Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Mechanisms of origin of...
3. Molecular heterogeneity...
4. Inter- and intra-tumor...
5. Heterogeneity of molecular...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(7):981-989. doi:10.7150/ijms.34739 This issue Cite
Review
Molecular heterogeneity in lung cancer: from mechanisms of origin to clinical implications
Federica Zito Marino1 ![]() , Roberto Bianco2, Marina Accardo1, Andrea Ronchi1, Immacolata Cozzolino1, Floriana Morgillo3, Giulio Rossi4, Renato Franco1
, Roberto Bianco2, Marina Accardo1, Andrea Ronchi1, Immacolata Cozzolino1, Floriana Morgillo3, Giulio Rossi4, Renato Franco1 ![]()
1. Pathology Unit, University of Campania “L. Vanvitelli”, Naples, Italy.
2. Department of Clinical Medicine and Surgery, Oncology Division, University of Naples Federico II, Naples, Italy.
3. Medical Oncology, Department of Precision Medicine, University of Campania “L. Vanvitelli”, Naples, Italy.
4. Pathology Unit, Hospital S. Maria delle Croci, Azienda Romagna, Ravenna, Italy.
Received 2019-3-9; Accepted 2019-5-5; Published 2019-6-10
Abstract
Molecular heterogeneity is a frequent event in cancer responsible of several critical issues in diagnosis and treatment of oncologic patients. Lung tumours are characterized by high degree of molecular heterogeneity associated to different mechanisms of origin including genetic, epigenetic and non-genetic source. In this review, we provide an overview of recognized mechanisms underlying molecular heterogeneity in lung cancer, including epigenetic mechanisms, mutant allele specific imbalance, genomic instability, chromosomal aberrations, tumor mutational burden, somatic mutations. We focus on the role of spatial and temporal molecular heterogeneity involved in therapeutic implications in lung cancer patients.
Keywords: lung cancer, molecular heterogeneity, therapy, driver mutations
1. Introduction
Tumor heterogeneity represents a well-known event in cancer, responsible of several critical issues in diagnosis and treatment of cancer patients. Different levels of heterogeneity have been recognized in cancer particularly interpatient, intratumor and intertumor.
Interpatient heterogeneity is related to genetic and phenotypic variations, observed among individuals with the same tumor type; it could explain the different treatment response of each patient. Intratumor heterogeneity refers to subclonal diversities of tumor cells observed within a single tumor, whereas intertumor heterogeneity is considered as diversity between primary tumor and its metastases [1-3].
Distinct cellular populations within a tumour could differ in a wide spectrum of features from the expression of cell markers to the genetic or epigenetic alterations which could cause heterogeneity [4].
Heterogeneity of molecular profile represents one of the most challenging issues in cancer, particularly in lung cancer, in the light of the resulting therapeutic implications.
In lung cancer, different levels of molecular heterogeneity have been recognized including inter-patients, intra- and inter-tumour variability. Molecular heterogeneity between lung cancer patients with the same histotype represents a proven biological process resulting frequently in different treatment response for each individual patient [1,5].
Furthermore, a high degree of genetic diversity between the primary lung tumor and corresponding metastatic lesions could play a pivotal role in the therapeutic context of lung cancer patients [6-14].
In this review, we provide an overview of recognized mechanisms underlying molecular heterogeneity in lung cancer, including genetic as well as epigenetic sources and non-genetic sources such as cancer stem cells (CSCs) and immune contexture. We focus on the role of spatial and temporal molecular heterogeneity involved in therapeutic implications in lung cancer patients.
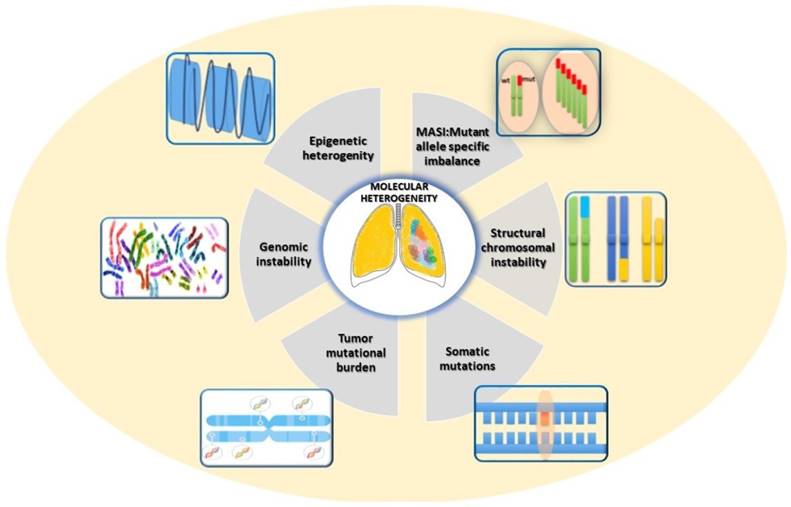
2. Mechanisms of origin of molecular heterogeneity in lung cancer
In lung cancer, heterogeneity could be attributed to several different sources [15, 16], related to genetic, epigenetic and non-genetic mechanisms (Fig. 1).
Mechanisms of origin of molecular heterogeneity in Lung Cancer.
Lung tumours are characterized by extensive genomic aberrations including aneusomy, gains and losses of large chromosome regions, gene rearrangements, copy number gain, amplifications [17].
Genomic instability represents one of the hallmarks in human cancer resulting in various genetic aberrations at different level from mutations in single or few nucleotides to changes of part or entire chromosomes [18].
The term chromosomal instability (CIN) defines a type of genomic instability associated to numerical and structural variations of part or whole chromosomes, for example gain or loss of chromosome fragments, translocations, deletions and amplifications of DNA [19, 20]. CIN could have clinical importance in lung cancer patients being generally associated with poor prognosis regardless of other conventional risk factors such as tumour stage, age and sex [21- 23].
Furthermore, CIN may frequently generate the intertumor heterogeneity resulting in a possible increase, before the treatment, of resistant pre-existing sub-clones. Consistent with the selective pressure related to drug treatment, tumor cells characterized by hight levels of CIN might promote drug resistence [24, 20]. Moreover, genomic diversity facilitates the adaptation of cancer cell populations in the context of tumor microenvironment resulting in tumor progression and poor prognosis [19].
Jamal‑Hanjani and colleagues have recently performed whole-exome sequencing on multiple regions in a cohort of 100 non-small cell lung cancer (NSCLC) patients who had not received previous systemic therapy. Their results showed widespread intratumor heterogeneity for both somatic copy-number alterations and mutations, particularly an elevated copy-number heterogeneity was associated with an increased risk of recurrence or death (hazard ratio, 4.9; P = 4.4×10-4), statistically significant in multivariate analysis. These finding demonstrate that intratumor heterogeneity due to CIN in NSCLC is strictly associated to increased risk of recurrence or death, suggesting its potential prognostic role [25].
Human malignancies are characterized by a high variable frequency of somatic mutations between and within tumor types, ranging from about 0.001 to 400 per megabase (Mb), suggesting the complexity mutational burden underlying the carcinogenesis [26]. Lung cancer is featured by a high tumor mutational burden (TMB) compared to other cancer type, probably related to smoking habits frequently observed in lung cancer patients. Recent finding have highlighted the pivotal role of the TMB as predictor of response to immunotherapies [27].
The tumorigenesis in lung cancer represents a multi-step process involving genetic alterations. Previous studies proposed a mathematical modeling related to a clonal mutation burden in several cancer types, suggesting that lung cancer reflects predominantly mutations accumulated early during tumorigenesis compared to others cancers with late mutation rate [28].
Mutant allele specific imbalance (MASI) represents another genetic mechanism that could promote heterogeneity and impact tumorigenesis, progression, metastasis, prognosis and potentially therapeutic responses in cancer. MASI could occur with copy neutral alteration defined as acquired uniparental disomy (UPD), or with loss of heterozygosity (LOH) due to the loss of the wild-type allele [29]. Previous studies reported that MASI is a frequent event in some major oncogenes, such as EGFR, KRAS, PIK3CA, and BRAF [29].
In lung cancer, EGFR MASI is a frequent event counted approximately in 26-37% of cases, more commonly associated with exon 19 deletion than with exon 21 mutation [30, 31]. Although related to poor disease-specific survival, EGFR MASI seems not to be associated with time to progression and overall survival, nor to sensitivity to treatment with EGFR specific inhibitors [32].
Although intratumoral molecular heterogeneity in human cancer has historically attributed to genetic alterations, to date a high degree of heterogeneity has been related to epigenetic mechanisms, including DNA methylation, chromatin remodeling, and post-translational modification of histones [16, 33].
Epigenetic modifications induce a variability in gene expression determining a remarkable diversity. Recently, several studies analyzed a potential predictive role of epigenetic modifications in lung cancer, particularly microRNA (miRNAs) and DNA methylation [34]. MiRNAs play a crucial role in post-transcriptional regulation of several genes expression by binding to messenger RNA (mRNA) through complementary sequences. Physiologically, a single miRNA can modulate cell growth, differentiation and apoptosis, therefore an altered expression of miRNAs in different cancer types can affect the deregulation of cellular activities [35]. Recent findings showed a promising predictive role of miRNA signatures for chemotherapy response and clinical outcome in NSCLC patients, particularly miR-1290, miR-196b, and miR-135a in tumor tissue and miR-25, miR-27b, miR-21, and miR-326 in blood [34]. Although preliminary results are encouraging, further prospective studies and clinical validation on large patient cohorts are needed in order to use these miRNAs as predictive biomarkers of the response to treatment to platinum-based chemotherapy in NSCLC patients.
Beyond strictly genetic and epigenetic mechanisms, the heterogeneity could result from various non-genetic mechanisms, including the lung stem cell populations and the immune contexture of lung cancer [15].
CSCs represent a crucial non-genetic source of heterogeneity providing different subclonal lineages dynamically maintained in various solid tumors, including lung cancer [36, 37]. Several studies showed that CSCs drive tumor formation and progression, metastasis, recurrence and drug resistance. CSCs have unique characteristics including capacity of self-renewal, multipotency, ability to initiate new tumors in vivo, increased capacity of proliferation and differentiation [38, 39]. Studies in genetically engineered mouse models have enabled to prove the existence of lung stem cells able to self-renew regenerating lung parenchima, bronchioles, alveoli and pulmonary vessels [40]. Moreover, the distinctive biology of pre-existing different lung cells could drive the distinct phenotypes and genotypes of tumors, resulting in heterogeneity since the tumor initiation. Historically, various lung stem cell populations in different anatomical sites lead to the development of different istotypes [41].
Increasing evidence has highlighted the key contribution of microenvironment in the initiation and progression of lung cancer, since cancer cells are closely interconnected with the milieu of the tumor. The immune contexture of lung cancer is composed of several elements including endothelial cells, fibroblasts, myeloid cells, including T cells, B cells, natural killer cells, mature and immature dendritic cells, tumor-associated macrophages, neutrophils, and mast cells.
Lung tumor heterogeneity could be caused by different acidity and oxygen conditions, or variable concentrations of growth factors that could generate different levels of selective pressure, which in turn could sustain the survival of some clones rather than others [42].
Furthermore, the microenvironment can affect drug resistance since a determinate tumor context could improve the formation of protective compartments in response to treatments. In NSCLC, a typical example is EGFR TKI resistance due to activating MET signaling pathway based on increased hepatocyte growth factor (HGF) secretion by stromal fibroblasts under the stimulation of tumor-derived factors [43].
The variable pressure of lung tumor environment could generate inter- and intra-tumoral heterogeneity that affects sensitivity to target- and immuno- therapy response [44].
Finally, in lung cancer the mixture of genetic aberrations, epigenetic features, differentiation hierarchies of lung stem cell populations and microenvironmental factors all contribute to outgrowth of subpopulations of cells that may have genetic, epigenetic, and/or phenotypic differences, resulting in a condition of heterogeneity.
3. Molecular heterogeneity between histotypes
Lung cancer is historically classified based on tumor histology into small cell (SCLC) and non-small cell lung cancer (NSCLC), the latter accounting of about 80% of cases. NSCLC include different histotypes such as adenocarcinoma (ADC), adenosquamous carcinoma, squamous cell carcinoma (SqCC), and large cell carcinoma. Lung neuroendocrine tumours (LNETs) are classified into different histological types including typical carcinoid, atypical carcinoid, large-cell neuroendocrine carcinoma (LCNEC), and SCLC [45]. The different histotypes are associated with specific different mutational profiles (Table 1) [46-73].
Molecular landascape in lung cancer associated to diverse histotypes.
| Histotype | Type of genomic aberrations | Gene | Frequency (%) | Currently available Target therapy | Ref. | ||
|---|---|---|---|---|---|---|---|
| NSCLC | ADC | Fusions | ALK | 3-7 | A | [46] | |
| ROS1 | 2-3 | A | [47] | ||||
| RET | 1-2 | NA | [47] | ||||
| NTRK1 | 1-2 | NA | [48] | ||||
| Mutations | EGFR | 30-40 | A | [49] | |||
| BRAF | 0.5-5 | NA | [47] | ||||
| KRAS | 20-30 | NA | [47] | ||||
| MET | 3-4 | NA | [50] | ||||
| PTEN | 1.7 | NA | [51] | ||||
| PDGFRA | 6-7 | NA | [52] | ||||
| PIK3CA | 5 | NA | [53, 54] | ||||
| TP53 | 52 | NA | [55] | ||||
| Copy number gene alterations | Gains | ERBB2 | 2-5 | NA | [56] | ||
| EGFR | 10 | NA | [57] | ||||
| MET | 2-5 | NA | [50] | ||||
| TERT | 75 | NA | [58] | ||||
| Losses | CDKN2A | 7 | NA | [59] | |||
| SqCCs | Fusions | FGFRs | 23 | NA | [60] | ||
| Mutations | TP53 | 79 | NA | [55] | |||
| NF1 | 10 | NA | [52] | ||||
| FGFR1 | 20 | NA | [60] | ||||
| FGFR2 | 3 | NA | [61] | ||||
| DDR2 | 2-3 | NA | [62] | ||||
| BRAF | 4-5 | NA | [63] | ||||
| KRAS | 1-2 | NA | [64] | ||||
| PDGFRA | 4 | NA | [52] | ||||
| PIK3CA | 15 | NA | [53, 54] | ||||
| PTEN | 10 | NA | [51] | ||||
| Copy number gene alterations | Gains | SOX2 | 65 | NA | [65] | ||
| PIK3CA | 15 | NA | [53, 54] | ||||
| TP53 | 79 | NA | [55] | ||||
| Losses | CDKN2A | 15 | NA | [59] | |||
| PTEN | 8 | NA | [66] | ||||
| SCLC | Mutations | TP53 | 90 | NA | [67, 68] | ||
| RB1 | 90 | NA | [67, 68] | ||||
| EP300 | 4-6 | NA | [69, 70] | ||||
| CREBBP | 4-6 | NA | [69, 70] | ||||
| PTEN | 10-18 | NA | [68] | ||||
| Copy number gene alterations | Gains | MYC | 20-30 | NA | [71] | ||
| MYCN | 20-30 | NA | [71] | ||||
| MYCL1 | 20-30 | NA | [71] | ||||
| SOX2 | 27 | NA | [72] | ||||
| FGFR1 | 5-6 | NA | [73] | ||||
Technological advances in molecular biology have provided a comprehensive means of molecular profile and the identification of driver oncogenes.
Oncogenes generally encode proteins that regulate several cellular processes including proliferation and survival. Mutations, gene rearrangements and gene amplification represent the most common genetic aberrations that could activate an oncogene, leading to a deregulated expression and/or function of the gene [5].
The definition of “driver” and “passenger” mutations represents a key point related to the tumorigenesis and the treatment with specific inhibitors. The term “driver” refers to somatic mutations that are able to increase the fitness of the cell, whereas “passenger” includes mutations that are biologically neutral and not confers growth advantage [74-76].
A “driver” mutation is causally related to cancer development, so in this view targeting a “driver” mutation with specific inhibitors represents generally a successful therapeutic strategy in cancer.
NSCLC is one of the tumors with a higher mutation rate of protein-altering mutations, particularly adenocarcinomas showed a rate of 3.5 per Mb and squamous cell carcinomas a rate of 3.9, compared to the rate of 1.8 across all tumor types [77]. Large-scale sequencing studies have shown a broad spectrum of genetic aberrations in NSCLC and a different genetic profile between lung adenocarcinomas and lung squamous cell carcinomas [25, 78-80].
NSCLC molecular profile is markedly distinct from other lung cancer histotypes: mainly in adenocarcinoma specific therapeutic targets have been defined. EGFR activating mutation, ALK rearrangements (ALK-R) and ROS1 rearrangements (ROS1-R) represent genetic hallmarks that predict a good response to treatment with specific tyrosine kinase inhibitor (TKI) in lung cancer with adenocarcinoma histology. Beyond these targetable alterations, other genomic aberrations have been reported in adenocarcinoma, including mutations, copy number gene alterations, as well as fusion mechanisms involving the receptor tyrosine kinase, such as ROS1, NTRK1 and RET (Table 1).
The updated molecular testing guidelines for the selection of lung cancer patients proposed by the College of American Pathologists (CAP), the International Association for the Study of Lung Cancer (IASLC), and the Association for Molecular Pathology (AMP) suggest the analysis of genetic alterations of additional genes such as ERBB2, MET, BRAF, KRAS, and RET not indicated as a routine stand-alone assay however as additional genes for laboratories that perform next-generation sequencing panels [47, 81].
Historically, a better understanding of the genetic aberrations was confined exclusively to adenocarcinoma, but more recently next-generation sequencing technologies are allowing a better molecular characterization also in other hystotypes.
Recently, increasing interest in comprehensive genome-wide characterization of SqCC has been reported, however, unfortunately no therapeutic targets have been yet identified. As it would be expected, molecular landscape in SqCC is distinct from the 'driver' mutations generally associated to adenocarcinoma. Several recurrent mutations have been found in SqCC, including DDR2 mutations, FGFR1 amplification, FGFR2,3,4 mutations and rearrangements (Table 1).
Recently, Devarakonda and colleagues analysed the molecular profile of 908 resected NSCLC specimens by sequencing a targeted panel consisting of 1,538 genes. The analyzed panel set of genes was selected based on knowledge of the most frequent genes involved in lung cancer pathogenesis, regardless of their clinical implications [27]. Sequencing results show that the genes most differentially mutated between ADC and SqCC were KRAS (19% versus 2%), TP53 (44% versus 69%), and STK11 (21% versus 2%); furthermore aberrations in receptor tyrosine kinase/RAS signaling were detected in approximately 70% of ADCs analyzed. As previously reported, activating mutations in KRAS, HRAS, NRAS, and EGFR were identified only in 3% of SqCCs [27].
Unfortunately, until now no molecular targets have been identified for the treatment with specific inhibitors of LNETs, thus surgery and/or conventional systemic therapy represents the treatment of choice for these tumors [82]. Previous studies analysing genomic aberrations in SCLC shown that the most frequent are inactivating mutations in TP53 and Rb1 genes, whereas activating mutations of EGFR, KRAS, as well mutations of PIK3CA, c-Myc amplification, c-KIT overexpression and PTEN mutation/loss are rare [83-85].
Recently, Simbolo and colleagues performed a comprehensive molecular analysis of LNETs, showing a prognostic impact of aberrations involved in RB1 and TERT in all histological subtypes, MEN1 mutations in SCLCs and KMT2D in ACs [86].
In the context of the predictive value of target therapies, preliminary data showed that the alterations involved in PI3K/AKT/mTOR pathway activation could be a potential therapeutic target, particularly PIK3CA mutations and copy gains of PIK3CA and RICTOR [87].
In conclusion, high heterogeneous genomic profiles between different histotypes of lung cancer could provide an explanation for great variable treatment response and prognostic stratification histotypes-related factors.
4. Inter- and intra-tumor heterogeneity of oncogenic driver mutations in NSCLC
In the last decade, the therapeutic decision-making approach based on the presence of oncogenic “driver” aberrations has incredibly changed the treatment of NSCLC patients with the development of target therapies, particularly specific inhibitor of EGFR, ALK and ROS1 aberrations.
In NSCLC, oncogenic driver mutations are frequently associated with specific clinical and pathological features, including histologic subtypes, gender, ethnic, age, past smoking history/status of other common oncogenes.
Dietz and colleagues investigated the spatial distribution of allele frequencies of KRAS and EGFR mutations in lung adenocarcinomas throughout whole tumor sections in correlation to all different histopathological patterns. The variant allele frequencies (VAFs) of KRAS and EGFR mutations were determined for all segments by digital PCR and their results showed that mutant allele frequencies were significantly higher in segments with a predominant solid pattern compared to all other histologies (p < 0.01) [88].
Heterogeneous distribution of EGFR mutations was observed within a primary tumor composed of mixed atypical adenomatous hyperplasia, bronchoalveolar carcinoma, and adenocarcinoma [89].
Previously, we demonstrated that homogeneity in EGFR aberrations occur within lung mixed ADCs regardless histological patterns, contrary to ALK rearrangements that are generally observed in solid patterns and exclusively in the adenocarcinoma areas of adenosquamous lung carcinomas [90].
In lung cancer, frequently cytologic samples or small biopsies represent the only specimens for tumor diagnosis and affect the choice of treatment, thus a potential genetic heterogeneity within a primary tumor could crucially affect clinical outcome to a specific treatment.
NSCLC patients harboring targetable driver mutations generally respond well to specific inhibitors, however some patients show short responses and TKIs resistance that could be frequently explained through molecular heterogeneity between the primary lung tumors and the metastases [91-93].
The intratumor genetic heterogeneity represents one of the most critical issues related to sensitivity to the treatment and ultimately to resistance to specific TKI. In literature, several studies in lung cancer series reported discrepancies in EGFR, ALK and KRAS mutational status between primary tumors and corresponding metastases [94-99]. Moreover, numerous studies have revealed the concordance of EGFR status in primary tumours and corresponding metastases, suggesting a possible explanation of the discordance due to technical limitations [6, 14, 90, 100, 101]. In contrast, several results demonstrated hetereogeneity in the EGFR mutation status between the primary lung tumor and the metastases [94, 102, 103].
Chen et al analyzed EGFR mutational condition in paired samples of primary lung adenocarcinoma and regional lymph nodes or distant metastases. Heterogeneity of EGFR mutations was higher (rate of 24.4%; 10 of 41) in patients with multiple pulmonary nodules resulting in significant clinical implications since the current guidelines recommend biopsy in only one lesion [93].
For ALK gene, some data revealed disconcordance between ALK rearrangement in primary NSCLC tumor and corresponding metastases [98, 105].
In conclusion, discordances between oncogenic driver mutations status in primary lesions and metastases may have significant implications in treatment with specific inhibitors of NSCLC patients.
5. Heterogeneity of molecular profile and potential value in clinical setting of lung cancer
Tailored therapies based on the identification of molecular targets represent currently a well-established therapeutic scenario in the treatment of NSCLC patients, however short responses and development of resistance are frequently observed in daily clinical practice. Although the optimal efficacy of specific TKIs, a subset of NSCLC patients often shows a mixed response to treatment. Patient-specific response and resistance can originate not only from secondary aberrations induced by targeted therapy but also from intratumoral genetic heterogeneity [106].
To date, different models have been proposed to explain the difference of genetic profile between primary tumour and corresponding metastases. Particularly, a classical model for development of metastases proposes that primary tumor cells have a low metastatic potential, thus the acquirement of enough genetic aberrations improve the metastatic progression. Another theory suggests a metastatic potential of primary tumor that leads a clonal progression from a non-malignant to malignant state, involving random metastases from tumor cells without any significant additional genetic aberrations [103].
Recently, a multicenter prospective cohort study, Tracking Non-Small-Cell Lung Cancer Evolution through Therapy (TRACERx), investigated the intratumor heterogeneity in surgically resected early-stage NSCLCs [25]. TRACERx analyzed the intratumor variability of several genetic aberrations including single or dinucleotide base substitutions, small insertions and deletions, somatic copy-number alterations [25].
Jamal‑Hanjani and colleagues demonstrated that some targetable driver mutations involved in EGFR, MET and BRAF are generally clonal and early, compared to other aberrations in genes such as PIK3CA, NF1, KRAS, TP53, and NOTCH family members that are subclonal and appear later in tumor evolution [25].
Beyond heterogeneity of druggable driver mutations, previous studies have analyzed the presence of mutational signatures across human cancer types, proving that specific mutational signatures could correlate with defined tumors [26].
In ADCs, SqCCs and SCLC a higher prevalence of mutational signature associated with smoking has been reported. Similarly, the signature associated to APOBEC, a family of cytidine deaminase enzymes involved in messenger RNA editing, exhibited strong correlations with ADCs and SqCCs [26].
Recently, a multicenter prospective study analyzed the expression clonal and subclonal of these validated mutational signatures suggesting that the signature associated to APOBEC could frequently induce subclonal mutations resulting in a spatial heterogeneity [25].
In lung cancer, another great biological variability was reported between smokers versus never-smokers, since several carcinogens of the tobacco smoke lead to a high mutational rate including both driver and passenger mutations [26, 107].
Recently, Soo and colleagues showed the clinical-pathological features typical of never-smokers analyzed in a wide series of NSCLC, in order to clarify their characteristics still not fully known. Never-smokers showed a higher rate of ALK-rearrangement (26% vs. 4%, p < .001) and EGFR mutations (36% vs. 8%, p < .001) [108].
Genome-wide studies identified several potential genetic marker of susceptibility in LCINS, such as chromosomal locus 5p15.33 comprising TERT and CLPTMIL genes, the hypoxia-inducible factor-2α EPAS1, specific SNPs in CSF1R, p63, TP63 genes, a functional polymorphism in CSF1R gene [109].
The biological differences between these two subsets result in differential response to therapies, including EGFR inhibitors, thus a better genetic characterization of lung cancer in non-smokers (LCINS) is needed [110].
Conclusion
Discordance of molecular profiles between primary lesions and their corresponding metastases in the context of druggable driver mutations could be the key point in personalized medicine of lung cancer patients. Indeed, intra-tumor molecular heterogeneity represents a great source of concern in mixed tumor responses to treatment, including treatment with specific TKI inhibitors but also chemotherapy.
In lung cancer patients the rebiopsy is rarely performed, however in the view of intratumor heterogeneity a single biopsy-based analyses for personalized medicine could be a great limitation.
Abbreviations
CSCs: cancer stem cells; CIN: chromosomal instability; NSCLC: non-small cell lung cancer; TMB: tumor mutational burden; MASI: mutant allele specific imbalance; miRNAs: microRNA; SCLC: small-cell lung carcinoma; ADC: adenocarcinoma; SqCC: squamous cell carcinoma; LNETs: lung neuroendocrine tumours; ALK-R: ALK rearrangements; ROS1-R: ROS1 rearrangements; TKI: tyrosine kinase inhibitor.
Acknowledgements
The manuscript was supported by 'Programma Valere', funded by Università Vanvitelli per la Ricerca.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jamal-Hanjani M, Quezada SA, Larkin J. et al. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21:1258-1266
2. Rich JN. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine (Baltimore). 2016;95(1 Suppl 1):S2-S7
3. Burrell RA, McGranahan N, Bartek J. et al. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338-345
4. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275-291
5. Vogelstein B, Papadopoulos N, Velculescu VE. et al. Cancer genome landscapes. Science. 2013;339:1546-58
6. de Bruin EC, McGranahan N, Mitter R. et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251-256
7. Walter MJ, Shen D, Ding L. et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090-1098
8. Boeckx N, Op de Beeck K, Deschoolmeester V. et al. Anti-EGFR resistance in colorectal cancer: current knowledge and future perspectives. Curr Colorectal Cancer Rep. 2014:1-15
9. Diaz LA, Williams RT, Wu J. et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:1-4
10. Fojo T, Mailankody S, Lo A. Unintended consequences of expensive cancer therapeutics-the pursuit of marginal indications and a me-too mentality that stifles innovation and creativity: the john conley lecture. JAMA Otolaryngol Head Neck Surg. 2014;140:1225-1236
11. Kleppe M, Levine RL. Tumor heterogeneity confounds and illuminates. Nat Med. 2014;20:342-344
12. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323-334
13. Vogelstein B, Papadopoulos N, Velculescu VE. et al. Cancer genome landscapes. Science. 2013;339:1546-1558
14. Zhang J, Fujimoto J, Zhang J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256-259
15. Neelakantan D, Drasin DJ, Ford HL. Intratumoral heterogeneity: Clonal cooperation in epithelial-to-mesenchymal transition and metastasis. Cell Adh Migr. 2015;9:265-276
16. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613-628
17. Varella-Garcia M. Chromosomal and genomic changes in lung cancer. Cell Adh Migr. 2010;4:100-106
18. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220-228
19. Lee AJ, Endesfelder D, Rowan AJ. et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71:1858-1870
20. McGranahan N, Burrell RA, Endesfelder D. et al. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528-358
21. Carter SL, Eklund AC, Kohane IS. et al. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043-1048
22. Mettu RK, Wan YW, Habermann JK. et al. A 12-gene genomic instability signature predicts clinical outcomes in multiple cancer types. Int J Biol Markers. 2010;25:219-228
23. Yoo JW, Seo KW, Jang SJ. et al. The relationship between the presence of chromosomal instability and prognosis of squamous cell carcinoma of the lung: fluorescence in situ hybridization analysis of paraffin-embedded tissue from 47 Korean patients. J Korean Med Sci. 2010;25:863-867
24. Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010;103:1139-1143
25. Jamal-Hanjani M, Wilson GA, McGranahan N. et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med. 2017;376:2109-2121
26. Alexandrov LB, Nik-Zainal S, Wedge DC. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-421
27. Devarakonda S, Rotolo F, Tsao MS. et al. Tumor Mutation Burden as a Biomarker in Resected Non-Small-Cell Lung Cancer. J Clin Oncol. 2018 [Epub ahead of print]
28. Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A. 2013;110:1999-2004
29. Soh J, Okumura N, Lockwood WW. et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009:7464
30. Oakley GJ, Chiosea SI. Higher dosage of the epidermal growth factor receptor mutant allele in lung adenocarcinoma correlates with younger age, stage IV at presentation, and poorer survival. J Thorac Oncol. 2011;6:1407-1412
31. Malapelle U, Vatrano S, Russo S. et al. EGFR mutant allelic-specific imbalance assessment in routine samples of non-small cell lung cancer. J Clin Pathol. 2015;68:739-741
32. Takano T, Ohe Y, Sakamoto H. et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23:6829-6837
33. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057-1068
34. Szejniuk WM, Robles AI, McCulloch T. et al. Epigenetic predictive biomarkers for response or outcome to platinum-based chemotherapy in non-small cell lung cancer, current state-of-art. Pharmacogenomics J. 2018;19:5-14
35. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857-66
36. Michor F, Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res (Phila). 2010;3:1361-1364
37. Codony-Servat J, Verlicchi A, Rosell R. Cancer stem cells in small cell lung cancer. Transl Lung Cancer Res. 2016;5:16-25
38. Wang P, Gao Q, Suo Z. et al. Identification and characterization of cells with cancer stem cell properties in human primary lung cancer cell lines. PLoS One. 2013;8:57020
39. Sales KM, Winslet MC, Seifalian AM. Stem cells and cancer: an overview. Stem Cell Rev. 2007;3:249-55
40. Kajstura J, Rota M, Hall SR. et al. Evidence for human lung stem cells. N Engl J Med. 2011;364:1795-806
41. Chen Z, Fillmore CM, Hammerman PS. et al. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535-546
42. Trédan O, Galmarini CM, Patel K. et al. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441-1454
43. Yamada T, Takeuchi S, Kita K. et al. Hepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancer. J Thorac Oncol. 2012;7:272-280
44. Zito Marino F, Ascierto PA, Rossi G. et al. Are tumor-infiltrating lymphocytes protagonists or background actors in patient selection for cancer immunotherapy? Expert Opin Biol Ther. 2017;17:735-746
45. Travis WD, Brambilla E, Nicholson AG. et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 2015;10:1243-1260
46. Franco R, Rocco G, Marino FZ. et al. Anaplastic lymphoma kinase: a glimmer of hope in lung cancer treatment? Expert Rev Anticancer Ther. 2013;13:407-20
47. Lindeman NI, Cagle PT, Aisner DL. et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med. 2018;142:321-346
48. Vaishnavi A, Capelletti M, Le AT. et al. Oncogenic ad drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19:1469-1472
49. Paez JG, Jänne PA, Lee JC. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497-500
50. Salgia R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol Cancer Ther. 2017;16:555-565
51. Jin G, Kim MJ, Jeon HS. et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer. 2010;69:279-83
52. Kandoth C, McLellan MD, Vandin F. et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333-339
53. Engelman JA, Chen L, Tan X. et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351-6
54. Kawano O, Sasaki H, Endo K. et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006;54:209-15
55. Mitsudomi T, Hamajima N, Ogawa M. et al. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: a meta-analysis. Clin. Cancer Res. 2000;6:4055-4063
56. Li BT, Ross DS, Aisner DL. et al. HER2 Amplification and HER2 Mutation Are Distinct Molecular Targets in Lung Cancers. J Thorac Oncol. 2016;11:414-9
57. Fiala O, Pesek M, Finek J. et al. Epidermal Growth Factor Receptor Gene Amplification in Patients with Advanced-stage NSCLC. Anticancer Res. 2016;36:455-60
58. Zhu CQ, Cutz JC, Liu N. et al. Amplification of telomerase (hTERT) gene is a poor prognostic marker in non-small-cell lung cancer. Br J Cancer. 2006;94:1452-9
59. Lou-Qian Z, Rong Y, Ming L. et al. The prognostic value of epigenetic silencing of p16 gene in NSCLC patients: a systematic review and meta-analysis. PLoS One. 2013;8:54970
60. Marek L, Ware KE, Fritzsche A. et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol. 2009;75:196-207
61. The Cancer Genome Atlas Research Network (TCGA). Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519-525
62. Hammerman PS, Sos ML, Ramos AH. et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011;1:78-89
63. Paik PK, Arcila ME, Fara M. et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046-51
64. Mascaux C, Iannino N, Martin B. et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92:131-9
65. Bass AJ, Watanabe H, Mermel CH. et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238-42
66. Soria JC, Lee HY, Lee JI. et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002;8:1178-84
67. Meuwissen R, Linn SC, Linnoila RI. et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181-9
68. Karachaliou N, Pilotto S, Lazzari C. et al. Cellular and molecular biology of small cell lung cancer: an overview. Transl Lung Cancer Res. 2016;5:2-15
69. Umemura S, Mimaki S, Makinoshima H. et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J Thorac Oncol. 2014;9:1324-1331
70. Ross JS, Wang K, Elkadi OR. et al. Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. J Clin Pathol. 2014;67:772-776
71. Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015;29:1447-62
72. Peifer M, Fernández-Cuesta L, Sos ML. et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104-1110
73. Schultheis AM, Bos M, Schmitz K. et al. Fibroblast growth factor receptor 1 (FGFR1) amplification is a potential therapeutic target in small-cell lung cancer. Mod Pathol. 2014;27:214-21
74. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108:479-85
75. Haber DA, Settleman J. Cancer: drivers and passengers. Nature. 2007;446:145-6
76. Greenman C, Stephens P, Smith R. et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153-8
77. Kan Z, Jaiswal BS, Stinson J. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869-873
78. Imielinski M, Berger AH, Hammerman PS. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107-20
79. Govindan R, Ding L, Griffith M. et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121-34
80. Campbell JD, Alexandrov A, Kim J. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607-16
81. Hanna N, Johnson D, Temin S. et al. Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2017;35:3484-3515
82. Gridelli C, Rossi A, Airoma G. et al. Treatment of pulmonary neuroendocrine tumours: state of the art and future developments. Cancer Treat Rev. 2013;39:466-472
83. Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol. 2001;28:3-13
84. Shibata T, Kokubu A, Tsuta K. et al. Oncogenic mutation of PIK3CA in small cell lung carcinoma: a potential therapeutic target pathway for chemotherapy-resistant lung cancer. Cancer Lett. 2009;283:203-11
85. Tatematsu A, Shimizu J, Murakami Y. et al. Epidermal growth factor receptor mutations in small cell lung cancer. Clin Cancer Res. 2008;14:6092-6096
86. Simbolo M, Mafficini A, O Sikora K. et al. Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J Pathol. 2017;241:488-500
87. Umemura S, Mimaki S, Makinoshima H. et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J Thorac Oncol. 2014;9:1324-1331
88. Dietz S, Harms A, Endris V. et al. Spatial distribution of EGFR and KRAS mutation frequencies correlates with histological growth patterns of lung adenocarcinomas. Int J Cancer. 2017;141:1841-1848
89. Nakano H, Soda H, Takasu M. et al. Heterogeneity of epidermal growth factor receptor mutations within a mixed adenocarcinoma lung nodule. Lung Cancer. 2008;60:136-140
90. Zito Marino F, Liguori G, Aquino G. et al. Intratumor Heterogeneity of ALK-Rearrangements and Homogeneity of EGFR-Mutations in Mixed Lung Adenocarcinoma. PLoS One. 2015;10:e0141521
91. Taniguchi K, Okami J, Kodama K. et al. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008;99:929-935
92. Jiang SX, Yamashita K, Yamamoto M. et al. EGFR genetic heterogeneity of nonsmall cell lung cancers contributing to acquired gefitinib resistance. Int J Cancer. 2008;123:2480-2486
93. Chen ZY, Zhong WZ, Zhang XC. et al. EGFR Mutation Heterogeneity and the Mixed Response to EGFR Tyrosine Kinase Inhibitors of Lung Adenocarcinomas. Oncologist. 2012;17:978-985
94. Gow CH, Chang YL, Hsu YC. et al. Comparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancer. Ann Oncol. 2009;20:696-702
95. Kalikaki A, Koutsopoulos A, Trypaki M. et al. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br J Cancer. 2008;99:923-9
96. Monaco SE, Nikiforova MN, Cieply K. et al. Teot LA, Khalbuss WE, Dacic S. A comparison of EGFR and KRAS status in primary lung carcinoma and matched metastases. Hum Pathol. 2010;41:94-102
97. Kim H, Xu X, Yoo SB. et al. Discordance between anaplastic lymphoma kinase status in primary non-small-cell lung cancers and their corresponding metastases. Histopathology. 2013;62:305-14
98. Wu C, Zhao C, Yang Y. et al. High discrepancy of driver mutations in patients with NSCLC and synchronous multiple lung ground-glass nodules. J Thorac Oncol. 2015;10:778-83
99. Abe H, Kawahara A, Azuma K. et al. Heterogeneity of anaplastic lymphoma kinase gene rearrangement in non small cell lung carcinomas: a comparative study between small biopsy and excision samples. J Thorac Oncol. 2015;10:800-5
100. Park S, Holmes-Tisch AJ, Cho EY. et al. Discordance of molecular biomarkers associated with epidermal growth factor receptor pathway between primary tumors and lymph node metastasis in non-small cell lung cancer. J Thorac Oncol. 2009;4:809-15
101. Hiley CT, Le Quesne J, Santis G. et al. Challenges in molecular testing in non-small-cell lung cancer patients with advanced disease. Lancet. 2016;388:1002-11
102. Matsumoto S, Takahashi K, Iwakawa R. et al. Frequent EGFR mutations in brain metastases of lung adenocarcinoma. Int J Cancer. 2006;119:1491-1494
103. Wang S, Wang Z. Meta-analysis of epidermal growth factor receptor and KRAS gene status between primary and corresponding metastatic tumours of non-small cell lung cancer. Clin Oncol (R Coll Radiol). 2015;27:30-9
104. Han HS, Eom DW, Kim JH. et al. EGFR mutation status in primary lung adenocarcinomas and corresponding metastatic lesions: discordance in pleural metastases. Clin Lung Cancer. 2011;12:380-386
105. Kim H, Xu X, Yoo SB. et al. Discordance between anaplastic lymphoma kinase status in primary non-small-cell lung cancers and their corresponding metastases. Histopathology. 2013;62:305-14
106. Dong ZY, Zhai HR, Hou QY. et al. Mixed Responses to Systemic Therapy Revealed Potential Genetic Heterogeneity and Poor Survival in Patients with Non-Small Cell Lung Cancer. Oncologist. 2017;22:61-69
107. Hudson AM, Wirth C, Stephenson NL. et al. Using large-scale genomics data to identify driver mutations in lung cancer: Methods and challenges. Pharmacogenomics. 2015;16:1149-60
108. Dias M, Linhas R, Campainha S. et al. Lung cancer in never-smokers - what are the differences? Acta Oncologica. 2017;7:931-935
109. Choi JR, Park SY, Noh OK. et al. Gene mutation discovery research of non-smoking lung cancer patients due to indoor radon exposure. Ann Occup Environ Med. 2016;28:13
110. Wakelee HA, Chang ET, Gomez SL. et al. Lung cancer incidence in never smokers. J Clin Oncol. 2007;25:472
Author contact
![]() Corresponding authors: Renato Franco; Pathology Unit, University of Campania “L. Vanvitelli”, Naples, Italy. Email: RENATO.FRANCOit; renfrcom and Federica Zito Marino; Pathology Unit, University of Campania “L. Vanvitelli”, Naples, Italy. Email: federicazito.marinoit
Corresponding authors: Renato Franco; Pathology Unit, University of Campania “L. Vanvitelli”, Naples, Italy. Email: RENATO.FRANCOit; renfrcom and Federica Zito Marino; Pathology Unit, University of Campania “L. Vanvitelli”, Naples, Italy. Email: federicazito.marinoit