Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(3):282-290. doi:10.7150/ijms.7634 This issue Cite
Research Paper
Protective Effect of Quercetin against Oxidative Stress and Brain Edema in an Experimental Rat Model of Subarachnoid Hemorrhage
Yu-shu Dong1,2*, Ju-lei Wang1,3*, Da-yun Feng1, Huai-zhou Qin1, Hua Wen2, Zhong-min Yin2, Guo-dong Gao1 ![]() , Chuan Li4
, Chuan Li4 ![]()
1. Department of Neurosurgery, Tangdu Hospital, Fourth Military Medical University, Xi'an 710032, PR China.
2. Department of Neurosurgery, 463rd Hospital of PLA, Shenyang 110042, PR China.
3. Department of Neurosurgery, Wuhan General Hospital of Guangzhou Military Command, Wuhan 430070, PR China.
4. Department of Medical Administration, General Hospital of Shenyang Military Command, Shenyang 110016, PR China.
* These authors contributed equally to this work.
Received 2013-9-11; Accepted 2014-1-6; Published 2014-1-28
Abstract
Quercetin has been demonstrated to play an important role in altering the progression of ischemic brain injuries and neurodegenerative diseases by protecting against oxidative stress. The effects of quercetin on brain damage after subarachnoid hemorrhage (SAH), however, have not been investigated. This study was designed to explore the effects of quercetin on oxidative stress and brain edema after experimental SAH using four equal groups (n = 16) of adult male Sprague-Dawley (SD) rats, including a sham group, an SAH + vehicle group, an SAH + quercetin10 group, and an SAH + quercetin50 group. The rat SAH model was induced by injection of 0.3 ml of non-heparinised arterial blood into the prechiasmatic cistern. In the SAH + quercetin10 and SAH + quercetin50 groups, doses of 10 mg/kg and 50 mg/kg quercetin, respectively, were directly administered by intraperitoneal injection at 30 min, 12 h, and 24 h after SAH induction. Cerebral tissue samples were extracted for enzymatic antioxidant determination, lipid peroxidation assay, caspase-3 activity and water content testing 48 h after SAH. Treatment with a high dose (50 mg/kg) of quercetin markedly enhanced the activities of copper/zinc superoxide dismutase (CuZn-SOD) and glutathione peroxidase (GSH-Px), and treatment with this dose significantly reduced the level of malondialdehyde (MDA). Caspase-3 and brain edema was ameliorated and neurobehavioral deficits improved in rats that received the high dose of quercetin. The findings suggest that the early administration of optimal dose of quercetin may ameliorate brain damage and provide neuroprotection in the SAH model, potentially by enhancing the activity of endogenous antioxidant enzymes and inhibiting free radical generation.
Keywords: Subarachnoid hemorrhage, Oxidative stress, Brain edema, Quercetin, Rat
Introduction
Subarachnoid hemorrhage (SAH) has a worldwide incidence of 10.5 cases per 100,000 person-years and accounts for 3% of all strokes [1]. In addition, SAH from a ruptured intracranial aneurysm is a serious medical problem because of its high mortality and morbidity [2]. Despite extensive research, patient outcomes post SAH remain poor. Morbidity following SAH are commonly defined in terms of neurological repercussions, including aneurysm rebleeding, intracranial hypertension, cerebral vasospasm, brain edema, cerebral infarction, and hydrocephalus [3]. Delayed vasospasm is not the sole determinant of a poor outcome in SAH patients. Brain injury begins at aneurysm rupture and contributes to the overall outcome [4]. The initial event associated with SAH is the cessation of blood flow, which either occurs due to a mechanical factor, such as elevated intracranial pressure, or due to a chemical factor that decreases nitric oxide (NO) availability. The sequence of events could be that global cerebral ischemia leads to neuronal apoptosis [5]. Among the complex factors involved in the neuronal death after SAH, oxidative stress has been consistently highlighted. Meanwhile, mounting evidence suggests that oxidative stress injury is intimately associated with brain edema [6].
Oxidative stress reflects an imbalance between systemic production of reactive oxygen species (ROS) and a biological system's ability to readily detoxify the reactive intermediates or repair the resulting damage. Disturbances in the normal redox state of cells can cause toxic effects through the production of peroxides and free radicals that damage all components of the cell, including proteins, lipids, and DNA [7]. Further, some reactive oxidative species act as cellular messengers in redox signalling. Thus, oxidative stress can cause disruptions in the normal mechanisms of cellular signalling. In humans, oxidative stress is thought to be involved in the development of many diseases, including SAH, and oxidative stress may exacerbate their symptoms [8]. Usually, ROS can be efficiently scavenged by the antioxidant defence system, which includes copper/zinc superoxide dismutase (CuZn-SOD) and glutathione peroxidase (GSH-Px). Malondialdehyde (MDA) is the end product of the major reactions leading to significant oxidation of polyunsaturated fatty acids in cellular membranes, and it thus serves as a reliable marker of oxidative stress [9]. Production of reactive oxygen species is a particularly destructive aspect of oxidative stress. Such species include free radicals and peroxides, which cause oxidative brain injury after cerebral hemorrhage [10].
Some antioxidative treatments improve neurological deficits in the rat hemorrhage model [11]. Quercetin is one of the most frequently studied bioflavonoids in its class of flavonols. Quercetin is present in high concentrations in fruits and vegetables such as tea, apples, mulberries, onions, potatoes, broccoli, soybeans, peanuts, and red wine. It has been shown to have highly potent antioxidant and cytoprotective effects in preventing endothelial apoptosis caused by oxidants [12]. Quercetin is a more potent antioxidant than other antioxidant nutrients, such as vitamin C, vitamin E, and β-carotene, and it can chelate transition metal ions, including iron, thus preventing the iron-catalysed Fenton reaction [13,14]. Treatment with quercetin has been shown to prevent liver damage and suppress overexpression of the inducible form of nitric oxide synthase (iNOS), which is induced by various inflammatory stimuli [15]. Epidemiological studies have suggested that the intake of food containing flavonoids may be associated with a reduced risk of coronary heart disease, hypercholesterolemia, atherosclerosis, and heart failure [16].
The role of quercetin in brain oxidative stress in the SAH rat model, however, has not been addressed previously. Therefore, this study was designed to elucidate whether quercetin can ameliorate oxidative stress-mediated cerebral dysfunction, including brain edema and neurobehavioral deficits.
Materials and methods
Animals and experimental design
Adult male Sprague-Dawley (SD) rats weighing 250 to 300 g were purchased from the Animal Centre of the Fourth Military Medical University. The rats were housed in temperature- and humidity-controlled animal quarters with a 12 h light/dark cycle and free access to food and water for 10 days prior to the experiment. All procedures were approved by the Animal Care and Use Committee of the Fourth Military Medical University and conformed to the Guide for the Care and Use of Laboratory Animals set forth by the National Institute of Health (NIH).
Male SD rats (n= 96) were randomly and equally assigned to one of four weight-matched groups: sham-operated (sham group), SAH treated with 0.9% saline as vehicle (SAH + vehicle group), SAH treated with 10 mg/kg quercetin (SAH + quercetin10 group), and SAH treated with 50 mg/kg quercetin (SAH + quercetin50 group). In the subjects of the SAH+quercetin10 and SAH+quercetin50 groups, quercetin (Sigma, USA, dissolved in 0.9% saline solution) was administered intraperitoneally at a dose of 10 mg/kg or 50 mg/kg at 30 min, 12 h, and 24 h after the SAH insult.
Animals that died either immediately or within the first 6 h after the operation were not allocated to any group. Upon death, animal subjects were replaced until the final group size reached the expected number, resulting in an equal number of subjects in each group. Animals in each group (n = 24) underwent neurological scoring and were subsequently anesthetised and decapitated 48 h after SAH for test of oxidative stress, caspase-3 activity and brain edema.
Experimental rat model of subarachnoid hemorrhage
The experimental SAH model was induced by prechiasmatic blood injection according to a published procedure [17]. The experimental rats were anesthetised by intraperitoneal injection of chloral hydrate (400 mg/kg). A medial incision of the scalp was made after proper positioning the rat in a stereotactic frame. A 1.0-mm burr hole was drilled 7.5 mm anterior to the Bregma at the midline. A rounded tip needle with a side hole was tilted 30º in the sagittal plane, manually directed through the burr hole, and pushed to the skull base until the tip reached 2.5 mm anterior to the chiasma. The burr hole was then sealed with bone wax after the insertion of the needle. A sample of 0.3 ml of autologous arterial blood from the right femoral artery was then injected aseptically into the prechiasmatic cistern over a period of approximately 60 s using a syringe pump. To permit blood distribution around the basal arteries, the rats were kept in a head-down position at 30º for 30 min. The sham-operated rats were injected with equal volumes of 0.9% saline solution. After the procedure, the rats were returned to their cages and allowed free access to food and water for 48 h.
Neurological scoring
The neurological function of all animals was recorded 48 h after SAH using a previously described scoring system modified for rodents [18]. The behaviour and activity scores were as follows: 1, appetite (finished meal 0; left meal unfinished 1; scarcely ate 2); 2, activity (walked and reach at least three corners of the cage 0; walked with some stimulation 1; almost always lying down 2); and 3, Deficits (no deficits 0; unstable walk 1; impossible to walk 2). The minimum neurological score is 0 and the maximum is 6, which means that neurological deficits were graded as a total score from 0 to 6. A higher score represents more serious neurological deficits. The scoring was conducted by two independent researchers who were blinded to the groups.
Enzymatic antioxidant determinations and lipid peroxidation assay
After examination of the neurological score, the rats were deeply anesthetised and sacrificed. The brains were dissected, weighed and kept chilled until homogenisation. These procedures lasted up to 3 min. The cerebral cortex was homogenised in 10 volumes (1:10, w/v) of cold saline. The brain samples were homogenised and centrifuged at 4000 rpm at 4 ℃ for 10 min. Supernatant aliquots were collected to assay the CuZn-SOD and GSH-Px activity and MDA levels. The protein concentration of the cerebral cortex supernatants was measured using the method of Bradford [19]. Bovine serum albumin was used as a standard.
The CuZn-SOD and GSH-Px activities were determined with a Shimadzu UV-2100 spectrophotometer (Shimadzu International Trading, Shanghai, China) at 25 ℃. The assay for CuZn-SOD was based on this enzyme's activity in the xanthine-xanthine oxidase system [20]. The change in absorbance at 550 nm was monitored using a spectrophotometer. The CuZn-SOD levels were expressed as units per mg protein. GSH-Px activity was determined using the method of Wheeler [21]. The assay measures the enzymatic reduction of peroxide (H2O2) by GSH-Px through the consumption of reduced glutathione (GSH) that is restored from oxidised glutathione (GSSG) in a coupled enzymatic reaction. In the presence of GSSG and NADPH, GR reduces GSSG and oxidises NADPH, resulting in a decrease of absorbance at 340 nm. The activity of GSH-Px was computed using the molar extinction coefficient of 6.22 mM-1 cm-1. One unit of GSH-Px was defined as the amount of enzyme that catalysed the oxidation of 1.0 mmol of NADPH to NADP+ per minute.
The level of lipid peroxidation in the cerebral cortex homogenate was indicated by the content of MDA in the brain tissue. The level of MDA in the brain tissue homogenates was determined using the thiobarbituric acid (TBA) reaction method. MDA levels, which can be measured at the wavelength of 532 nm due to a reaction with TBA to form a pink chromogen, were expressed as nanomoles per milligram of brain protein [22].
Immunohistochemical staining and analysis
After deparaffinization, the brain sections were microwaved for 10 min to promote antigen retrieval and then allowed to cool. The sections were incubated for 15 min in 3% H2O2 to quench endogenous peroxidases and then placed in PBS and blocked with normal goat serum at 37°C for 20 min. Rabbit anti-caspase-3 (diluted to 1:1000, Abcam, Ltd., USA) were used as the primary antibodies. After primary antibody treatment at 4°C overnight, the sections were incubated with peroxidase-conjugated goat anti-rabbit IgG (1:1000) for 1 h at 37°C. The sections were coverslipped with glycerin, and images were captured using an Olympus digital camera (Olympus DP70).
The CA1 area of the hippocampus was observed at 400 ×. The integral optical density (IOD) values that indicated the expression levels of the proteins were measured using Image-Pro Plus 5.0 (Media Cybernetics, Silver Spring, MD, USA). A neuron with a nucleus stained dark brown was considered a positive cell. By counterstaining with hematoxylin, viable neurons were also observed. The number of active caspase-3-positive cells in the hippocampus was counted. Data from eight animals per group at each time point were analyzed.
Western blotting analysis
The rats were intracardially perfused with 200 ml of ice-cold 0.9% saline solution at 48 h post-SAH under deep anesthesia. The cerebral cortex was carefully isolated, frozen in liquid nitrogen, and stored at -80ºC until use. Frozen tissues were homogenized for 20 min at 4ºC using an ultrasonic wave (10 s × 3 times) in 100 ml of extraction buffer containing 50 mM Tris-HCl (pH 7.5), 1% nonylphenol ethoxylate, 0.25% sodium deoxycholate, 150 mmol/L NaCl, 1 mmol/L EGTA, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mg/mL aprotinin plus leupeptin and pepstatin, 1 mmol/L Na3VO4, and 1 mmol/L NaF. The insoluble material was removed by centrifugation at 16,000 rpm for 15 min at 4ºC. The protein-containing supernatants were then transferred and stored at -80ºC until further use.
Protein concentrations were determined via the Bradford method using a Protein Assay Kit according to the manufacturer's instructions (Bio-Rad Laboratories, Hercules, CA). A total of 50 µg of each lysate sample was denatured for 5 min in sample buffer and then separated using 10% SDS-polyacrylamide gel electrophoresis. After electrophoretic transfer (100 V for 90 min) of the separated polypeptides to polyvinylidene difluoride (PVDF) membranes (Bio-Rad), the membranes were blocked with 1% bovine serum albumin in Tween/Tris-buffered saline for 1 h at room temperature. The membranes were then washed and incubated with the primary antibody at 4ºC overnight. The following primary antibodie was used: rabbit polyclonal anti-active caspase-3 (1:1500) (Santa Cruz Biotechnology, Inc., USA). The membranes were probed with horseradish peroxidase-conjugated secondary antibodies (1:1000) (Santa Cruz Biotechnology, Inc., USA) for 2 h at room temperature. The blotted protein bands were visualized using enhanced chemiluminescence (ECL) western blotting detection reagents (Amersham, Arlington Heights, IL, USA) and exposed to X-ray film. Developed films were digitized using an Epson Perfection 2480 scanner (Seiko Corp, Nagano, Japan). Optical densities were obtained using Glyko Bandscan software (Glyko, Novato, CA, USA).
Brain water content test
The brain water content was determined using the wet/dry method previously described [23]. Briefly, the brain water content (%) was calculated as [(wet weight-dry weight)/wet weight] × 100%. After examination of the neurological score, the rats (n = 8) were deeply anesthetised and sacrificed. Each cerebral tissue sample was removed and weighed immediately to obtain the wet weight. The samples were then dried at 105 ℃ for 48 h and weighed to determine the dry weight.
Statistical analysis
All data were expressed as the mean +/- standard deviation (SD). Statistical analyses were performed using the Statistical Package for the Social Sciences software, version 16.0 (SPSS, Chicago, IL, USA). Differences between all groups were compared by one-way analysis of variance (ANOVA). If a significant difference was determined by ANOVA, additional analysis by the Tukey-Kramer multiple comparison procedure was applied. A probability value of P < 0.05 was considered statistically significant.
Results
Effects of quercetin on neurobehavioral scores
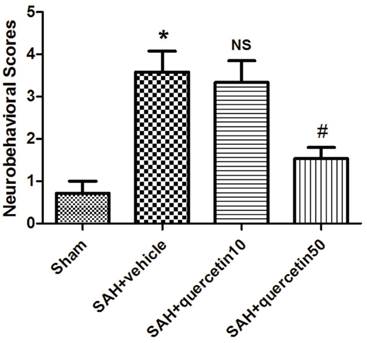
In comparison with the sham group, neurobehavioral deficits were observed in the SAH+vehicle group (3.63 ± 0.96 versus 0.63 ± 0.47, P < 0.01). No significant difference was observed between the SAH+quercetin10 group and the SAH+vehicle group (3.38 ± 0.81 versus 3.63 ± 0.96, P > 0.05). Neurobehavioral scores were significantly decreased in the SAH+quercetin50 group compared to the SAH+vehicle group (1.63 ± 0.62 versus 3.63 ± 0.96, P < 0.05) (Fig. 1).
Effects of quercetin on neurobehavioral scores at 48h after SAH. (n=16, *P<0.01 vs. the sham group, NSP>0.05 vs. the SAH + vehicle group, #P<0.05 vs. the SAH + vehicle group).
Effects of quercetin on oxidative stress
In comparison with the sham group, CuZn-SOD activity in the rat brain cortices was markedly decreased in the SAH+vehicle group (27.65 ± 3.19 versus 48.35 ± 5.32, P < 0.01). No significant difference was observed between the SAH+quercetin10 group and the SAH+vehicle group (28.44 ± 3.31 versus 27.65 ± 3.19, P > 0.05). CuZn-SOD activity in rat brain cortices was significantly increased in the SAH+quercetin50 group compared to the SAH+vehicle group (40.71 ± 4.25 versus 27.65 ± 3.19, P < 0.05) (Fig. 2 A).
Effects of quercetin on the activities of antioxidant enzymes and the content of MDA in cerebral cortex at 48 h after SAH. The effects of quercetin on (A) SOD, (B) GSH-PX, (C) MDA activities in cerebral cortex, respectively, are shown. (n=8, *P<0.01 vs. the sham group, NSP>0.05 vs. the SAH + vehicle group, #P<0.05 vs. the SAH + vehicle group).
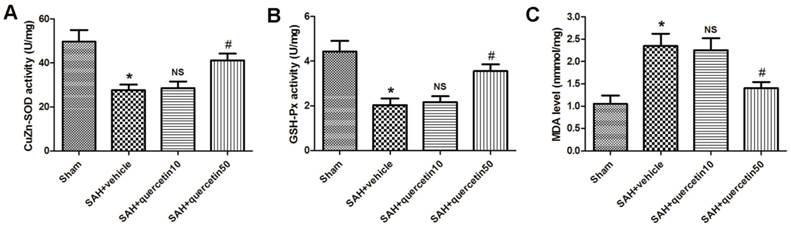
In comparison with the sham group, GSH-Px activity in the rat brain cortices was markedly decreased in the SAH+vehicle group (2.09 ± 0.34 versus 4.57 ± 0.63, P < 0.01). No significant difference was observed between the SAH+quercetin10 group and the SAH+vehicle group (2.21 ± 0.37 versus 2.09 ± 0.34, P > 0.05). GSH-Px activity in the rat brain cortices was significantly and statistically increased in the SAH+quercetin50 group compared to the SAH+vehicle group (3.79 ± 0.51 versus 2.09 ± 0.34, P < 0.05) (Fig. 2 B).
The level of MDA in the rat brain cortices was significantly higher in the SAH+vehicle group than the sham group (2.25 ± 0.35 versus 1.07 ± 0.16, P < 0.01). No significant difference was observed between the SAH+quercetin10 group and the SAH+vehicle group (2.19 ± 0.33 versus 2.25 ± 0.35, P > 0.05). The level of MDA in the rat brain cortices was significantly lower in the SAH+quercetin50 group than the SAH+vehicle group (1.38 ± 0.19 versus 2.25 ± 0.35, P < 0.05) (Fig. 2 C).
Effects of quercetin on caspase-3 in the CA1 region of the hippocampus
Immunohistochemical staining of active caspase-3 in the CA1 region of the hippocampus is shown in Fig. 3 A. At 48 h after SAH, few active caspase-3-positive cells were detected in samples from the sham group. A number of active caspase-3-positive cells were observed in the SAH + vehicle and SAH+quercetin10 group, and a greater number of positive cells were observed in the SAH + quercetin50 group. The apoptotic index of the CA1 region of the hippocampus is shown in Fig. 3 B. The apoptotic index was increased in the SAH + vehicle group and SAH + quercetin10 compared with the sham group (P < 0.01) and decreased in the SAH + quercetin50 group compared with the SAH + vehicle group (P < 0.01).
Effects of quercetin on caspase-3 activity in the CA1 region of the hippocampus at 48 h after SAH. (A) Representative sections of immunohistochemical staining of caspase-3. (B) Quantitative analysis of integral optical density (IOD). (C) Representative autoradiogram of caspase-3 protein expression. Lane 1, the sham group; Lane 2, the SAH + vehicle group; Lane 3, the SAH + quercetin10 group; Lane 4, the SAH + quercetin50 group. (D) Quantitative analysis of western blotting results for the expressions of active caspase-3. Scale bars represent 50μm. (n=8, *P<0.01 vs. the sham group, NSP>0.05 vs. the SAH + vehicle group, #P<0.05 vs. the SAH + vehicle group).
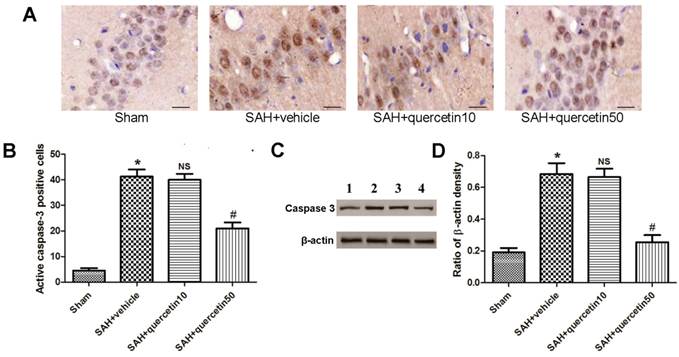
Representative autoradiograms of western blots of active caspase-3 expression in the hippocampus are shown in Fig. 3 C. Quantitative analyses of the ratios to β-actin density are shown in Fig. 3 D. The expression of active caspase-3 was increased in the SAH + vehicle group and SAH + quercetin10 compared with the sham group (P < 0.01) and decreased in the SAH + quercetin50 group compared with the SAH + vehicle group (P < 0.01).
Effects of quercetin on brain edema
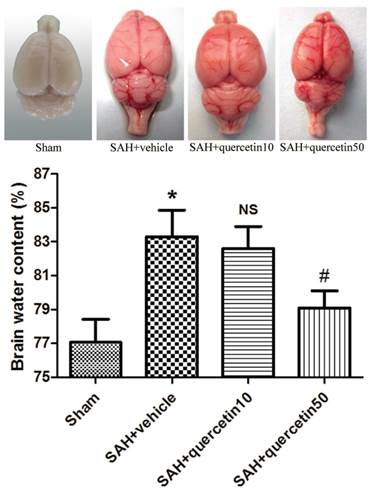
In comparison with the sham group, the brain water content in the SAH+vehicle group was significantly increased 48 h after SAH (83.19 ± 1.63% versus 77.28 ± 1.45%, P<0.01). No significant difference was observed between the SAH+quercetin10 group and the SAH+vehicle group (82.87 ± 1.59 versus 83.19 ± 1.63%, P > 0.05). The brain water content was, however, significantly decreased in the SAH+quercetin50 group compared with the SAH+vehicle group (78.83±1.51% versus 83.19 ± 1.63%, P < 0.05) (Fig. 4).
Effects of quercetin on brain water content at 48h after SAH. (n=8, *P<0.01 vs. the sham group, NSP>0.05 vs. the SAH + vehicle group, #P<0.05 vs. the SAH + vehicle group).
Discussion
In this study, we examined the protective effect of quercetin against oxidative stress and brain edema in an experimental rat model of SAH. Our data showed that 48 h after SAH, (1) brain oxidative enzymatic activity was decreased and lipid peroxidation increased, (2) quercetin improved neurobehavioral deficits and ameliorated brain edema, (3) brain oxidative enzymatic activity was enhanced and lipid peroxidation was alleviated by quercetin, (4) hippocampal apoptosis was apparent 48 h after SAH but was alleviated by quercetin, (5) quercetin played a protective role at a dose of 50 mg/kg, but not at a dose of 10 mg/kg. These findings suggest that early treatment with the optimal dose of quercetin may provide an effective method for protecting the brain against damage following SAH in rats.
Previous studies demonstrate that early generation of ROS and oxidative stress after SAH is associated with early brain injury, including brain edema [24,25]. Animal studies show that the activities of enzymatic antioxidant systems decrease and the products of lipid peroxidation increase after SAH [26,27]. Our results were in accordance with these above reports. In humans, a decrease in antioxidant systems [28] and an increase in lipid peroxidation products is found within 72 h from ictus and correlates well with a poor clinical status and outcome [29]. The concentration of ROS is determined by the balance between the rate of production and the rate of clearance of ROS by various antioxidant compounds and enzymes. Animal studies show that the majority of ROS are generated by the auto-oxidation of haemoglobin following erythrocytes lysis in the subarachnoid space [30]. The consequences of oxidative stress after SAH may include injury to the vascular wall endothelium and smooth muscle, disruption of the blood-brain barrier (BBB), and the production of strong spasmogens, such as prostaglandin D2 and leukotriene C4 from the lipoxygenase and cyclo-oxygenase pathways of arachidonic acid metabolism [27]. In addition, oxidative stress induces enzymes of the apoptotic pathway, including p53 and caspase-3 and 9, which promote apoptotic cell death. [31]. Neuronal apoptosis results in blood brain barrier dysfunction, inflammation, and oxidative cascades that lead to brain edema [32]. Brain edema is an abnormal accumulation of fluid within the brain parenchyma, producing a volumetric enlargement of the brain cells or tissue [33]. Brain edema has been demonstrated to be an independent risk factor for mortality and poor outcome after SAH [34]. In this study, the water content of the brain was measured to examine the effect of quercetin on brain edema after SAH.
CuZn-SOD is the first enzyme of the enzymatic antioxidative pathway that converts superoxide anions into peroxides, which are then converted into GSH-Px. Overexpression of CuZn-SOD in transgenic mice prevents apoptotic cell death and reduces mortality after SAH [35,36]. In our study, the activities of CuZn-SOD and GSH-Px were designated as indicators to determine the oxidative damage in the brains of SAH rats. Lipid peroxidation is one of the major consequences of free radical-mediated injury in the brain. The peroxidation of polyunsaturated fatty acids, mostly in membrane phospholipids, is a chain reaction that can continue until the substrate is completely consumed or termination occurs due to antioxidants. If not terminated fast enough, there will be damage to the cell membrane, which consists mainly of lipids. Lipid peroxidation produces structural and functional damage to membranes as well as several secondary products. Phototherapy may cause haemolysis by rupturing red blood cell membranes via lipid peroxidation [37]. Certain diagnostic tests are available for the quantification of the end-products of lipid peroxidation. Most studies have used MDA as an index of lipid peroxidation [38].
Some antioxidants have been used to prevent oxidative stress and decrease early SAH-induced brain injury in animals [39]. However, these antioxidants have had little success in improving outcomes in clinical trials, so researchers are trying to find a more effective antioxidant, including natural medicines [40]. Several previous studies suggested that flavonoids may act as antioxidants, free-radical scavengers and radioprotectors [41]. Quercetin is a ubiquitous flavonoid that is present in numerous plants that are utilised in many different cultures for their nervous system and anticancer effects [42,43]. Quercetin plays an important role in altering the progression of neurodegenerative diseases by its protective effect against oxidative stress [44]. Pre-treatment of primary hippocampal cultures with quercetin significantly attenuated amyloid β-induced cytotoxicity, protein oxidation, lipid-peroxidation, and apoptosis [45]. Studies indicated the potential benefit of quercetin treatment for conditions involving mitochondrial dysfunction associated with increased oxidative stress [46]. The central role of oxidative stress in brain pathology is the basis for the therapeutic potential of quercetin. Its potent ROS scavenging activity is attributed to its high number of hydroxyl substitutions, which correlates with its electron-donating ability [47]. In addition to the direct scavenging of free radicals, other antioxidant mechanisms contribute to the potent action of quercetin against oxidative stress, such as the chelation of iron and calcium and the inhibition of lipid-peroxidation, as well as other biochemical mechanisms, such as the inhibition of enzymes, including xanthine oxidase and nitric oxide synthase [48,49]. Quercetin has been shown to alter the expression of the Nrf2 gene. A key function of quercetin is the activation of Nrf2 in cerebellar granule cells after oxidative stress, resulting in the induction of genes encoding g-glutamylcysteine syntheses and increased neuronal glutathione levels that restore redox homeostasis. The result of these antioxidant activities allows quercetin to re-establish the redox regulation of proteins, transcription factors, and survival signalling cascades that are otherwise inhibited by elevated ROS. Pre-treatment of primary hippocampal cultures with quercetin significantly attenuated lipid-peroxidation and apoptosis [50]. In the present study, quercetin played a protective role by reducing brain oxidative enzymatic activity ,decreasing lipid peroxidation and inhibiting cell apoptosis. This result was in accordance with previous reports.
Several factors could be responsible for the induction of edema in the brain [51]. The excessive production of free radicals in conjunction with a weakened scavenger system contributes to both cytotoxic and vasogenic edema by directly disrupting the BBB and by triggering the pathways that mediate brain edema [52]. Caspase-3 is one member of the caspase family and it plays a central role in the execution-phase of cell apoptosis. It has been reported that the expression of caspase-3 was intensified in the cortical neurons after SAH [53] and its inhibition could reduce the neuron loss and brain edema. The next step in our experimental program will be to determine whether and how quercetin protects from BBB impairment and cerebral vasospasm in our experimental rat model of SAH.
Conclusions
In summary, this is the first in vivo experiment to demonstrate that quercetin has a protective effect against the damage following SAH in experimental rats. Neurobehavioral deficits were improved, brain edema was reduced, oxidative stress and hippocampus apoptosis was significantly protected following quercetin administration. The mechanism could be partly attributed to antioxidant enzymes scavenging free radicals. Recently, much attention has been focused on the protective biochemical functions of naturally occurring antioxidants following SAH. However, further investigations are essential to elucidate the precise mechanisms of quercetin protection before evaluating it as a neuroprotective agent useful in the treatment in SAH.
Acknowledgements
We thank American Journal Experts for assisting in the preparation of this manuscript. This work was supported by grants from the National Natural Science Foundation of China (No. 81171086; 81300956) and Medical Science Youth Breeding Project of PLA (No. 13QNP007).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rosamond W, Flegal K, Furie K. et al. Heart disease and stroke statistics-2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:25-146
2. Lai L, Morgan MK. Incidence of subarachnoid hemorrhage: an Australian national hospital morbidity database analysis. J Clin Neurosci. 2012;19:733-739
3. Bakker AM, Dorhout Mees SM, Algra A. et al. Extent of acute hydrocephalus after aneurismal subarachnoid hemorrhage as a risk factor for delayed cerebral infarction. Stroke. 2007;38:2496-2499
4. Fatima A. Sehba, Jack Hou, Ryszard M. Pluta, et al. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol. 2012;97:14-37
5. Yushu Dong, Yue Li, Dayun Feng. et al. Protective effect of HIF-1α against hippocampal apoptosis and cognitive dysfunction in an experimental rat model of subarachnoid hemorrhage. Brain Res. 2013;1517:114-121
6. Doczi T, Joo F, Adam G. et al. Blood-brain barrier damage during the acute stage of subarachnoid hemorrhage, as exemplified by a new animal model. Neurosurgery. 1986;18:733-739
7. Hua Lu, Dong-Mei Zhang, Hui-Ling Chen. et al. N-acetylcysteine suppresses oxidative stress in experimental rats with subarachnoid hemorrhage. J Clin Neurosci. 2009;16:684-648
8. Peter H. Proctor. Free Radicals and Human Disease. CRC Handbook of Free Radicals and Antioxidants. 1989;1:209-221
9. Rossi R, Dalle-Donne I, Milzani A. et al. Oxidized forms of glutathione in peripheral blood as biomarkers of oxidative stress. Clin Chem. 2006;52:1406-1414
10. Wu J, Hua Y, Keep RF. et al. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 2009;953:45-52
11. Hall NC, Packard BA, Hall CL. et al. Protein oxidation and enzyme susceptibility in white and gray matter with in vitro oxidative stress: relevance to brain injury from intracerebral hemorrhage. Cell Mol Biol. 2000;46:673-683
12. Choi YJ, Kang JS, Park JH. et al. Polyphenolic flavonoids differ in their antiapoptotic efficacy in hydrogen peroxide treated human vascular endothelial cells. J Nutr. 2003;133:985-991
13. Rice-Evans CA, Miller NJ, Bolwell PG. et al. The relative antioxidant activities of plant derived polyphenolic flavonoids. Free Radic Res. 1995;22:375-383
14. Ferrali M, Signorini C, Ciccoli L. et al. Protection of erythrocytes against oxidative damage and autologous immunoglobulin G (IgG) binding by iron chelator fluor-benzoil-pyridoxal hydrazone. Biochem Pharmacol. 2000;59:1365-1373
15. Pavanato A, Tunon MJ, Camposs S. et al. Effects of quercetin on liver damage in rats with carbon tetrachloride-induced cirrhosis. Dig Dis Sci. 2003;48:824-829
16. Okamoto T. Safety of quercetin for clinical application. Int J Mol Med. 2005;16:275-278
17. Prunell GF, Mathiesen T, Diemer NH. et al. Experimental subarachnoid hemorrhage: subarachnoid blood volume, mortality rate, neuronal death, cerebral blood flow, and perfusion pressure in three different rat models. Neurosurgery. 2003;52:165-175
18. Yamaguchi M, Zhou C, Nanda A. et al. Ras protein contributes to cerebral vasospasm in a canine double-hemorrhage model. Stroke. 2004;35:1750-1755
19. Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-254
20. Oyanagui Y. Revaluation of assay methods and establishment of kit for superoxide dismutase activity. Anal Biochem. 1984;142:290-296
21. Wheeler CR, Salzman JA, Elsayed NM. et al. Automated assays for superoxide dismutase, catalase, glutathione peroxidase and glutathione reductase activity. Anal Biochem. 1990;184:193-199
22. Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351-358
23. Xi G, Hua Y, Bhasin RR. et al. Mechanisms of edema formation after intracerebral hemorrhage: effects of extravasated red blood cells on blood flow and blood-brain barrier integrity. Stroke. 2001;32:2932-2938
24. Liu S, Tang J, Ostrowski RP. et al. Oxidative stress after subarachnoid hemorrhage in gp91phox knockout mice. Can J Neurol Sci. 2007;34:356-361
25. Pyne-Geithman GJ, Caudell DN, Prakash P. et al. Glutathione peroxidase and subarachnoid hemorrhage: implications for the role of oxidative stress in cerebral vasospasm. Neurol Res. 2009;31:195-199
26. Marzatico F, Gaetani P, Cafe C. et al. Antioxidant enzymatic activities after experimental subarachnoid hemorrhage in rats. Acta Neurol Scand. 1993;87:62-66
27. Gaetani P, Marzatico F, Rodriguezy Baena R. et al. Arachidonic acid metabolism and pathophysiologic aspects of subarachnoid hemorrhage in rats. Stroke. 1990;21:328-332
28. Lin CL, Hsu YT, Lin TK. et al. Increased levels of F2-isoprostanes following aneurismal subarachnoid hemorrhage in humans. Free Radic Biol Med. 2006;40:1466-1473
29. Hsieh YP, Lin CL, Shiue AL. et al. Correlation of F4-neuroprostanes levels in cerebrospinal fluid with outcome of aneurysmal subarachnoid hemorrhage in humans. Free Radic Biol Med. 2009;47:814-824
30. Sercombe R, Dinh YR, Gomis P. Cerebrovascular inflammation following subarachnoid hemorrhage. Jpn J Pharmacol. 2002;88:227-249
31. Ayer RE, Zhang JH. Oxidative stress in subarachnoid hemorrhage: significance in acute brain injury and vasospasm. Acta Neurochir. 2008;104:33-41
32. Bosoi CR, Yang X, Huynh J. et al. Systemic oxidative stress is implicated in the pathogenesis of brain edema in rats with chronic liver failure. Free Radic Biol Med. 2012;52:1228-1235
33. Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22:1323-1329
34. Claassen J, Carhuapoma JR, Kreiter KT. et al. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33:1225-1232
35. Matz PG, Copin JC, Chan PH. Cell death after exposure to subarachnoid hemolysate correlates inversely with expression of CuZn-superoxide dismutase. Stroke. 2000;31:2450-2459
36. Endo H, Nito C, Kamada H. et al. Reduction in oxidative stress by superoxide dismutase overexpression attenuates acute brain injury after subarachnoid hemorrhage via activation of Akt/glycogen synthase kinase-3beta survival signaling. J Cereber Blood F Met. 2007;27:975-982
37. Marnett LJ. Lipid peroxidation-DNA damage by malondialdehyde. Muta Res. 1999;424:83-95
38. Cini M, Fariello RG, Bianchetti A. et al. Studies on lipid peroxidation in the rat brain. Neurochem Res. 1994;19:283-288
39. Gul S, Bahadir B, Hanci V, Bektas S. et al. Effect of vardenafil on cerebral vasospasm following experimental subarachnoid hemorrhage in rats. J Clin Neurosci. 2010;17:1038-1041
40. Gomis P, Graftieaux JP, Sercombe R. et al. Double-blind, placebo-controlled, pilot trial of high-dose methylprednisolone in aneurysmal subarachnoid hemorrhage. J Neurosurg. 2010;112:681-688
41. Shimoi K, Masuda S, Furugori M. et al. Radio protective effect of antioxidative flavonoids in γ-ray irradiated mice. Carcinogenesis. 1994;75:2668-2672
42. Zhang ZJ, Cheang LC, Wang MW. et al. Quercetin exerts a neuroprotective effect through inhibition of the iNOS/NO system and pro-inflammation gene expression in PC12 cells and in zebrafish. Int J Mol Med. 2011;27:195-203
43. Federico Dajas. Life or death: Neuroprotective and anticancer effects of quercetin. J Ethnopharmacol. 2012;143:383-396
44. Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58:39-46
45. Ansari MA, Abdul HM, Joshi G. et al. Protective effect of quercetin in primary neurons against Alzheimer's disease. J Nutr Biochem. 2009;20:269-275
46. Carrasco-Pozo C, Mizgier M, Speisky L. et al. Differential protective effects of quercetin, resveratrol, rutin and epigallocatechin gallate against mitochondrial dysfunction induced by indomethacin in Caco-2 cells. Chem Biol Interact. 2012;195:199-205
47. Boots AW, Haenen GR, Bast A. Health effects of quercetin: from antioxidant to nutraceutical. Eur J Pharmacol. 2008;585:325-337
48. Mira L, Fernandez MT, Santos M. et al. Interactions of flavonoids with iron and copper ions: a mechanism for their antioxidant activity. Free Radical Res. 2002;36:1199-1208
49. Bindoli A, Valente M, Cavallin L. Inhibitory action of quercetin on xanthine oxidase and xanthine dehydrogenase activity. Pharmacol Res Commun. 1985;17:831-839
50. Ishikawa Y, Sugiyama H, Stylianou E. et al. Bioflavonoid quercetin inhibits interleukin-1-induced transcriptional expression of monocyte chemoattractant protein-1 in glomerular cells via suppression of nuclear factor-kappaB. J Am Soc Nephrol. 1999;10:2290-2296
51. Germano A, Avella D, Imperatore C. et al. Time-course of blood-brain barrier permeability changes after experimental subarachnoid hemorrhage. Acta Neurochir. 2000;142:575-580
52. Park S, Yamaguchi M, Zhou C. et al. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412-2417
53. Simard JM, Geng Z, Woo SK. et al. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009;29:317-330
Author contact
![]() Corresponding author: Guo-dong Gao, Department of Neurosurgery, Tangdu Hospital, Fourth Military Medical University, No. 1, Xinshi Road, Xi'an 710032, PR China. Tel: +86 29-84777902 Fax +86 29-84777435 Email: gguodongfmmucom; Chuan Li, Department of Medical Administration, General Hospital of Shenyang Military Region, No. 63, Wenhua Road, Shenyang 110016, PR China. Tel: +86 24-28851861 Fax +86 24-28851861 Email: jk_lc1977com.
Corresponding author: Guo-dong Gao, Department of Neurosurgery, Tangdu Hospital, Fourth Military Medical University, No. 1, Xinshi Road, Xi'an 710032, PR China. Tel: +86 29-84777902 Fax +86 29-84777435 Email: gguodongfmmucom; Chuan Li, Department of Medical Administration, General Hospital of Shenyang Military Region, No. 63, Wenhua Road, Shenyang 110016, PR China. Tel: +86 24-28851861 Fax +86 24-28851861 Email: jk_lc1977com.