Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(7):990-997. doi:10.7150/ijms.34297 This issue Cite
Research Paper
Locus 5p13.1 may be associated with the selection of cancer-related HBV core promoter mutations
Qin-Yan Chen1,#, Yan-Ling Hu2,#, Xue-Yan Wang1, Tim J. Harrison3, Chao Wang1, Li-Ping Hu1, Qing-Li Yang1, Chuang-Chuang Ren1,4, Hui-Hua Jia1,4, Zhong-Liao Fang1 ![]()
1. Guangxi Zhuang Autonomous Region Center for Disease Prevention and Control, Guangxi Key Laboratory for the Prevention and Control of Viral Hepatitis, Nanning, Guangxi 530028, China.
2. Center for Genomic and Personalized Medicine, Guangxi Medical University, 22 ShuangYong Road, Nanning, Guangxi 530021, China.
3. Division of Medicine, UCL Medical School, London, UK.
4. School of Preclinical Medicine, Guangxi Medical University, 22 ShuangYong Road, Nanning, Guangxi 530021, China.
# These authors contributed equally to this work.
Received 2019-2-20; Accepted 2019-5-21; Published 2019-6-10
Abstract
Background: The basal core promoter (BCP) double mutations (A1762T and G1764A) of hepatitis B virus (HBV) have been reported to be an aetiological factor of hepatocellular carcinoma (HCC). What distinguishes the subset of HBV carriers in whom these mutations are selected?
Methods: A genome-wide association study (GWAS) was carried out on 218 asymptomatic HBsAg carriers infected with HBV with BCP double mutations and 191 controls infected with HBV with the wild type BCP. The highest ranking nucleotide polymorphisms (SNPs) were validated with other study subjects, 203 cases and 181 controls. The expression of the gene nearest a SNP found to be significant was examined using RT-PCR.
Results: Forty-five candidate SNPs were identified in the GWAS. Three SNPs were found to be associated with the selection of HBV BCP double mutations in the replication stage, including rs7717457 at 5p13.1, rs670011 at 17q21.2, rs2071611 at 6p22.2. Especially, rs7717457 (P= 4.57×10-5 combined P) reached the potential GWAS significance level. The expression of gene complement component 7 (C7), nearest to SNP rs7717457, differed significantly between the case and control groups (t=2.045, P=0.04), suggesting that SNP rs7717457 was associated with the expression of its nearest gene.
Conclusions: SNP rs7717457 is associated with the selection of HBV BCP double mutations, providing an important clue to understanding the mechanisms of oncogenesis of HBV BCP double mutations.
Keywords: Genome-wide association study (GWAS), hepatitis B virus (HBV), basal core promoter (BCP), mutations, single nucleotide polymorphisms (SNPs).
Introduction
Worldwide, hepatocellular carcinoma (HCC) is the fifth most common cancer in males and the seventh in females and is the third most common cause of cancer death [1]. The incidence of HCC varies greatly according to the geographic area; the highest incidence of HCC in the world is reported by registries in Asia and Africa. Approximately 85% of all liver cancers occur in these areas, with Chinese registries alone reporting over 50% [2]. HCC in China ranks as the second most common cause of cancer death in males and the third in females. The mortality rate from HCC is higher in males (37.4/100,000) than in females (14.3/100,000) [3]. The major risk factors for HCC in Asia and Africa are chronic hepatitis B virus (HBV) infection and aflatoxin B1 (AFB1) exposure. HBV is responsible for 75 to 80% of virus-associated HCC [4].
However, the mechanisms of the oncogenesis of HBV remain obscure. Nonetheless, mutations in the viral genome associated with tumour development recently have become a major focus of research. The precore mutation (G1896A), mutations in enhancer II (C1653T) and the BCP (T1753V and the double mutations, A1762T, G1764A), and deletions in the pre-S region have been reported to be associated with the development of HCC [5-11]. Perhaps the most convincing association is with HBV with the double mutations in the BCP; this has been confirmed by several cohort studies, suggesting that the double mutations are an aetiological factor of HCC [8, 12-13].
In addition to HBV and AFB1 exposure, host factors may play a role in the development of HCC. There have been a few genome-wide association studies (GWAS) conducted on the genetic susceptibility to HBV-related HCC. Various single nucleotide polymorphisms (SNPs), such as rs7574865 at STAT4, rs9275319 at HLA-DQ and rs12682266, rs7821974, rs2275959, rs1573266 at chromosome 8p12, have been found to be associated with the development of HBV-related HCC [14-15]. Combined analyses of copy number variation (CNV), individual SNPs, and pathways suggests that HCC susceptibility is mediated by germline factors affecting the immune response and differences in T-cell receptor processing [16].
When we established the Long An cohort in 2004, we found that about half of the HBV-infected individuals have BCP double mutations (A1762T, G1764A) in the viral genome and more than 93% of HCC cases occurred in those with BCP double mutations[8]. Why are BCP double mutations selected in a subset of HBV carriers? The answers may be helpful in understanding the pathogenesis of HCC. It has been reported from candidate-gene studies that host genetic polymorphisms are associated with the immune selection of HBV mutations [17]. This phenomenon may also be seen in other viruses, such as HIV-1 [18]. Therefore, we carried out a genome-wide association study (GWAS), based on the Long An cohort, to search for a genetic basis of the selection of HCC-related, HBV BCP mutations and which may potentially identify novel related SNPs.
Materials and Methods
Study subjects
The study subjects were recruited from the Long An cohort, which was described previously [8]. The cohort was recruited in early 2004 from agricultural workers aged 30-55 living in the rural area of Long An county, Guangxi, China, using stratified sampling. This cohort comprises 2258 HBsAg-positive study subjects, including a group (1261) with BCP double mutations and a wild type BCP group (997). They were further stratified into the male mutant (702) and wild type (561) groups and female mutant (559) and wild type (436) groups. When we recruited study subjects for this study, we retested BCP sequence of HBV of each subject in 2014. The selection criterion is that they were infected with HBV with the same BCP sequence as at baseline.
Informed consent in writing was obtained from each individual. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and has been approved by the Guangxi Institutional Review Board.
Serological Testing
Sera were tested for HBV serological markers using enzyme immunoassays and AFP using a Diagnostic Kit for the Quantitative Determination of Alpha-feto-protein (ELISA) (Beijing Wantai Biological Pharmacy Enterprise Co., Ltd., Beijing, China) according to the manufacturer's instructions. The cut-off value of AFP for HCC was set at 20 ng/mL. Alanine aminotransferase (ALT) concentrations were determined using a kinetic method (Zhejiang Elikan Biological Technology Company, Limited, Wenzhou, Zhejiang, China).
Nested PCR for HBV DNA and nucleotide sequencing
HBV DNA was extracted from 85 μl serum by pronase digestion followed by phenol/chloroform extraction. The method for amplification and sequencing of the BCP region has been reported previously [8].
Genotyping in GWAS
Peripheral blood mononuclear cell (PBMC) DNA was extracted from 200 μl blood using a QIAamp®DNA Mini Kit. PBMC DNA was sent to the CapitalBio Corporation (Beijing 102206, China) for genotyping. The Infinium® HumanCore BeadChips (Illumina Inc.) was used for genotyping 306670 SNPs in the GWAS stage. For the genotyping reactions, 250 ng of genomic DNA was analyzed using the Infinium® Human Core Bead Chips according to the manufacturer's recommendations and using their reagents [19]. Infinium® HumanCore BeadChips Genotype data were generated using GenomeStudio Genotyping Module v1.0. The genotyping was performed by laboratory personnel blinded to the study subjects.
SNP selection and genotyping in the replication study
If a locus had a SNP with a P value <1.0 × 10-4 in the GWAS stage, it was chosen for replication. If several SNPs were in the linkage disequilibrium with R2>0.6, the SNP with the lowest P value was selected. The iPLEX MassARRAY platform (Sequenom Inc.) was used in the replication stage. 50 ng of genomic DNA was analyzed using the iPLEX MassARRAY platform according to the manufacturer's recommendations and using their reagents [19]. iPLEX MassARRAY platform Genotype data were generated using MassARRAY® Typer 4.0 software. The genotyping was performed by laboratory personnel blinded to the study subjects.
Functional annotation and differential expression analysis
Whole blood was collected in EDTA tubes and RNALock Reagent (TIANGEN, China) was added immediately. Total RNA was extracted from the PBMC using RNAprep Pure Blood Kit (TIANGEN, China) according to manufacturer's instructions. The RNA was reverse transcribed as PCR template using a PrimeScriptTM II 1st Strand cDNA Synthesis Kit (TaKaRa, China), followed by PCR with SYBR Premix Ex TaqTM II (TaKaRa, China). The expression of mRNA was detected by quantitative real-time reverse transcriptase PCR (qRT-PCR) on CFX96 (BioRad). The primers used for GAPDH, CARD6, PTGER4 and C7 were GAPDH-2F (5' GAAGGTGAAGGTCGGAGTC 3') and GAPDH-2R (5' GAAGATGGTGATGGGATTTC 3'), CARD6-F (5' CCCACTGTGCTTGTATCTGC 3') and CARD6-R (5' CGGTAGCCATTGTTCCTGT 3'), PTGER4-F (5' CGCAAGGAGCAGAAGGAGAC 3') and PTGER4-R (5'CAGGCTGAAGAAGAGCAGAATGAA 3'), C7-2F (5' AACGGCAAGGAGCAGACG 3') and C7-2R (5' TGTCCAGTGCCCAGTTGTG 3'), respectively. GAPDH was chosen as an endogenous control to normalize the relative mRNA expression levels. Experiments were performed in duplicate for each sample and fold changes were calculated by the equation 2-ΔΔCt.
Statistical analysis
The PLINK package [20] was used to carry out the quality control procedures and association analyses. Quality control (QC) procedures were carried out using GenomeStudio Genotyping Module v1.0. The exclusion criteria were minor allele frequency (MAF) <0.04, SNP call rate <90%, and deviation from Hardy-Weinberg Equilibrium (p <0.05). Genetic association tests were carried out by multivariate analysis using logistic regression by entering variables in the PLINK software. Statistical comparisons of gene expression between cases and controls were performed carried out using a non-parametric paired t test (SPSS v.16.0). All P values were two-tailed and P<0.05 was considered to be significant.
Results
Genome-wide association analysis
In the initial discovery stage, we conducted a GWAS using samples from 218 asymptomatic HBsAg carriers with BCP double mutations (cases) and 191 asymptomatic HBsAg carriers with the wild type BCP (controls). There are 122 males in the case group (56.0%) and 102 males in control group (53.4%). The average ages of the case and control groups are 50.8±6.3 and 51.0±6.5, respectively. There was no difference between the two groups in terms of sex (χ2 =0.274, P>0.05) and age (F =1.974, P>0.05) (Table 1).
General characteristics of the study subjects in the GWAS
| Variables | Total | Cases | Control | P value |
|---|---|---|---|---|
| Number | 409 | 218 | 191 | |
| Male | 224 | 122 | 102 | P=0.604 |
| Female | 185 | 96 | 89 | |
| Age, Years | 50.9±6.4 | 50.8±6.3 | 51.0±6.6 | P=0.782 |
| Abnormal ALT, % | 9.3 (38/409) | 10.1 (22/218) | 8.4 (16/191) | P=0.551 |
| AFP (+), % | 4.9 (20/409) | 5.5 (12/218) | 4.1 (8/191) | P=0.538 |
Abnormal ALT: ≥40 IU/ml, AFP (+): >20 μg/L.
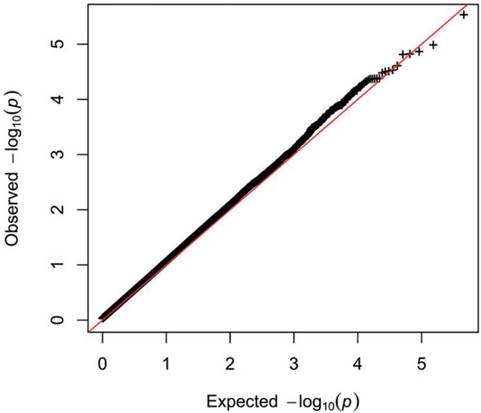
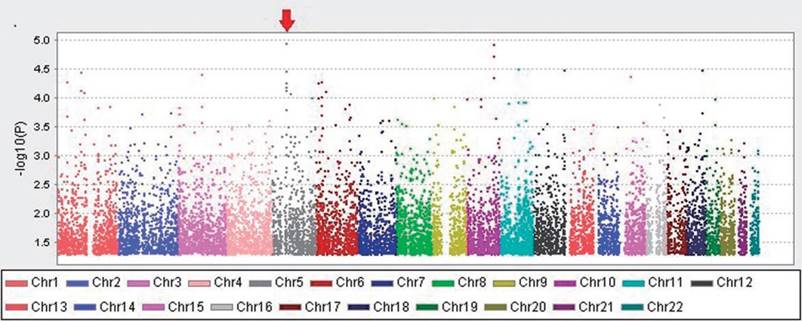
Quality control (QC) procedures were first applied to 409 individuals. All 409 study subjects passed the call rate of 90% and were used in the final statistical analysis. The exclusion criteria were minor allele frequency (MAF) <0.05, SNP call rate <90%, and deviation from Hardy-Weinberg Equilibrium (p <0.05). Based on these criteria, 25296 SNPs were retained. The quantile-quantile plot for the cases and controls is shown (Figure 1). The genomic inflation factor for the cases and controls was 1.147, indicating adequate control of population stratification and systematic bias in our GWAS population. In the GWAS stage, we assessed genome-wide associations for the cases and controls using multivariate linear models adjusted with age and sex. We found that there are two regions with trends of significant difference. They located in Chromosomes 5 and 10.The strongest association signals was SNP rs2910830 in phosphodiesterase 4D (PDE4D), located on chromosome 5q12 (P=1.136 × 10-5) (Figure 2).
Quantile-Quantile plot of genome-wide quantitative trait loci mapping for log-transformation.
Manhattan plot of genome-wide association analysis, adjusted with sex and age. The X-axis shows chromosomal positions. The Y-axis shows -log10 P-values from the linear regression.
SNP selection and genotyping in the replication study
A P value <10-4 was considered to be statistically significant and selected for the replication stage. When several SNPs were in linkage disequilibrium with R2 >0.6, the SNP with the lowest P value was selected. These parameters led to the identification of 45 candidate SNPs which were taken forward to the replication stage. The study subjects were 203 asymptomatic HBsAg carriers infected with HBV with BCP double mutations (case group) and 181 asymptomatic HBsAg carriers infected with HBV with wild-type BCP (control group). There are 102 males in the case group (50.2%) and 100 males in the control group (55.2%). The average ages of the case and control groups are 50.4±7.0 and 47.5±7.0, respectively. There are no significant differences between the two groups in terms of sex and age (Table 2). Genotyping was carried out in the replication stage using the iPLEX MassARRAY platform (Sequenom Inc.). The primers and probes are available upon request. The laboratory technicians who performed the genotyping experiments were blinded to the status of case and control. Three SNPs were found to be differ significantly, rs7717457 (P=0.01387), rs670011 (P=0.04085) and rs2071611 (P=0.04627) (Table 3).
General characteristics of the study subjects in the replication study
| Variables | Total | Cases | Control | P value |
|---|---|---|---|---|
| Number | 384 | 203 | 181 | |
| Male | 202 | 102 | 100 | P=0.327 |
| Female | 182 | 101 | 81 | |
| Age, Years | 49.0±7.1 | 50.4±7.0 | 47.5±7.0 | P=0.001 |
| Abnormal ALT, % | 2.3 (9/384) | 1.5 (3/203) | 3.3 (6/181) | P=0.235 |
| AFP (+), % | 3.4 (13/384) | 4.9 (10/203) | 1.7 (3/181) | P=0.08 |
| HBeAg (+) | 6.0 (23/384) | 0 | 12.7 (23/181) | P=0.001 |
Abnormal ALT: ≥40 IU/ml, AFP (+): >20 μg/L.
Results of replication study for forty-five significant SNPs.
| CHR | SNP | BP | OR(GWAS) | OR | P(GWAS) | P value | P-hwe | A1 | A2 |
|---|---|---|---|---|---|---|---|---|---|
| 5 | rs2935623 | 2767442 | 0.3544 | 1.448 | 0.0006732 | 0.2235 | 1 | G | A |
| 5 | rs7717457 | 40887679 | 1.725 | 1.466 | 0.000469 | 0.01387 | 0.305 | G | A |
| 5 | rs16887016 | 57501466 | 1.829 | 1.095 | 0.0009644 | 0.6009 | 0.04854 | C | T |
| 5 | rs7703245 | 60034662 | 0.482 | 0.8695 | 0.00006463 | 0.4721 | 0.8108 | T | C |
| 5 | rs10940659 | 60064514 | 0.49 | 0.8695 | 0.0001595 | 0.4721 | 0.8108 | A | G |
| 5 | rs1588265 | 60073967 | 0.482 | 1.018 | 0.00006463 | 0.9259 | 1 | A | G |
| 5 | rs4700365 | 60131915 | 0.5 | 0.8132 | 0.0007128 | 0.3381 | 0.577 | A | C |
| 5 | rs1544791 | 60143255 | 0.5108 | 0.87 | 0.0004244 | 0.5163 | 0.5777 | C | T |
| 5 | rs983280 | 60149310 | 0.4767 | 0.9854 | 0.00005486 | 0.9397 | 1 | T | C |
| 5 | rs2910830 | 60171370 | 0.4365 | 0.9605 | 0.00001136 | 0.8298 | 0.6397 | A | G |
| 5 | rs2910829 | 60174072 | 0.4606 | 0.8797 | 0.00007343 | 0.5162 | 1 | A | G |
| 5 | rs4235479 | 60248826 | 0.5541 | 1.186 | 0.0009699 | 0.3299 | 0.4829 | C | T |
| 5 | rs35247 | 68715157 | 2.161 | 1.252 | 0.0003169 | 0.3384 | 0.6982 | T | C |
| 5 | rs10057967 | 75701931 | 1.701 | 1.105 | 0.0005652 | 0.492 | 0.7575 | T | C |
| 5 | rs27135 | 76717018 | 0.5228 | 1.064 | 0.00008475 | 0.7675 | 4.36E-07 | G | A |
| 5 | rs253061 | 76718175 | 0.5718 | 1.006 | 0.0008246 | 0.973 | 0.4289 | C | A |
| 5 | rs9293505 | 88890652 | 1.96 | 1.268 | 0.0006696 | 0.2523 | 0.7528 | T | G |
| 5 | rs17085231 | 95886452 | 1.662 | 1.023 | 0.0004618 | 0.8803 | 0.5238 | T | C |
| 5 | rs11741590 | 95905423 | 1.627 | 0.8981 | 0.0006059 | 0.4685 | 0.8801 | T | C |
| 5 | rs7707391 | 103613967 | 0.5803 | 1.219 | 0.000974 | 0.2356 | 0.007409 | T | G |
| 5 | rs246430 | 143602616 | 0.2891 | 1.321 | 0.0004727 | 0.3539 | 1 | A | C |
| 5 | rs3756309 | 150126061 | 1.93 | 1.102 | 0.0008312 | 0.6101 | 0.2895 | C | T |
| 5 | rs13166904 | 160940790 | 0.5477 | 0.9373 | 0.0003813 | 0.7185 | 0.4022 | T | C |
| 5 | rs17066036 | 165873965 | 0.5401 | 1.061 | 0.0001008 | 0.7235 | 0.452 | A | C |
| 5 | rs1445844 | 179080269 | 2.914 | 1.129 | 0.0003181 | 0.5991 | 0.4727 | A | G |
| 5 | rs1136377 | 179086140 | 2.892 | 1.135 | 0.000349 | 0.5798 | 0.4727 | C | T |
| 6 | rs4712415 | 19524844 | 0.6175 | 0.9775 | 0.0006521 | 0.8843 | 0.2958 | T | C |
| 6 | rs2743582 | 19525822 | 0.5086 | 0.853 | 0.0002772 | 0.4193 | 1 | T | C |
| 6 | rs1165159 | 25864397 | 2.093 | 1.322 | 0.00004903 | 0.3529 | 1 | G | A |
| 6 | rs670011 | 25887731 | 0.6324 | 1.378 | 0.0002081 | 0.04085 | 0.4132 | A | C |
| 6 | rs1150658 | 26098527 | 0.5641 | 0.8705 | 0.0001346 | 0.4634 | 0.03195 | T | C |
| 6 | rs707898 | 26116992 | 0.5634 | 0.8873 | 0.0001342 | 0.5196 | 0.08859 | C | T |
| 6 | rs198828 | 26119231 | 0.559 | 1.042 | 0.000004639 | 0.7854 | 0.7536 | A | G |
| 6 | rs6457736 | 33596635 | 2.135 | 1.042 | 0.000546 | 0.9035 | 0.335 | C | T |
| 6 | rs9365246 | 161056632 | 1.571 | 0.9879 | 0.0003458 | 0.938 | 0.5261 | G | A |
| 6 | rs2981977 | 167336662 | 0.582 | 1.045 | 0.00102 | 0.82 | 1 | G | A |
| 10 | rs11253241 | 5579665 | 0.5802 | 0.9532 | 0.0007793 | 0.7643 | 0.3643 | T | C |
| 10 | rs11005046 | 55827304 | 1.786 | 0.7658 | 0.0001032 | 0.08212 | 0.8746 | C | A |
| 10 | rs7908845 | 55887821 | 1.817 | 0.8838 | 0.0001392 | 0.4034 | 0.5379 | C | T |
| 10 | rs1245907 | 109768722 | 2.036 | 1.08 | 0.00001174 | 0.6255 | 0.4795 | A | G |
| 10 | rs7916801 | 109872687 | 1.941 | 1.088 | 0.00001864 | 0.59 | 0.8661 | A | G |
| 10 | rs9422853 | 126909164 | 1.649 | 1.073 | 0.0006043 | 0.6393 | 0.2301 | G | A |
| 10 | rs10781564 | 131944418 | 0.5955 | 0.9054 | 0.0005059 | 0.5081 | 0.8805 | G | A |
| 10 | rs7098827 | 131984924 | 0.4858 | 0.8068 | 0.0007101 | 0.3734 | 0.7227 | T | G |
| 17 | rs2071611 | 41439409 | 0.482 | 0.6775 | 0.00006463 | 0.04627 | 0.8168 | T | A |
CHR: Chromosome; SNP: single nucleotide polymorphism; BP: base-pair position; OR: odds ratio; P-hwe: P value for Hardy-Weinberg equilibrium; A1 and A2 are Allele, A1 is mutant and A2 is wild type. MAF: minor allele frequency. GWAS: OR and P from genome-wide association study.
We also carried out a combined analysis of data of the GWAS and replication studies, using multivariate linear models adjusted with age and sex. We found that rs7717457 (P= 4.57×10-5) reached the potential GWAS significance level. However, rs2910830 (P= 1.136×10-5), which had the most significant association at the GWAS stage, showed a P value of 6.53×10-4 when the data from the two stages were combined.
Differential expression analysis
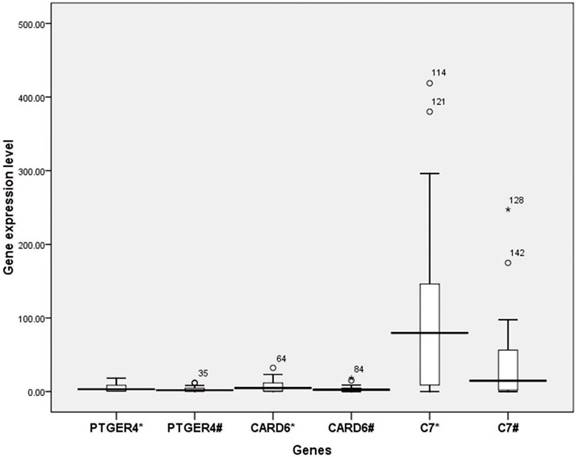
To determine whether the SNPs found to be associated with BCP double mutations in the GWAS stage and replication stage influence the expression of the corresponding genes, rs7717457, with the lowest P value among the three SNPs above, was selected for the analysis. The position of rs7717457 is near gene CARD6, gene complement component 7 (C7) and gene PTGER4 of 5p13.1. (https://genome.ucsc.edu and https://snpinfo.niehs.nih.gov/snpinfo/snpfunc.htm). Whole blood samples were collected from the Long An cohort, including 23 individuals infected with HBV with BCP double mutations (case group) and 28 individuals infected with BCP wild type (control group) (Table 4). These study subjects differ from those in the GWAS stage and replication stage. We found that the expression of genes CARD6 and PTGER4 did not differ significantly between the two groups. However, the difference in the expression of gene C7 between the case group (1.99) and control group (4.10) was significant (t=2.045, P=0.04) (Figure 3), suggesting that human genes are involved in selecting viral mutations.
Differential expression analysis of PTGER4, CARD6 and C7. *: Group infected with BCP wild type, #: Group infected with HBV with BCP double mutations.
General characteristics of the study subjects in the differential expression study
| Variables | Total | Cases | Control | P value |
|---|---|---|---|---|
| Number | 51 | 23 | 28 | |
| Male | 35 | 17 | 18 | P=0.46 |
| Female | 16 | 6 | 10 | |
| Age, Years | 46.0±4.7 | 49.5±4.5 | 46.0±4.7 | P=0.01 |
| Abnormal ALT, % | 3.9 (2/51) | 8.7 (2/23) | 0 | P=0.111 |
| AFP (+), % | 0 | 0 | 0 | P=1 |
| Viral loads | 8.49×104 | 1.06×105 | 6.72×104 | P=0.58 |
Clinical significance of the SNPs
We randomly tested the serological parameters of HBV, ALT and AFP for 196 study subjects from the GWAS and replication stages. No association between the rs7717457 mutations and sex, HBeAg, ALT or AFP was found (Table 5).
The distribution of SNP rs7717457 according to the characteristics of the study subjects
| Number of study subjects | Allele (A+A*) | Allele (A+G or G+G) | Rate of (A+G or G+G) (%) | X2 | P value | |
|---|---|---|---|---|---|---|
| Sex | ||||||
| Male | 102 | 78 | 24 | 23.5 | 0.439 | P=0.508 |
| Female | 94 | 68 | 26 | 27.7 | ||
| HBeAg(-) | 184 | 137 | 47 | 25.5 | 0.002 | P=0.967 |
| HBeAg(+) | 12 | 9 | 3 | 25.0 | ||
| ALT <40 IU/ml | 191 | 145 | 46 | 24.1 | 8.017 | P=0.005 |
| ALT≥40 IU/ml | 5 | 1 | 4 | 80.0 | ||
| AFP <20 μg/L | 189 | 141 | 48 | 25.4 | 0.036 | P=0.967 |
| AFP ≥20 μg/L | 7 | 5 | 2 | 28.6 |
* Allele (A+A) is wild type and A or G signifies the nucleotide.
Discussion
The major findings of this study are that three SNPs were found to be associated with HBV BCP double mutations in the replication stage, rs7717457, rs670011, rs 2071611. rs7717457 may influence the expression of its nearest gene, C7, suggesting that human genes are involved in selecting viral mutations. No association was found between rs7717457 and sex, HBeAg, ALT or AFP. A strength of this study is that the study subjects in the GWAS were recruited from a long-term cohort, which provides reliable information for each study subject, such as the status of the BCP sequence of HBV. A weakness of the study is that the sample size is small, which may prevent some interesting SNPs being found. Another weakness is that the subjects of the GWAS and replication studies are all from the same ethnic minority, although they are not the same subjects. Therefore, we do not know whether the findings are applicable to other ethnic populations.
The lack of a proof-reading activity of the viral polymerase leads to a high rate of mutation during replication of the HBV genome. Some of these mutants may become predominant strains but others not, and some predominant strains have clinic significance. The question is which mutants can become predominant strains; more than 60% of the mutations are subject to selection forces from host immune surveillance, antiviral therapy and replication fitness [21]. So the common explanation is the active adaptive evolution of mutant strains under various selection pressures, such as from immunoglobulin [22], immunization [23] or antiviral therapy [24]. However, these mutants may also occur naturally [25]. Clearly, the mechanism remains obscure. It also has been reported that HBV adapts to increasing immune pressure through preferential mutations in B-cell epitopes and by replicative attenuation [26]. The human leukocyte antigen (HLA) class I was found to be involved in this selection [27]. A candidate-gene study reported that rs2233406 variant genotypes significantly increased the frequencies of BCP double mutations and rs28362491 significantly increased the frequency of BCP double mutations but reduced the frequency of preS2 start codon mutations [17]. In this study, we are the first to use GWAS to find another SNP associated with double mutations in the core promoter of HBV. Furthermore, we found that this SNP influenced the expression of its nearest gene.
In this study, we found in the second stage three SNPs, rs7717457, rs670011 and rs2071611, are associated with the selection of double mutations in the core promoter of HBV. SNP rs2071611 is located in the intron region of gene KRT38 of 17q21.2. The protein encoded by gene KRT38 is a member of the keratin gene family [28]. The rs670011 was located between gene HIST1H2APS2 and gene SLC17A2 of 6p22.2. As a type I hair keratin, it is an acidic protein which heterodimerizes with type II keratins to form hair and nails. Gene HIST1H2APS2 is a histone pseudogene [29]. Gene SLC17A2 encodes an Na (+)-phosphate cotransporter 3 (NPT3) [30]. It seems that these SNPs are unlikely to influence the selection of double mutations in core promoter of HBV, considering the proteins encoded by the nearby genes.
rs7717457 is near gene CARD6, gene C7 and gene PTGER4 of 5p13.1. The expression of genes CARD6 and PTGER4 were not found to differ significantly between the groups with BCP double mutations (cases) and BCP wild type (controls), suggesting that the genes CARD6 and PTGER4 could not influence the selection of BCP double mutations. However, the difference in the expression of gene C7 between the two groups was significant, suggesting that rs7717457 is involved in selecting viral mutations. It has been reported that SNP can alter gene expression by affecting transcription rate because of altered transcription factor binding [31]. Therefore, the mechanism by which rs7717457 influences the expression of C7 gene requires study, which is important to understand the mechanisms of oncogenesis of HBV.
Gene C7 encodes a serum glycoprotein that forms a membrane attack complex, together with complement components C5b, C6, C8, and C9, as part of the terminal complement pathway of the innate immune system. The protein encoded by this gene contains a cholesterol-dependent cytolysin/membrane attack complex/perforin-like (CDC/MACPF) domain and belongs to a large family of structurally related molecules that form pores involved in host immunity and bacterial pathogenesis. This protein initiates membrane attack complex formation by binding the C5b-C6 subcomplex and inserts into the phospholipid bilayer, serving as a membrane anchor [32-34]. Mutations in this gene are associated with a rare genetic disorder, C7 deficiency [35]. It has been reported that complement component 7 (C7) is a potential tumor suppressor [36]. The reduced expression of C7 mRNAs may be associated with oesophageal tumorigenesis [37]. Complement proteins C7 and complement factor H (CFH) may control the stem of liver cancer cells via LSF-1[38]. Therefore, clearly, on one hand, SNP rs7717457 is associated with in the selection of BCP double mutations. On another hand, it may be involved in liver tumorigenesis. This may be an important finding towards understanding the mechanisms of oncogenesis of HBV BCP double mutations. This is also important because only a small fraction of asymptomatic HBsAg carriers with BCP double mutations go on to develop HCC, so the ability to predict those at highest risk may permit a more 'personalized' screening strategy, and probably earlier intervention or treatment, and hence will be of great clinical relevance.
Although no association was found between rs7717457 and sex, HBeAg, ALT or AFP in our study, more clinical markers could be used for exploring for association between rs7717457 and HBV viral load, HCC status, cirrhosis, end-stage liver disease, etc.
In summary, our study provides evidence using GWAS that host genetic polymorphisms are associated with the immune selection of HCC-related double mutations (A1762T and G1764A) in the basal core promoter of HBV. We also found that this SNP, rs7717457, influenced the expression of its nearest gene, which has been reported to be involved in the control stemness of liver cancer cells. These results are important in furthering our understanding of the mechanisms of oncogenesis of HBV. In the future, the rates of SNP rs7717457 should be determined among patients with HCC, liver cirrhosis and chronic hepatitis, which will be helpful to understand further the mechanisms of oncogenesis.
Acknowledgements
We are indebted to staff members of Centre for Disease Prevention and Control of Long An and local town hospitals in Long An county, Guangxi, who assisted in recruiting the study subjects, sample collection. This study was supported by the Wellcome Trust (WT072058MA) and the National Natural Science Foundation of China (Grant No. 81260439/H2609).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bosetti C, Turati F, La Vecchia C. Hepatocellular carcinoma epidemiology. Best Pract Res Clin Gastroenterol. 2014;28:753-70
2. Ferlay J, Parkin DM, Curado MP, et al. Cancer Incidence in Five Continents, Volumes I to IX: IARC CancerBase No. 9. http://ci5.iarc.fr
3. Tanaka M, Katayama F, Kato H. et al. Hepatitis B and C virus infection and hepatocellular carcinoma in China: a review of epidemiology and control measures. J Epidemiol. 2011;21:401-16
4. McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015;19:223-38
5. Fang ZL, Ling R, Wang SS. et al. HBV core promoter mutations prevail in patients with hepatocellular carcinoma from Guangxi, China. J Med Virol. 1998;56:18-24
6. Fang ZL, Yang J, Ge X. et al. Core promoter mutations (A(1762)T and G(1764)A) and viral genotype in chronic hepatitis B and hepatocellular carcinoma in Guangxi, China. J Med Virol. 2002;68:33-40
7. Lyu H, Lee D, Chung YH. et al. Synergistic effects of A1896, T1653 and T1762/A1764 mutations in genotype c2 hepatitis B virus on development of hepatocellular carcinoma. J Viral Hepat. 2013;20:219-24
8. Fang ZL, Sabin CA, Dong BQ. et al. HBV A1762T, G1764A mutations are a valuable biomarker for identifying a subset of male HBsAg carriers at extremely high risk of hepatocellular carcinoma: A prospective study. American J Gastroenterol. 2008;103:2254-2262
9. Fang ZL, Sabin CA, Dong BQ. et al. Hepatitis B virus pre-S deletion mutations are a risk factor for hepatocellular carcinoma: a matched nested case-control study. J Gen Virol. 2008;89(Pt 11):2882-90
10. Qu LS, Liu JX, Liu TT. et al. Association of hepatitis B virus pre-S deletions with the development of hepatocellular carcinoma in Qidong, China. PLoS One. 2014;9:e98257
11. Qu LS, Zhu J, Liu TT. et al. Effect of combined mutations in the enhancer II and basal core promoter of hepatitis B virus on development of hepatocellular carcinoma in Qidong, China. Hepatol Res. 2014;44:1186-95
12. Yuen MF, Tanaka Y, Fong DY. et al. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J Hepatol. 2009;50:80-8
13. Chu CM, Lin CC, Lin SM. et al. Viral Load, Genotypes, and Mutants in Hepatitis B Virus-Related Hepatocellular Carcinoma: Special Emphasis on Patients with Early Hepatocellular Carcinoma. Dig Dis Sci. 2012;57:232-8
14. Chan KY, Wong CM, Kwan JS. et al. Genome-wide association study of hepatocellular carcinoma in Southern Chinese patients withchronic hepatitis B virus infection. PLoS One. 2011;6:e28798
15. Jiang DK, Sun J, Cao G. et al. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet. 2013;45:72-5
16. Clifford RJ, Zhang J, Meerzaman DM. et al. Genetic variations at loci involved in the immune response are risk factors for hepatocellular carcinoma. Hepatology. 2010;52:2034-43
17. Zhang Q, Ji XW, Hou XM. et al. Effect of functional nuclear factor-kappaB genetic polymorphisms on hepatitis B virus persistence and their interactions with viral mutations on the risk of hepatocellular carcinoma. Ann Oncol. 2014;25:2413-9
18. Bartha I, Carlson JM, Brumme CJ. et al. A genome-to-genome analysis of associations between human genetic variation, HIV-1 sequence diversity, and viral control. Elife. 2013;2:e01123
19. Oka R, Sasagawa T, Ninomiya I. et al. Reduction in the local expression of complement component 6 (C6) and 7 (C7) mRNAs in oesophageal carcinoma. Eur J Cancer. 2001;37(9):1158-65
20. Seol HS, Lee SE, Song JS. et al. Complement proteins C7 and CFH control the stemness of liver cancer cells via LSF-1. Cancer Lett. 2016;372:24-35
21. Sun Y, Huang Y, Yin A. et al. Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat Commun. 2015;6:6414
22. Purcell S, Neale B, Todd-Brown K. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559-575
23. Xu Z, Wu G, Li F. et al. Positive selection signals of hepatitis B virus and their association with disease stages and viral genotypes. Infect Genet Evol. 2013;19:176-87
24. Terrault NA, Zhou S, McCory RW. et al. Incidence and clinical consequences of surface and polymerase gene mutations in liver transplant recipients on hepatitis B immunoglobulin. Hepatology. 1998;28:555-561
25. Carman WF, Zanetti AR, Karayiannis P. et al. Vaccine-induced escape mutant of hepatitis B virus. Lancet. 1990;336:325-329
26. Sheldon J, Camino N, Rodés B. et al. Selection of hepatitis B virus polymerase mutations in HIV-coinfected patients treated with tenofovir. Antiviral Ther. 2005;10:727-734
27. Yamamoto K, Horikita M, Tsuda F. et al. Naturally occurring escape mutants of hepatitis B virus with various mutations in the S gene in carriers seropositive for antibody to hepatitis B surface antigen. Journal of Virology. 1994;68:2671-6
28. Mondal RK, Khatun M, Ghosh S. et al. Immune-driven adaptation of hepatitis B virus genotype D involves preferential alteration in B-cell epitopes and replicative attenuation-an insight from human immunodeficiency virus/hepatitis B virus coinfection. Clin Microbiol Infect. 2015;21:710.e11-20
29. Kefalakes H, Budeus B, Walker A. et al. Adaptation of the hepatitis B virus core protein to CD8(+) T-cell selection pressure. Hepatology. 2015;62:47-56
30. Rogers MA, Winter H, Langbein L. et al. The human type I keratin gene family: characterization of new hair follicle specific members and evaluation of the chromosome 17q21.2 gene domain. Differentiation. 2004;72:527-40
31. Stevens A, Ray DW, Worthington J. et al. Polymorphisms of the human prolactin gene-implications for production of lymphocyte prolactin and systemic lupus erythematosus. Lupus. 2001;10(10):676-83
32. Database: RefSeq. Available from:https://ghr.nlm.nih.gov/gene/C7
33. Togawa N, Juge N, Miyaji T. et al. Wide expression of type I a+-phosphate cotransporter 3 (NPT3/SLC17A2), a membrane potential-driven organic anion transporter. Am J Physiol Cell Physiol. 2015;309:C71-80
34. Müller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321-47
35. Sonnen AF, Henneke P. Structural biology of the membrane attack complex. Subcell Biochem. 2014;80:83-116
36. Serna M, Giles JL, Morgan BP. et al. Structural basis of complement membrane attack complex formation. Nat Commun. 2016;7:10587
37. Fernie BA, Hobart MJ. Complement C7 deficiency: seven further molecular defects and their associated marker haplotypes. Hum Genet. 1998;103:513-9
38. Ying L, Zhang F, Pan X. et al. Complement component 7 (C7), a potential tumor suppressor, is correlated with tumor progression and prognosis. Oncotarget. 2016;7:86536-86546
Author contact
![]() Corresponding author: Zhong-Liao Fang, Guangxi Zhuang Autonomous Region Center for Disease Prevention and Control, 18 Jin Zhou Road, Nanning, Guangxi, China, 530028. Tel: 0086 771 2518306; Fax: 0086 771 2518768; Email: zhongliaofangcom
Corresponding author: Zhong-Liao Fang, Guangxi Zhuang Autonomous Region Center for Disease Prevention and Control, 18 Jin Zhou Road, Nanning, Guangxi, China, 530028. Tel: 0086 771 2518306; Fax: 0086 771 2518768; Email: zhongliaofangcom