Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(7):960-966. doi:10.7150/ijms.32157 This issue Cite
Research Paper
Metformin Inhibits Epithelial-to-Mesenchymal Transition of Keloid Fibroblasts via the HIF-1α/PKM2 Signaling Pathway
Rui Lei1, Shizhen Zhang2, Yuming Wang1, Siya Dai1, Jiaqi Sun1, Chaoqun Zhu3 ![]()
1. Department of Plastic Surgery, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, China;
2. Institute of Translational Medicine, Zhejiang University School of Medicine, Hangzhou, Zhejiang, China.
3. Department of Surgery, Family Planning Service Center Of YiWu Maternity And Child Health Care Hospital, Yiwu, China
Received 2018-12-10; Accepted 2019-4-24; Published 2019-6-10
Abstract
Background: Epithelial-to-mesenchymal transition (EMT) is a process whereby epithelial cells lose cell-cell contacts and acquire expression of mesenchymal components and manifest a migratory phenotype. Recent studies indicated that EMT is involved in the development of keloids. Therefore, this study aims to investigate the mechanisms of the effects of metformin in hypoxia-induced EMT in keloid fibroblasts (KFs).
Methods: KFs were cultured in a hypoxia incubator to induce EMT and were treated with or without metformin. Cell viability was evaluated by a cell counting kit 8 (CCK-8), and cell migration was measured by the transwell assay. The expression levels of HIF-1α, E-cadherin, vimentin, phosphorylated p70s6k (p-p70s6k) and pyruvate kinase M2 (PKM2) were evaluated by western blotting.
Results: Hypoxia promoted EMT in KFs. Metformin significantly inhibited the expression of HIF-1α and partially abolished hypoxia-induced EMT. PKM2 is involved in hypoxia-induced EMT of KFs and metformin decreased the expression of p-p70s6k and PKM2.
Conclusions: Metformin abolishes hypoxia-induced EMT in KFs by inhibiting the HIF-1α/PKM2 signaling pathway. Our study provides a novel mechanistic insight into potential use of metformin for treatment of keloids.
Keywords: Metformin, EMT, HIF-1α, PKM2, Keloid.
Introduction
Keloids are typically formed during abnormal wound healing that extends beyond the site of the original skin injury [1, 2]. The formation of a keloid always results in physical discomfort, functional limitation, and psychological impairment [3]. The inside of keloids frequently exhibit ischemic-hypoxic states due to the destruction of the vascular network and excessive oxygen consumption during inflammation and repair [4]. Hypoxia-inducible factor-1 (HIF-1) is one of the key mediators of the hypoxic stress response [5]. Our previous studies have demonstrated that HIF-1α activates the TGF-β1/Smad signaling pathway and increases collagen deposition in the dermal fibroblasts in response to hypoxic conditions [6].
The epithelial-to-mesenchymal transition (EMT) is a process associated with the loss of polarity of epithelial cells and their development into mesenchymal cells with invasive and migratory properties [7]; EMT is ubiquitous in wound healing, organ fibrosis and development of cancer [7]. During EMT, the expression of epithelial markers, such as E-cadherin and zonula occludens-1 (ZO-1), are decreased and the expression of mesenchymal markers, such as vimentin and fibronectin, are increased, thus resulting in nonpolar mobilizable cells. Recently, EMT was detected in the keloid tissues. Hypoxia-rich environments in keloids induced elevated levels of HIF-1α and promoted the transformation of the keloid-derived keratinocytes into fibroblast-like cells via the activation of EMT [8].
Metformin is a typical antidiabetic drug that has significant antitumor effects in several types of human cancer [9, 10]. Metformin is recently reported to regulate cell energy metabolism [11] and repress EMT by inhibiting the mTOR signaling pathway [12]. Furthermore, metformin regulates the EMT process by inhibiting the expression of Vimentin, thus destroying the invasive phenotype induced by TGF-β1 in human cervical cancer cells [13]. Most importantly, metformin is suggested to exert an anti-keloid effect in vitro [14]. However, the complete mechanism of metformin-induced inhibition of keloid growth remains unknown.
The aim of the current study was to explore the mechanism of the effects of metformin in keloid fibroblasts. We found that metformin can reverse EMT by downregulation of HIF-1α, p-p70s6k and PKM2, suggesting that metformin is involved in the HIF-1α/PKM2 signaling pathway to inhibit keloid development.
Materials and Methods
Clinical samples
Keloid specimens and the adjacent normal skin tissues were collected at the First Affiliated Hospital of Zhejiang University. The information of the keloid patients are listed in Table 1. All procedures performed in this study were approved by the audit department of the hospital in compliance with the requirements of the Ethics Committee. All subjects included in this study have provided informed consent.
Keloid patients information.
| Patients Number | Human Race | Gender | Age | Keloid Location |
|---|---|---|---|---|
| 1 | Yellow race | Female | 25 | earlobe |
| 2 | Yellow race | Female | 32 | earlobe |
| 3 | Yellow race | Female | 22 | earlobe |
| 4 | Yellow race | Female | 36 | earlobe |
Cell culture
Primary human keloid fibroblasts (KFs) and normal skin fibroblasts (NFs) were isolated according to the protocol as previously described [14]. Briefly, the surgically excised human keloid tissues and adjacent normal skin tissues were cut away of epidermis and fat manually. Only the dermal layer of the fibrous tissues were reserved and then cut into smaller pieces. These small pieces were digested in Dulbecco's modified Eagle's medium (DMEM) lysis buffer with 1x collagenase for 2 hours. After centrifugation, the precipitates were collected and cultured in DMEM (Gibco, Gaithersburg, MD) supplemented with 1% antibiotic-antimycotic and 15% fetal bovine serum (10099-141; Gibco) in 5% CO2 at 37°C. Metformin (PeproTech, Rocky Hill, NJ, USA) was stocked at a concentration of 50 mM at 4 °C. The cells were treated with 10, 15, 20 mM metformin and collected for CCK8. The cells treated with 10 mM metformin were collected for migration assay, PCR, and Western blot.
Hypoxia culture model
Hypoxic conditions of 1% oxygen concentration (37°C, 1% O2, 5% CO2, 94% N2) were established by mixed gas hypoxia. Cells were detected after they were cultured in the hypoxic incubator for 24 h.
Small interfering RNA and cell transfection
The siRNA of PKM2 was purchased from GenePharma (Shanghai, China), the sequence is 5'-GATTATCAGCAAAATCGAG-3'. Cells were transfected using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The transfected cells were cultured for 48 hr and collected for quantitative real-time PCR and western blot analysis.
Cell viability assay
A cell counting kit-8 (CCK-8) was used to measure cell viability. The cells (1×104 per well) were seeded into 96-well plates and cultured in a hypoxic incubator. After treatment with metformin for various periods of time, 10 μL CCK-8 was added into each well and incubated for 2 h at 37°C. The absorbance was measured at 450 nm using a microplate reader.
Quantitative Real-time PCR
Total RNA was extracted using Trizol Reagent. The RNA was reverse transcribed by using the PrimeScript RT Master Mix according to the manufacturer's instruction. The subsequent Quantitative Real-time PCR was carried out in the 7500 Real-time PCR System (Applied Biosystems) using SYBR Premix Ex Taq reagents. The specific primers were as follows: HIF1α: Forward: 5'-GTAGTGCTG- ACCCTGCACTCAA-3' Reverse: 3'-CCATCGGAAGGACTAGGTGTCT-5'; β-actin: Forward: 5'-ACCGAGCGCGGCTACA-3', Reverse: 3'-CAGCCGTGG- CCATCTCTT-5'. According to the manufacture's protocol, RT-PCR was carried out in a total volume of 20 ul reaction mixture, and amplified as following steps: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 60 s. Samples were analyzed in triplicate as biological replicate. 2-ΔΔCt method was used for calculations, where ΔCt = Cttarget gene - Ctβ-actin and ΔΔCt = ΔCtinduced -ΔCtreference.
Western Blot Analysis
The KFs with different treatment were harvested by centrifugation and washed with PBS. Cells were lysed in RIPA buffer containing protease inhibitors. The lysate were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred to Polyvinylidene Fluoride (PVDF) membranes by electroblotting. The membranes were incubated overnight at 4 °C with the primary antibodies after blocking with 5% nonfat milk. The anti-human E-cadherin, anti-human vimentin, anti-human PKM2, anti-human HIF-1α, and anti-human phospho-p70s6k primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The blots were then incubated with the secondary antibody for 2 h at room temperature. Each experiment was repeated for 3 times. Specific proteins were visualized using the ECL system (GE Healthcare) and the FUJIFILM Luminescent Image Analyzer LAS3000 (Fuji Film).
Statistical Analysis
The statistical analyses were performed using SPSS 21.0 (SPSS Inc., Chicago, IL, USA). The values were expressed as the means ± SD. Significant differences among groups were analyzed by T-test and one-way analysis of variance (ANOVA). And p-value of <0.05 was defined as statistically significant.
Results
Metformin inhibits hypoxia-induced proliferation and migration
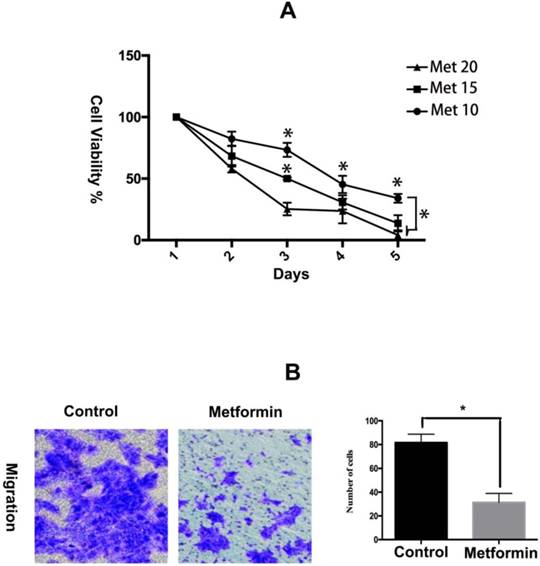
Metformin has been showed to inhibit the proliferation and collagen synthesis of the human keloids fibroblasts [15]. Here, we further evaluated the effects of metformin on viability and migration of KFs in a hypoxic environment. The results of the CCK-8 assay showed that metformin inhibited the growth of KFs in a dose- and time-dependent manner (Figure 1A). Furthermore, metformin significantly impaired the migration ability of KFs (p<0.05) (Figure 1B).
Metformin inhibits hypoxia-induced proliferation and migration in KFs. A. KFs were treated with metformin (10, 15, 20 mM) for the indicated times and cell viability was measured by the CCK-8 assay. B. The representative images of cell migration assay were obtained at 40x magnification. Graphs show the number of migrating cells after 24 h incubation.
Metformin inhibits HIF-1α expression in KFs
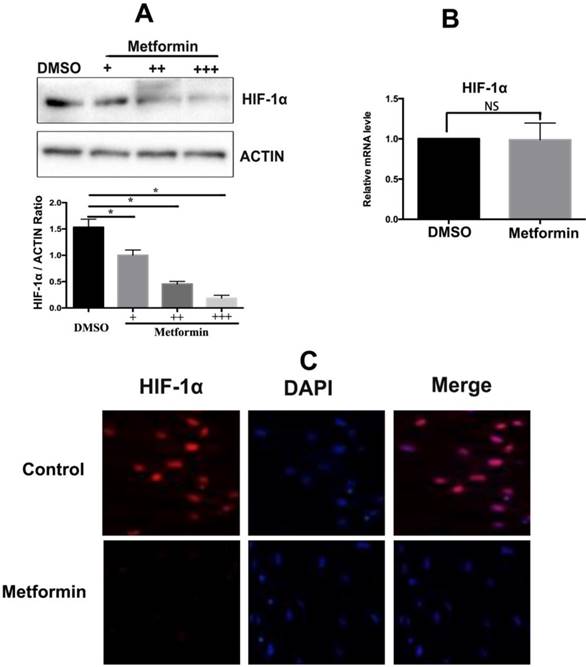
Metformin repression of HIF-1α expression was analyzed by western blotting; the data indicate that HIF-1α expression in KFs under hypoxic condition was significantly decreased in a dose-dependent manner in the presence of metformin (10mM), the results were confirmed by densitometric quantified analysis (Figure 2A). However, metformin did not affect the mRNA levels of HIF-1α, suggesting that metformin decreased the posttranslational levels of HIF-1α (p>0.05) (Figure 2B). Moreover, immunofluorescence staining confirmed that metformin inhibited the expression of the HIF-1α protein (Figure 2C).
Metformin downregulates HIF-1α expression at a posttranscriptional level. A. Metformin inhibited HIF-1α expression in a time- and dose-dependent manner, Columns represent the mean of at least three independent experiments; error bars represent the SD of the mean. *p<0.05. B. Metformin did not influence the mRNA levels of HIF-1α (NC. Not significant, p>0.05). C. Immunofluorescent staining showed that metformin inhibited HIF-1α expression.
PKM2 is required for HIF-1α-induced EMT
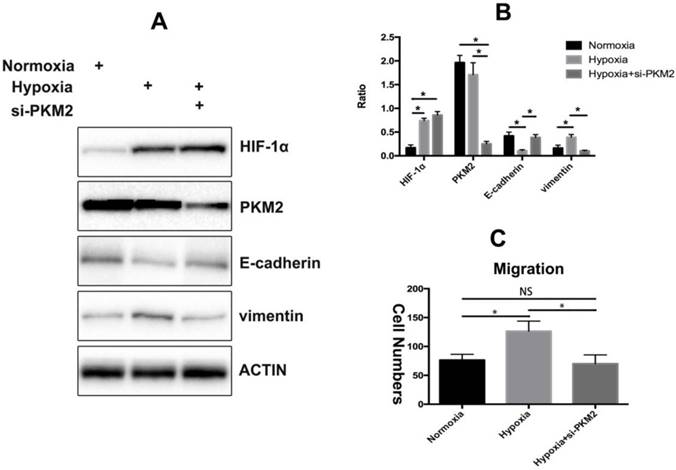
PKM2 is essential for TGF-β1-induced EMT in cancer cells and knockdown of PKM2 failed to induce spindle-shaped morphological changes and EMT-related alterations in the TGF-β1-treated colorectal cancer cells[16]. We have previously proved that HIF-1α could activate TGF-β1 expression to promote collagen deposition in dermal fibroblasts[6]. Therefore, we tested whether PKM2 plays a critical role in HIF-1-induced EMT in KFs. We found that PKM2 knockdown reduced the HIF-1α-induced loss of E-cadherin and gain of vimentin in KFs under hypoxic condition (Figure 3A). We also confirmed the results by densitometric quantified analysis (Figure 3B). Furthermore, PKM2 silencing decreased the migratory ability of KFs compared with that of the control group (p<0.05) (Figure 3C).
PKM2 is involved in hypoxia-induced EMT. A. Hypoxia induced the expression of vimentin and decreased the levels of E-cadherin while silencing of PKM2 reversed this effect, the results were confirmed by densitometric quantified analysis (B), Columns represent the mean of at least three independent experiments; error bars represent the SD of the mean. *p<0.05. C. Knockdown of PKM2 impaired hypoxia-induced EMT (NC. Not significant, *p<0.05).
Metformin reverses EMT in keloids through inhibition of the HIF-1α/PKM2 pathway
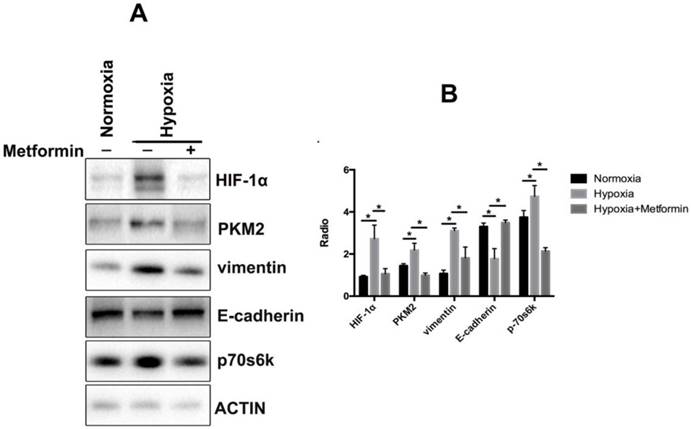
We further investigated the mechanism of metformin effects involved in the regulation of EMT in keloid. The results showed that metformin rescued the accumulation of vimentin and decreased the levels of E-cadherin in hypoxia-treated KFs (Figure 4A, 4B). Additionally, we found that stimulation by hypoxia increased the phosphorylation of p70s6k and upregulated the expression of PKM2 in KFs. Hence, metformin reversed hypoxia-induced EMT by inhibiting the phosphorylation of p70s6k and suppressing expression of PKM2 in KFs (Figure 4A, 4B).
Metformin reverses EMT by inhibiting the HIF-1α/ PKM2 pathway. A. Metformin rescued the accumulation of vimentin and decreased the levels of E-cadherin in hypoxia-treated KFs. Metformin also inhibited phosphorylation of p70s6k and upregulated the expression of PKM2 in KFs under hypoxia condition; B. Columns represent the mean of at least three independent experiments; error bars represent the SD of the mean. *p<0.05.
Discussion
Numerous studies have demonstrated that HIF-1α plays a pivotal role in promoting cancer metastasis [17, 18]. HIF-1α promotes fibrogenesis and ECM accumulation in a wide range of tumors through EMT [19-21]. Keloid predominantly develops after a trauma; the cutaneous vascular network is always disrupted leading to a hypoxic environment in the local tissue. Moreover, high metabolic state during tissue injury-induced inflammation and repair dramatically increases oxygen consumption and exacerbates the hypoxia [4]. As a result, HIF-1α is always upregulated in keloids due to the hypoxic microenvironment. HIF-1α induced the transformation of keloid-derived keratinocytes into fibroblast-like cells through EMT allowing the keloid fibroblasts to extend beyond the wound margin [8]. Our results proved that metformin can significantly inhibit HIF-1α expression and subsequently impair EMT.
PKM2 is a downstream effector of mTOR that plays a crucial role in aerobic glycolysis and tumor growth [22]. PKM2 is essential for TGF-induced EMT in several human cancers [16, 23]. The HIF-1α and c-Myc-hnRNP cascades are essential mediators of mTOR-dependent regulation of PKM2 [22]. PKM2 is a critical downstream effector of the HIF-1α-related signaling pathways and is the key glycolytic enzyme [24]. In this study, we observed that PKM2 is critical for HIF-1α-induced EMT in KFs because silencing of PKM2 reversed hypoxia-induced EMT. The phosphorylation of the p70 ribosomal S6 kinase 1 (S6K1), a downstream effector of mTOR, plays a critical role in controlling cell growth. Deactivation of S6K1 leads to cell proliferation at a smaller size and at a reduced rate [25]. Our results indicate that hypoxia induced the expression of PKM2 and p70s6k, suggesting that PKM2 and p70s6k are involved in EMT in keloids. Moreover, we demonstrated that metformin inhibited the HIF-1α/PKM2 signaling pathway and reversed hypoxia-induced EMT in KFs.
Conclusion
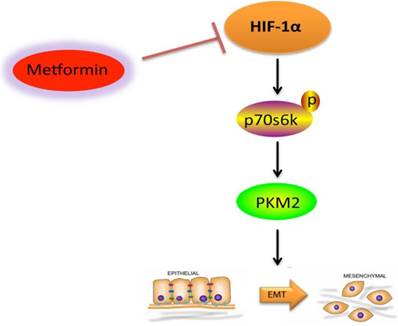
In the current study, we proved that hypoxic environment promoted KF migration by activating HIF-1α-induced EMT. Metformin inhibits hypoxia-induced EMT of KFs via the HIF-1α/PKM2 signaling pathway (Figure 5). Based on these data, we propose that the HIF-1α/PKM2 pathway is involved in hypoxia-induced EMT in keloids. Therefore, metformin may be a potential therapeutic agent for the treatment of keloids.
Graphical abstract of this study. Metformin inhibits hypoxia-induced EMT of KFs via the HIF-1α/PKM2 signaling pathway.
Abbreviations
Epithelial-to-mesenchymal transition (EMT); Keloid fibroblasts (KFs); Cell counting kit 8 (CCK-8); Pyruvate kinase M2 (PKM2); Hypoxia-inducible factor-1 (HIF-1); Zonula occludens-1 (ZO-1); Phosphorylation of the p70 ribosomal S6 kinase 1 (p70S6K1).
Acknowledgements
Conceptualization, R.L. and C.Z.; Experimental Performing: R.L., S.Z. Y.W. S.D. and J.S.; Writing-Original Draft Preparation, R.L.; Writing - Review & Editing, S.Z. Y.W. and C.Z.; Visualization, X.X.; Supervision, C.Z.
Ethics approval and consent to participate
This clinical trial was approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University. All procedures performed in this study were approved by the audit department of the hospital. The study further meets the requirements set out by the ICH-GCP, Declaration of Helsinki and the German Drug Law (AMG). Written informed consent was obtained from the patients.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Halder RM. Pseudofolliculitis barbae and related disorders. Dermatologic clinics. 1988;6:407-12
2. Marneros AG, Krieg T. Keloids-clinical diagnosis, pathogenesis, and treatment options. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology: JDDG. 2004;2:905-13
3. Lahiri A, Tsiliboti D, Gaze NR. Experience with difficult keloids. British journal of plastic surgery. 2001;54:633-5
4. Schreml S, Szeimies RM, Prantl L, Karrer S, Landthaler M, Babilas P. Oxygen in acute and chronic wound healing. Br J Dermatol. 2010;163:257-68
5. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437-43
6. Mingyuan X, Qianqian P, Shengquan X, Chenyi Y, Rui L, Yichen S. et al. Hypoxia-inducible factor-1alpha activates transforming growth factor-beta1/Smad signaling and increases collagen deposition in dermal fibroblasts. Oncotarget. 2018;9:3188-97
7. Kong D, Li Y, Wang Z, Sarkar FH. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins? Cancers. 2011;3:716-29
8. Ma X, Chen J, Xu B, Long X, Qin H, Zhao RC. et al. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxic microenvironment in vitro. International journal of molecular medicine. 2015;35:1246-56
9. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer research. 2006;66:10269-73
10. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clinical science (London, England: 1979). 2012;122:253-70
11. Xing Y, Meng Q, Chen X, Zhao Y, Liu W, Hu J. et al. TRIM44 promotes proliferation and metastasis in nonsmall cell lung cancer via mTOR signaling pathway. Oncotarget. 2016;7:30479-91
12. Li L, Han R, Xiao H, Lin C, Wang Y, Liu H. et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:2714-26
13. Tyszka-Czochara M, Lasota M, Majka M. Caffeic Acid and Metformin Inhibit Invasive Phenotype Induced by TGF-beta1 in C-4I and HTB-35/SiHa Human Cervical Squamous Carcinoma Cells by Acting on Different Molecular Targets. Int J Mol Sci. 2018:19
14. Lei R, Shen J, Zhang S, Liu A, Chen X, Wang Y. et al. Inactivating the ubiquitin ligase Parkin suppresses cell proliferation and induces apoptosis in human keloids. Journal of cellular physiology. 2019
15. Yi S, Weiyuan M. [The effect of Metformin on the proliferation and collagen synthesis of human keloids fibroblasts]. Zhonghua zheng xing wai ke za zhi = Zhonghua zhengxing waike zazhi = Chinese journal of plastic surgery. 2015;31:291-5
16. Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K. et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:15526-31
17. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399-408
18. LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nature cell biology. 2016;18:356-65
19. Jiang J, Tang YL, Liang XH. EMT: a new vision of hypoxia promoting cancer progression. Cancer biology & therapy. 2011;11:714-23
20. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B. et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. The Journal of clinical investigation. 2007;117:3810-20
21. Du R, Xia L, Ning X, Liu L, Sun W, Huang C. et al. Hypoxia-induced Bmi1 promotes renal tubular epithelial cell-mesenchymal transition and renal fibrosis via PI3K/Akt signal. Molecular biology of the cell. 2014;25:2650-9
22. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X. et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4129-34
23. Cheng K, Hao M. Metformin Inhibits TGF-beta1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells. Int J Mol Sci. 2016:17
24. Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends in biochemical sciences. 2012;37:309-16
25. Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285:2126-9
Author contact
![]() Corresponding author: Chaoqun Zhu. E-mail: zcq1123wcom.
Corresponding author: Chaoqun Zhu. E-mail: zcq1123wcom.