates endothelial cell proliferation and inhibits endothelial cell apoptosis, thus promoting angiogenesis [23-25]. Integrin can also participate in the transformation, growth, invasion, metastasis and apoptosis of cancer cells by transmitting specific signals or inducing gene expression [26,27]. Xu, H.M. et al. found that ES-2 had good anti-angiogenetic activity in vitro, but its activity in vivo was moderated [28]. In order for ES-2 to target tumor tissues, Xu and her colleagues designed AP25 by connecting RGD-4C (RGD-4C contains four cysteines, which form two disulfide bridges and confer the molecule higher stability [29,30]) and ES-2. AP25 targets integrin αvβ3 and α5β1 on the surface of vascular endothelial cells and several animal experiments showed that it has significant anti-tumor effect [1-3].
IgG-Fc fusion proteins are the combination of a bioactive protein with the Fc fragment of an IgG (CH2, CH3 functional region), generally prepared by gene recombination technology [31]. Because of high level of the proteases in tissues and the glomerular filtration, the plasma half-life of many bioactive proteins is relatively short, which limits their clinical application. However, the half-life of IgG in human body can reach 19-21 days. The Fc fragment of an Fc fusion protein increases the relative molecular weight of a fusion protein and avoids its filtration by the kidney. At the same time, Fc fragment can bind to the neonatal Fc receptor (FcRn) to avoid the degradation of the fusion protein, thus significantly prolongs the plasma half-life of the fusion protein, thereby reducing the frequency of drug injection and improving patients' compliance and tolerance to the treatment [32,33]. In addition to the long-action, Fc fragments can also improve the stability of molecules. Fc fusion proteins can form stable dimers through disulfide bonds in Fc hinges and Fc regions can be folded independently to ensure the stability of chaperone molecules in vivo and in vitro [34]. Fc fragments can also bind to the Fc gamma R (FcγR) on the surface of immune cells and play a variety of biological functions, such as mediating the transport through placental and mucosal barriers, inflammatory response, antibody-dependent cell-mediated phagocytosis (ADCP), antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [35,36]. In addition, fusion with Fc can improve the expression of protein in mammalian cells and can specifically bind to the Protein A affinity column, simplifying the purification process of Fc fusion protein, which is of great significance in the development of biological products [37,38].
In order to extend the half-life and preserve the anti-tumor activity of AP25, four different AP25-Fc fusion proteins were designed in this study, in two of them AP25 was located at the N-terminus and in the other two Fc segment was located at the N-terminus. Two were ligated with a helical linker AEAAAKEAAAKEAAAKEAAAKEAAAKA and the other two were ligated with a flexible linker GGGGSGGGSGGGGGS [39-41]. Considering that integrin is widely expressed in vivo, most cells can express more than one integrin on the surface, which plays a key role in many physiological processes. They not only participate in angiogenesis, invasion and metastasis of tumors, but also in physiological and pathological processes such as inflammation, wound healing and coagulation. In order to prevent the influence of the effect induced by the Fc fragment (ADCP, ADCC, CDD) and other physiological processes, the Fc segment used in this study was IgG4 Fc with lower affinity to Fc gamma Receptor, rather than IgG1 Fc with higher affinity to Fc gamma Receptor.
HUVEC cell migration inhibition assay was used to evaluate the anti-angiogenetic activity of the fusion protein [42] and HUVEC, HCT116, HeLa cell proliferation inhibition assay was used to evaluate the ability of the fusion protein to directly inhibit tumor cell proliferation. The results showed that PSG4R and PRKN3 had a higher anti-tumor activity in vitro. These results indicate that the arrangement of Fc segment and AP25 has a great influence on the anti-tumor activity of AP25, while the difference of linker sequence has no significant effect on the anti-tumor activity of AP25, suggesting that the arrangement of Fc segment and AP25 is related to the steric configuration of AP25.
Taking in consideration that the expression and purification yields of PPDN1, PPRT2, PRKN3 and PSG4R were about 15, 20, 10 and 30 mg/L, respectively, PSG4R was selected for large scale expression and purification. Pharmacokinetic study showed that the elimination half-life (t1/2β) of PSG4R in SD rats was 56.27 ± 15.40 h after tail vein injection of 5 mg/kg PSG4R, which was more than 60 times longer than that of AP25. Its in vivo anti-tumor activity was evaluated on the HCT-116 xenograft model in nude mice. The results showed that PSG4R (40 mg/kg or 1.33 × 10-6 mol/kg) administrated every 2 days could inhibit the xenograft growth of HCT116 in nude mice significantly in a dose-dependent manner. The inhibiting rate of PSG4R was similar to that of AP25 (10 mg/kg or 3.95 × 10-6 mol/kg) once a day because PSG4R had a longer half-life.
In conclusion, the fusion protein PSG4R has a long half-life, high anti-tumor activity in vivo and in vitro, and high expression and purification yield. It can be used as a candidate anti-cancer drug for further research and may expand the therapeutic spectrum of polypeptide AP25. Its development as a new anti-cancer drug may have significant social and market value.
Abbreviations
HUVEC: Human Umbilical Vein Endothelial Cells; IgG: immunoglobulin G; HEK293: Human Embryo Kidney 293 Cells; SPF: specific pathogen free; ECM: endothelial cell medium; FBS: fetal bovine serum; ECGS: endothelial cell growth supplement; MI: Migration inhibition rate; DMEM: Dulbecco's modified eagle medium; MTT: thiazolyl blue tetrazolium bromide; DMSO: dimethyl sulfoxide; IC50: half maximal inhibitory concentration; TV: tumor volume; RTV: relative tumor volume; ELISA: enzyme-linked immunosorbent assay; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; MRT: mean residence time; FcRn: neonatal Fc receptor; FcγR: Fc gamma R; ADCP: antibody-dependent cell-mediated phagocytosis; ADCC: antibody-dependent cell-mediated cytotoxiccity; CDC: complement-dependent cytotoxicity.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Dr. Wanzhou Zhao for his support for the In vivo bioactivity evaluation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang C, Yang YL, Pan L. et al. In vitro activity of anti-tumor peptide AP25. Chin Pharmacol Bull. 2012;28:1375-1378
2. Zhang XJ, Wang WJ, Wang JJ. et al. Anti-tumor activity of peptide AP25. Chin Pharmacol Bull. 2013;29:1225-1229
3. Wang JY, He JJ, Hao JC. et al. Inhibitory effect of combined usage of AP25 and Docetaxel in the growth of human breast cancer cell line on a xenograft model in nude mice. Chin Pharmacol Bull. 2015;31:1233-1238
4. Hu JL, Cheng T, Zhang LJ. et al. Anti-tumor peptide AP25 decreases cyclin D1 expression and inhibits MGC-803 proliferation via phospho-extracellular signal-regulated kinase-, Src-, c-Jun N-terminal kinase- and phosphoinositide 3-kinase-associated pathways. Mol Med Rep. 2015;12:4396-4402
5. Li Y, Xu H, Hu J. et al. Quantitative analysis and pharmacokinetics study of integrin antagonist AP25 in rat plasma. Protein Pept Lett. 2013;20:584-590
6. van Witteloostuijn SB, Pedersen SL, Jensen KJ. Half-Life Extension of Biopharmaceuticals using Chemical Methods: Alternatives to PEGylation. ChemMedChem. 2016;11:2474-2495
7. Kontermann RE. Half-life extended biotherapeutics. Expert Opin Biol Ther. 2016;16:903-915
8. Steiner D, Merz FW, Sonderegger I. et al. Half-life extension using serum albumin-binding DARPin(R) domains. Protein Eng Des Sel. 2017;30:583-591
9. Joo K, Park SJ, Choi Y. et al. Role of the Fc Region in the Vitreous Half-Life of Anti-VEGF Drugs. Invest Ophthalmol Vis Sci. 2017;58:4261-4267
10. Schafer K, Munn J, Khair K. et al. Pharmacokinetics, Safety, and Efficacy of Recombinant Factor VIII Fc Fusion Protein: A Practical Review. J Infus Nurs. 2017;40:65-75
11. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-1186
12. Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175:409-416
13. Kabbinavar F, Irl C, Zurlo A. et al. Bevacizumab improves the overall and progression-free survival of patients with metastatic colorectal cancer treated with 5-fluorouracil-based regimens irrespective of baseline risk. Oncol-Basel. 2008;75:215-223
14. Miller KD, Chap LI, Holmes FA. et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792-799
15. Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016;15:385-403
16. MacDonald DA, Martin J, Muthusamy KK. et al. Aflibercept exhibits VEGF binding stoichiometry distinct from bevacizumab and does not support formation of immune-like complexes. Angiogenesis. 2016;19:389-406
17. Funakoshi T, Latif A, Galsky MD. Risk of hematologic toxicities in cancer patients treated with sunitinib: a systematic review and meta-analysis. Cancer Treat Rev. 2013;39:818-830
18. Karamouzis MV, Moschos SJ. The use of endostatin in the treatment of solid tumors. Expert Opin Biol Ther. 2009;9:641-648
19. N Q, Ji H Zhao Z. et al. Endostar, a modified endostatin inhibits non small cell lung cancer cell in vitro invasion through osteopontin-related mechanism. EUR J Pharmacol. 2009;614:1-6
20. Mould AP. Analyzing Integrin-Dependent Adhesion. Curr Protoc Cell Biol. 2018 e69
21. Humphries JD, Chastney MR, Askari JA. et al. Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol. 2018;56:14-21
22. Galbraith CG, Davidson MW, Galbraith JA. Coupling integrin dynamics to cellular adhesion behaviors. Biol Open. 2018;7:e036806
23. Wang T, Huang J, Vue M. et al. alphavbeta3 Intergrin Mediates Radioresistance of Prostate Cancer Cells Through Regulation of Survivin. Mol Cancer Res. 2018 e101158
24. Vicente-Manzanares M, Sanchez-Madrid F. Targeting the integrin interactome in human disease. Curr Opin Cell Biol. 2018;55:17-23
25. Arun AS, Tepper CG, Lam KS. Identification of integrin drug targets for 17 solid tumor types. Oncotarget. 2018;9:30146-30162
26. Marsico G, Russo L, Quondamatteo F. et al. Glycosylation and Integrin Regulation in Cancer. Trends Cancer. 2018;4:537-552
27. Majumder P. Integrin-Mediated Delivery of Drugs and Nucleic Acids for Anti-Angiogenic Cancer Therapy: Current Landscape and Remaining Challenges. Bioengineering (Basel). 2018;5:e5040076
28. Xu HM, Chen LS, Yin RT. Biological activity of human endostatin derived synthetic peptide—Es-2. J China Pharm. 2006;37:346-348
29. Holtke C. isoDGR-Peptides for Integrin Targeting: Is the Time Up for RGD? J Med Chem. 2018;61:7471-7473
30. Zapp C, Minsky BB, Boehm H. Tuning RGD Motif and Hyaluronan Density to Study Integrin Binding. Front Physiol. 2018;9:1022
31. Lv LL, Yan ZH, Shi X. et al. Recombinant human tumor necrosis factor receptor type -IgG Fc fusion protein for treatment of occupational medicamentosa-like dermatitis induced by trichloroethylene. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2017;35:257-260
32. Kim NA, An IB, Lim HS. et al. Biophysical evaluation of hybrid Fc fusion protein of hGH to achieve basal buffer system. Int J Pharm. 2016;513:421-430
33. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715-725
34. Chakrabarti S, Barrow CJ, Kanwar RK. et al. Studies to Prevent Degradation of Recombinant Fc-Fusion Protein Expressed in Mammalian Cell Line and Protein Characterization. Int J Mol Sci. 2016;17:e17060913
35. Kim SJ, Kwak HH, Cho SY. et al. Pharmacokinetics, Pharmacodynamics, and Efficacy of a Novel Long-Acting Human Growth Hormone: Fc Fusion Protein. Mol Pharm. 2015;12:3759-3765
36. Zaharatos GJ, Yu J, Pace C. et al. HIV-1 and influenza antigens synthetically linked to IgG2a Fc elicit superior humoral responses compared to unmodified antigens in mice. Vaccine. 2011;30:42-50
37. Greene DG, Traylor SJ, Guo J. et al. Mechanisms of precipitate formation during the purification of an Fc-fusion protein. Biotechnol Bioeng. 2018;115:2489-2503
38. McCue J, Osborne D, Dumont J. et al. Validation of the manufacturing process used to produce long-acting recombinant factor IX Fc fusion protein. Haemophilia. 2014;20:e327-35
39. Gustavsson M, Lehtio J, Denman S. et al. Stable linker peptides for a cellulose-binding domain-lipase fusion protein expressed in Pichia pastoris. Protein Eng. 2001;14:711-715
40. Crasto CJ, Feng JA. LINKER: a program to generate linker sequences for fusion proteins. Protein Eng. 2000;13:309-312
41. Arai R, Ueda H, Kitayama A. et al. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001;14:529-532
42. Cerezo AB, Hornedo-Ortega R, Alvarez-Fernandez MA. et al. Inhibition of VEGF-Induced VEGFR-2 Activation and HUVEC Migration by Melatonin and Other Bioactive Indolic Compounds. Nutrients. 2017;9:e9030249
Author contact
![]() Corresponding authors: Dr. Junzhi Wang, Key Laboratory of the Ministry of Health for Research on Quality and Standardization of Biotech Products, National Institutes for Food and Drug Control, 2 Tiantanxili Road, Dongcheng District, Beijing 100050, China. Tel.: +86-010-6709-5782; E-mail: wangjzorg.cn; & Dr. Hanmei Xu, The Engineering Research Center of Peptide Drug Discovery and Development, China Pharmaceutical University, 639 Longmian Avenue, Jiangning District, Nanjing 211198, China. Tel.: +86-025-8327-1007; E-mail: xuhanmeiedu.cn.
Corresponding authors: Dr. Junzhi Wang, Key Laboratory of the Ministry of Health for Research on Quality and Standardization of Biotech Products, National Institutes for Food and Drug Control, 2 Tiantanxili Road, Dongcheng District, Beijing 100050, China. Tel.: +86-010-6709-5782; E-mail: wangjzorg.cn; & Dr. Hanmei Xu, The Engineering Research Center of Peptide Drug Discovery and Development, China Pharmaceutical University, 639 Longmian Avenue, Jiangning District, Nanjing 211198, China. Tel.: +86-025-8327-1007; E-mail: xuhanmeiedu.cn.