Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(11):1485-1496. doi:10.7150/ijms.6308 This issue Cite
This article has been retracted. See Int J Med Sci 2014; 11(3):246.
Research Paper
Cadmium Induced Cell Apoptosis, DNA Damage, Decreased DNA Repair Capacity, and Genomic Instability during Malignant Transformation of Human Bronchial Epithelial Cells
Zhiheng Zhou1*, Caixia Wang2*, Haibai Liu1, Qinhai Huang1, Min Wang1, Yixiong Lei1 ![]()
1. School of public health, Guangzhou Medical University, Guangzhou 510182, People's Republic of China
2. Department of internal medicine of Guangzhou First People's Hospital affiliated to Guangzhou Medical University, Guangzhou 510180, People's Republic of China
* These authors contributed equally to this work.
Received 2013-3-23; Accepted 2013-8-12; Published 2013-8-30
Abstract
Cadmium and its compounds are well-known human carcinogens, but the mechanisms underlying the carcinogenesis are not entirely understood. Our study was designed to elucidate the mechanisms of DNA damage in cadmium-induced malignant transformation of human bronchial epithelial cells. We analyzed cell cycle, apoptosis, DNA damage, gene expression, genomic instability, and the sequence of exons in DNA repair genes in several kinds of cells. These cells consisted of untreated control cells, cells in the fifth, 15th, and 35th passage of cadmium-treated cells, and tumorigenic cells from nude mice using flow cytometry, Hoechst 33258 staining, comet assay, quantitative real-time polymerase chain reaction (PCR), Western blot analysis, random amplified polymorphic DNA (RAPD)-PCR, and sequence analysis. We observed a progressive increase in cell population of the G0/G1 phase of the cell cycle and the rate of apoptosis, DNA damage, and cadmium-induced apoptotic morphological changes in cerebral cortical neurons during malignant transformation. Gene expression analysis revealed increased expression of cell proliferation (PCNA), cell cycle (CyclinD1), pro-apoptotic activity (Bax), and DNA damage of the checkpoint genes ATM, ATR, Chk1, Chk2, Cdc25A. Decreased expression of the anti-apoptotic gene Bcl-2 and the DNA repair genes hMSH2, hMLH1, ERCC1, ERCC2, and hOGG1 was observed. RAPD-PCR revealed genomic instability in cadmium-exposed cells, and sequence analysis showed mutation of exons in hMSH2, ERCC1, XRCC1, and hOGG1 in tumorigenic cells. This study suggests that Cadmium can increase cell apoptosis and DNA damage, decrease DNA repair capacity, and cause mutations, and genomic instability leading to malignant transformation. This process could be a viable mechanism for cadmium-induced cancers.
Keywords: Cadmium chloride, DNA damage, DNA repair genes, genomic instability.
INTRODUCTION
Cadmium is a heavy metal with widespread industrial applications. However, it is toxic, and occupational and environmental exposure to it harms human health [1,2,3]. The toxicological responses to exposure to cadmium include kidney, liver, and stomach damage; respiratory and bone disease; and neurological disorders [4,5,6]. Experimental and epidemiological data have shown that cadmium and its compounds are carcinogenic to animals and humans[7]. On the basis of the results of epidemiological and experimental studies, cadmium and its compounds were classified as human carcinogens in 1993 by the International Agency for Research on Cancer [8].
Short- and long-term health effects of cadmium exposure have been reported in humans. Inorganic cadmium has been shown to induce malignant transformation of human bladder urothelial cells [9]. Studies with in vitro models have shown that cadmium can induce malignant transformation of prostate epithelial cells from both humans [10] and rodents [11]. Thus, the carcinogenic potential of cadmium is well established; however, the mechanisms of cadmium-induced carcinogenesis remain unclear. In the past, cadmium has been shown to induce apoptosis in vivo [12-13] and in vitro [14-15] at concentrations from 1 to 300 micromoles per liter (μmol/L).
Therefore, Cadmium-mediated toxicity is thought to involve, at least in part, the induction of apoptosis [16]. Cadmium carcinogenesis involves multiple mechanisms, including DNA strand breakage and inhibition of DNA repair. Cadmium induces p53-dependent G1/S and/or G2/M cell cycle arrest in various cell lines expressing tumor suppressor protein p53 [17-18]. Previous reports suggest that Cadmium exposure can induce mutations in the genome [19]. Cadmium-induced hypermutability due to inhibition of mismatch repair in yeast has been reported [20]. These studies clearly indicate that cadmium exposure may cause genomic instability through mutation. Abrogation of the function of DNA repair genes could potentially increase instability in the genome, which may lead to the development of cancer [21].
Published reports suggest that the respiratory system is an important target for cadmium-induced toxicity and carcinogenicity. However, the effects of chronic exposure to cadmium at doses that cause overt lesions are not well understood. Moreover, the effects of chronic exposure to cadmium on cell cycle, cell apoptosis, DNA damage, DNA repair capacity, and expression of genes related to the vital function of maintaining genomic stability in respiratory cells are also unclear. We previously established a model system of morphological cell transformation with cadmium chloride (CdCl2) in human bronchial epithelial cells (16HBE). This can provide a human model for the molecular events of cadmium carcinogenesis [22].
Our objective was to determine the effects of cadmium on cell cycle, cell apoptosis, DNA damage, DNA repair capacity, and expression of associated genes at different stages of CdCl2-induced malignant transformation of 16HBE cells. The effects of cadmium on genomic stability and mutation of exons in the DNA repair genes hMSH2, ERCC1, XRCC1, and hOGG1 were also investigated.
MATERIALS AND METHODS
Chemicals and reagents
CdCl2, RPMI-1640 medium, procainamide, and L-glutamine were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The model for CdCl2-induced morphological transformation of 16HBE cells was provided by Dr. Lei [22] of the School of Preventive Medicine, Guangzhou Medical University, China. Keratinocyte serum-free medium, epidermal growth factor, bovine pituitary extract, a 100x antibiotic-antimycotic mixture, and TRIzol® reagent were purchased from Life Technologies, Inc. (Grand Island, NY, USA). Anti-hMSH2, anti-ERCC1, anti-XRCC1, and anti-hOGG1 were purchased from Calbiochem (San Diego, CA, USA). Monoclonal actin antibody was purchased from Oncogene Research Products (Cambridge, MA, USA). The Bradford Protein Assay Kit was purchased from Bio-Rad Laboratories (Hercules, CA, USA).
Cell culture and treatments
16HBE cells were morphologically transformed using CdCl2, as previously described (Lei, et al., 2008) [22]. Untransformed 16HBE cells (controls); cadmium-transformed cells at the 5th (5 μmol L-1 cadmium for 2 weeks), 15th (5 μmol L-1 cadmium for 6 weeks), and 35th (5 μmol L-1 cadmium for 14 weeks) passage; and tumorigenic cells from nude mice were cultured in RPMI-1640 medium containing L-glutamine and supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Life Technologies) at 37°C in a 5% CO2 humidified atmosphere. The cells were passaged twice a week and maintained in the log phase of growth at 2×105 to 5×105 cells per milliliter (mL) to avoid spontaneous differentiation.
Flow cytometric analysis of cell cycle
16HBE cells at different stages of cadmium-induced malignant transformation were harvested through trypsin digestion and fixed in 70% ethanol at -20°C. Before analysis, cells were incubated with RNase A (20 micrograms [μg] per mL) and stained with propidium iodide (PI) (50 μg/mL) for 5 minutes. Samples were immediately analyzed using flow cytometry with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). A total of 10,000 events were recorded per sample at FL2 peak emission values (FL2-H) (wavelength 575 ±26 nanometers [nm]), and the cell fractions in the sub-G1, G1, S, and G2/M cell cycle phases were quantified in histograms with WinMDI software (version 2.9, Joseph Trotter).
Hoechst 33258 staining
Apoptotic morphological changes in the nuclear chromatin of cells were detected using Hoechst 33258 staining. 16HBE cells at different stages of cadmium-induced malignant transformation were seeded on sterile cover glasses placed in 6-well plates. After overnight growth, cells were treated with N-Acetylcysteine (NAC) (500 micrometers [μM]) or selenium (20 μM) for 30 minutes. Cells were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 10 minutes, and incubated with 50 μM Hoechst 33258 staining solution for 10 minutes. After three washes with PBS, the cells were viewed under a fluorescence microscope (Olympus, IX-70, Japan).
Low cytometric assessment of apoptosis
Measurement of phosphatidylserine redistribution in the plasma membrane was conducted according to the protocol of the manufacturer of the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences). Briefly, after pre-treatment with NAC (500 μM) or selenium (20 μM) for 30 minutes, harvested cells were suspended in a binding buffer (1x). An aliquot of 100 μl was incubated with 5 microliters (μl) of Annexin V-FITC and 5μl of PI for 15 minutes in the dark, and 400 μl binding buffer (1x) was added to each sample. The stained cells were analyzed with flow cytometry using BD CellQuest™ Pro Software (BD, Franklin, NJ, USA).
Comet assay
DNA damage was investigated using the comet assay [23], with minor modifications. 16HBE cells at different stages of cadmium-induced malignant transformation were diluted in PBS so that 3 or 4 cells could be seen in a single field at 400x magnification. The resulting cell suspension (cell viability was found to be over 90% with trypan blue exclusion analysis) was mixed with 0.6% low-melting-point agarose (kept in a 37°C water bath). It was then rapidly spread onto a layer of 80 μL normal-melting-point agarose on a completely frosted microscope slide and covered with a 24x24 millimeter (mm) cover slip. The cover slip was gently removed, and the slide was submerged in lysing solution (2.5 molar [M] NaCl, 100 mM EDTA, 10 mM Tris, pH 10, 1% sodium lauryl sarcosinate, 10% dimethyl sulfoxide (DMSO), and 1% Triton™ X-100 added just before use at 4°C for 2 hours. The slides were then placed in the electrophoresis solution (300 mM NaOH, 1 mM EDTA, pH>13, 4°C) for 20 minutes to facilitate DNA unwinding before electrophoresis was conducted for 20 minutes at 25 volts and 300 milliamps (mA). After electrophoresis, the slides were drained and neutralized for 5 minutes 3 times with neutralization buffer (0.4 M Tris hydrochloride, pH 7.5) and stained with ethidium bromide (5 μg/mL). The fluorescently stained nucleoids were scored visually using a fluorescence microscope (Nikon) with an excitation filter of 510-560 nm and a barrier filter of 590 nm at 400x magnification.
Quantitative real-time polymerase chain reaction
Total RNA was isolated using TRIzol® reagent. Reverse transcription was performed using a Titanium® RT-PCR Kit (Clontech, CA, USA) according to the manufacturer's instructions. Gene expression was quantified using a fluorescence-based real-time polymerase chain reaction (PCR) according to manufacturer's instructions (Bio-Rad Laboratories). The sequences of primers used for real-time PCR are shown in Table 1.
Primers and product sizes for selected genes with real-time PCR.
| Genes | Primers | PCR product size (bp) |
|---|---|---|
| PCNA | Forward: 5´- GCCCTCAAAGACCTCATCAA -3' Reverse: 5´- TCTGGGATTCCAAGTTGCTC -3' | 338 |
| CyclinD1 | Forward: 5´- GCACAACGCACTTTCTTTCTTTCCA -3´ Reverse: 5´- CGCAGGCTTGACTCCAGAAG -3' | 97 |
| Bcl2 | Forward:5´- ATGGGGTGAACTGGGGGATTG -3´ Reverse: 5´- TTCCGAATTTGTTTGGGGCAGGTC -3´ | 328 |
| Bax | Forward:5´- GGGTGGTTGCCCTTTTCTACT -3´ Reverse: 5´- CCCGGAGGAAGTCCAGTGTC-3´ | 110 |
| ATM | Forward:5´- TGCCAGACAGCCGTGACTTAC -3´ Reverse: 5´- ACCTCCACCTGCTCATACACAAG -3´ | 98 |
| ATR | Forward:5´- GGGATGCCACTGCTTGTTATGAC -3´ Reverse: 5´- CTGTCCACTCGGACCTGTTAGC -3´ | 156 |
| Chk1 | Forward:5´- CGATTCTGCTCCTCTAGCTCTGC -3´ Reverse: 5´- TGACACACCACCTGAAGTGACTC -3´ | 151 |
| Chk2 | Forward:5´- CAGGTTCTAGCCCAGCCTTCTAC -3´ Reverse: 5´- GGAGTTCACAACACAGCAGCAC -3´ | 110 |
| Cdc25A | Forward:5´- CAACCACTGGAGGTGAAGAACAAC -3´ Reverse: 5´- CCCAACAGCTTCTGAGGTAGGG -3´ | 161 |
| hMSH2 | Forward: 5'- AAGCCCAGGATGCCATTG-3' Reverse:5'- CATTTGACACGTGAGCAAAGC--3' | 126 |
| ERCC1 | Forward:5'- CCCTGGGAATTTGGCGACGTAA-3' Reverse: 5'- CTCCAGGTAccGCCCAGCTTCC-3' | 223 |
| hOGG1 | Forward: 5'-ATCTGTTCTTCCAACAACAAC-3' Reverse: 5'-GCCAGCATAAGGTCCCCACAG-3' | 504 |
| XRCC1 | Reverse: 5'-AGCTGTCGCCATCTGTTC-3' Reverse:5'-AGCACCCACTCCTTACGC-3' | 370 |
| β-Actin | Forward: 5'-ACACTGTGCCCATCTACGAGG-3' Reverse: 5'-AGGGGCCGGACTCGTCATACT-3' | 250 |
Western blot analysis
Total protein was isolated, electrophoresed on NuPAGE® 4%-12% Bis-Tris gel, and transferred to nitrocellulose membranes according to the manufacturer's instructions (Invitrogen, CA, USA). Immunoblotting was performed using mouse antibodies against human hMSH2, ERCC1, XRCC1, and hOGG1 (Abcam, Cambridge, MA, USA) and horseradish peroxidase-conjugated anti-mouse secondary antibody (Amersham Biosciences, Piscataway, NJ, USA) according to the kit's instructions.
Random amplified polymorphic DNA (RAPD)-PCR
RAPD-PCR was performed on the DNA of 16HBE cells at different stages of cadmium-induced malignant transformation using a previously described method [24]. Briefly, PCR amplifications were performed in 25μl reaction mixture containing 2.5 μl 10x enzyme assay buffer, 100 μM each of dATP, dCTP, dGTP, and dTTP, 100 μM random (10-bp) primer, 1.5 mM MgCl2, 1.5 units AmpliTaq DNA polymerase (Stoffel fragment) and 75 nanograms (ng) DNA as a template. The amplification was performed in a Perkin-Elmer Cetus DNA thermal cycler programmed for 45 cycles as follows: first cycle: 3.5 minutes at 9°C, 1 minute at 34°C, and 2 minutes at 72°C; 44 additional cycles: 1 minute at 9°C, 1 minute at 34°C, 2 minutes at 72°C; followed by a final extension of 15 minutes at 72°C. Amplified products were resolved on a 1.5% agarose gel and visualized with ethidium bromide staining. Each experiment was repeated 3 or 4 times.
Analysis of DNA repair gene sequence
The sequence of exon6 in hMSH2, exon7 in hMSH2, exon8 in hMSH2, exon9 in hMSH2, exonl2 in hMSH2, exon3 in ERCC1, exon4 in ERCC1, exon6 in XRCC1, exon9 in XRCC1, and exon7 in hOGG1 in untransformed control cells and tumorigenic cells induced by CdCl2 were detected by sequencing amplified PCR and primers described at http://variantgps.nci.nih.gov/cgfseq/pages/home.do. In our study, DNA sequence variation was named according to den Dunnen and Antonarakis . Sequence analysis was performed on a CEQ 8000 (Beckman Coulter, Fullerton, CA, USA) sequencing device.
Statistical analysis
All data are represented as rate (%) or mean±SD (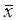 ± s) of three or more independent experiments. Statistical significance was determined using the chi square test for rate (%) comparison in several independent experiments and Student's t-test or by analysis of variance (ANOVA) followed by Dunnett's multiple comparison test. Significance was set at P≤0.05 in all cases.
± s) of three or more independent experiments. Statistical significance was determined using the chi square test for rate (%) comparison in several independent experiments and Student's t-test or by analysis of variance (ANOVA) followed by Dunnett's multiple comparison test. Significance was set at P≤0.05 in all cases.
RESULTS
Cadmium-induced cell cycle
As described previously, 16HBE cells were malignantly transformed through continuous treatment with CdCl2 and formed tumors in nude mice [21]. Cells isolated from the xenograft tumors were established as cell lines and tumorigenic cells. To ascertain whether abnormal genetic alteration is involved and has an important role in cadmium-induced transformation, we set up a parallel experiment with control 16HBE cells, 16HBE cells treated with CdCl2, and tumorigenic cells. 16HBE cells treated with CdCl2 for 35 passages can form robust colonies in soft agar and initiate xenograft tumors and malignant transformation in nude mice (data not shown). 16HBE cells treated with CdCl2 for 5 passages and 15 passages were used to represent the early and middle stages of transformation. The tumorigenic cells were the 35-passaged cells initiating xenograft tumors in nude mice. All examined cells in each group were at the same passage.
Cell cycle distribution of untreated control cells in the 5th, 15th, and 35th passage of CdCl2- treated cells and tumorigenic cells were measured with flow cytometric analysis. Figure 1 shows the flow analysis data (10,000 events). As the CdCl2-treated cell passages increased, the cell population increased in the G0/G1 phase and decreased in the S and G2 phases during cadmium exposure. The percentage of cells in G0/G1 in the 35th passage of CdCl2-treated cells (60.04%) and tumorigenic cells from nude mice (64.12%) significantly increased compared with untreated control cells (P<0.05). The percentage of cells in the S and G2 phases in the 35th passage of CdCl2-treated cells (35.03% and 4.93%) and tumorigenic cells (31.83% and 4.01%) significantly decreased in CdCl2-treated cells compared with the untreated control group (P<0.05) (Figure 1). We observed that cadmium inhibited cell cycle arrest by shifting to the G1 phase from the G2 and S phases.
Effect of cadmium on cell cycle phase during cadmium-induced malignant transformation of human bronchial epithelial cells. Cell cycle phase was determined in untreated control cells, 5th, 15th, and 35th passage of cadmium-treated cells, and tumorigenic cells from nude mice through flow cytometric analysis. All values are mean±SD (n=5). Statistical (one-way ANOVA and Dunnett's multiple comparison test) differences between control and cadmium-treated cells (*P<0.05) are marked.

Cadmium-induced apoptosis
In this study, flow cytometric measurement (Annexin V/FITC and PI double staining) was used to quantify the extent of apoptosis in the total cell population. After incubation of different lengths with CdCl2 (5th, 15th, and 35th passages of CdCl2-treated cells and tumorigenic cells), the percentage of total (early + late) apoptotic cells increased to 7.24%, 11.05%, 15.87%, and 16.21%, respectively, compared with 5.82% in controls (Figure 2), which indicated cadmium-induced apoptosis in 16HBE.
Apoptosis induced by cadmium during malignant transformation of human bronchial epithelial cells. Apoptosis was measured in untreated control cells, 5th, 15th, and 35th passage of cadmium-treated cells, and tumorigenic cells from nude mice through flow cytometry. Results are expressed as percentages of apoptosis with regard to the total cells. Data are mean±SD of three separate experiments, with each performed in triplicate. *P<0.05 compared with control using one-way ANOVA and Dunnett's multiple comparison test.

Effect of cadmium on apoptotic morphological changes
Cadmium-induced apoptotic morphological changes were assessed with Hoechst 33258 staining (Figure 3). In the control group, most cells had uniformly stained nuclei, and the chromatin of normal nuclei was unaltered and spread uniformly throughout the entire nucleus (Figure 3A). After exposure to CdCl2 for the 5th, 15th, and 35th passage, nuclear morphological changes typical of apoptosis were observed, nuclear chromatin was condensed, and fragmented chromatin was characterized by a scattered, drop-like structure. The nuclei of apoptotic cells appeared smaller and shrunken compared with intact cells (Figure 3B-D).
Effects of cadmium on apoptotic morphological changes during malignant transformation of human bronchial epithelial cells. Cells were incubated with cadmium (5th, 15th, and 35th passage) and tumorigenic cells. Nuclear chromatin changes (apoptosis) were assessed with Hoechst 33258 staining. Arrows identify apoptotic nuclei. Among the groups, (A) control; (B) 15th passage-transformed cells treated with cadmium; (C) 35th passage-transformed cells treated with cadmium; (D) tumorigenic cells induced by cadmium.

Increased cadmium-induced DNA damage
During cadmium-induced malignant transformation of 16HBE cells, the tail lengths of DNA comets were all longer than those in untransformed 16HBE cells (P<0.05), and the tail lengths of DNA comets in the cadmium-treated 35th passage-transformed cells and tumorigenic cells were longer than those in untransformed 16HBE cells (P<0.05) (Table 2, Figure 4). The cadmium-induced DNA damage rates in the 5th, 15th, and 35th passage-transformed cells and the tumorigenic cells were 10.45%, 22.00%, 46.75%, and 49.00%, respectively, compared with untransformed 16HBE cells, which had a DNA damage rate of only 4.75% (P<0.05).
DNA damage and its suppression during cadmium-induced malignant transformation of 16HBE cells determined using the comet assay
| Cell type | DNA damage rate (%) | Tail length (μm) |
|---|---|---|
| Untransformed cells | 4.75 | 12.8 ± 1.76 |
| 5th passage | 10.45 | 15.2 ± 3.54 |
| 15th passage | 22.00 * | 31.6 ± 2.80 * |
| 35th passage | 46.75 ** | 47.8 ± 2.36 ** |
| Tumorigenic cells | 49.00 ** | 48.5 ± 2.47 ** |
Tail lengths of cells in which DNA damage was induced and suppressed were determined using comet assay in untransformed controls; 5th, 15th, and 35th passage-transformed cells; and tumorigenic cells. *P<0.05 and **P<0.01 compared with untransformed controls.
DNA damage to 16HBE cells treated with cadmium. (A) DNA damage to untransformed 16HBE cells with the comet assay; (B) DNA damage of 15th passage-transformed cells by comet assay; (C) DNA damage of 35th passage-transformed cells by comet assay; and (D) DNA damage of tumorigenic cells by comet assay.

Cadmium-induced changes in gene expression
Biological effects of any factors are the consequence of gene expression changes. Therefore, to understand the role of cadmium-induced changes that might be involved in gene expression, changes were analyzed in untransformed controls; 5th, 15th, and 35th passage-transformed cells; and tumorigenic cells. Total RNA was isolated, and gene expression was measured with quantitative real-time PCR. Selected genes associated with cell proliferation (PCNA), cell cycle (CyclinD1), anti-apoptotic activity (Bcl-2), pro-apoptotic activity (Bax), DNA damage checkpoint-related genes (ATM, ATR, Chk1, Chk2, Cdc25A), and DNA repair genes (hMSH2, hMLH1, ERCC1, ERCC2 , XRCC1 ,hOGG1, and MGMT) were used in this study.
Cell cycle-related genes
The effect of cadmium on the cell cycle and cell proliferation was further determined by measuring the expression of a cell proliferation marker gene, PCNA, and a positive cell cycle regulator, CyclinD1. The expression of PCNA, and CyclinD1 mRNA were progressively increased during cadmium exposure. Furthermore, the expression levels of PCNA in 35th passage-transformed cells and tumorigenic cells from nude mice were increased 1.98 fold and 2.01 fold compared with controls (P<0.05). Expression levels of CyclinD1 in 35th passage-transformed cells and tumorigenic cells were increased 2.34 fold and 2.33 fold, respectively, compared with controls (P<0.05) (Figure 5A).
Cadmium-induced changes in gene mRNA expression during cadmium-induced malignant transformation of 16HBE cells. The mRNA expression of selected cell cycle-related genes. (A) PCNA and CyclinD1, apoptosis-related genes; (B) Bal-2 and Bax, DNA damage checkpoint genes; (C) ATM, ATR, Chk1m, Chk2, and Cdc25A were determined in untransformed controls; 5th, 15th, and 35th passage-transformed cells; and tumorigenic cells using quantitative real-time PCR. The threshold cycle number (Ct value) for each gene obtained with real-time PCR was normalized to Ct value of β-actin from same sample, and fold changes in expression for each gene were obtained with delta-delta Ct method. The graph shows the mean±SD of triplicate values. *Statistically significant differences relative to the control group (P<0.05) analyzed using one-way ANOVA.

Anti-apoptotic and pro-apoptotic genes
To determine the role of the anti-apoptotic gene Bcl-2 and the pro-apoptotic gene Bax in cadmium-induced cell proliferation, expression of these genes was measured with quantitative real-time PCR. Increases of 1.01, 1.22, 1.52, and 1.98 fold were observed for Bax expression, and decreases of 0.88, 0.66, 0.50, and 0.54 fold, respectively, were seen in Bal-2 expression at 5th, 15th, and 35th passage-transformed cells and tumorigenic cells compared with controls (P<0.05) (Figure 5B).
DNA damage checkpoint genes
To examine DNA damage during the cadmium-induced malignant transformation of 16HBE cells, we measured the expression of the DNA damage checkpoint genes ATM, ATR, Chk1m, Chk2, and Cdc25A with quantitative real-time PCR. The expression levels of these 5 genes in 35th passage-transformed cells increased 1.52, 1.75, 1.61, 1.95, 2.34 fold, respectively, compared with controls (P<0.05). The expression levels of these genes in tumorigenic cells increased 1.44, 1.8, 1.75, 1.96, 2.35 fold, respectively, compared with controls (P<0.05) (Figure 5C).
mRNA and protein expression of DNA repair genes
To identify alterations in DNA repair genes during the malignant transformation of 16HBE cells, we determined expression levels of the corresponding mRNA and proteins for the genes hMSH2, hMLH1, ERCC1, ERCC2, hOGG1g, and MGMT with real-time PCR and Western blot. The results showed no detectable changes in hMLH1, ERCC2, and MGMT expression, but the expression of hMSH2, ERCC1, XRCC1, and hOGG1 mRNA and protein was progressively reduced during cadmium exposure. Furthermore, the expression levels of these 4 genes in 35th passage-transformed cells (P<0.05) and tumorigenic cells (P<0.01) were significantly decreased compared with controls (Table 3) (Figure 6).
mRNA and protein expressions of hMSH2, ERCC1, XRCC1, and hOGG1 mRNA during cadmium-induced malignant transformation of 16HBE cells.
| Genes | Control cells | 5th passage | 15th passage | 35th passage | Tumorigenic cells |
|---|---|---|---|---|---|
| mRNA expressions in genes during 16HBE cells treated with cadmium | |||||
| hMSH2 | 0.406±0.008 | 0.403±0.007 | 0.396±0.007 | 0.198±0.015** | 0.198±0.008** |
| ERCC1 | 0.703±0.017 | 0.686±0.045 | 0.675±0.029 | 0.348±0.045** | 0.357±0.041** |
| XRCC1 | 0.385±0.013 | 0.386±0.014 | 0.377±0.021 | 0.200±0.020** | 0.191±0.007** |
| hOGG1 | 0.535±0.008 | 0.523±0.018 | 0.401±0.036* | 0.196±0.005** | 0.197±0.005** |
| The protein expressions in genes during 16HBE cells treated with cadmium | |||||
| hMSH2 | 0.538±0.014 | 0.442±0.081 | 0.425±0.097 | 0.214±0.045** | 0.189±0.081** |
| ERCC1 | 0.654±0.057 | 0.612±0.057 | 0.591±0.184 | 0.312±0.068** | 0.281±0.081** |
| XRCC1 | 0.451±0.049 | 0.397±0.051 | 0.384±0.034 | 0.241±0.034** | 0.191±0.027** |
| hOGG1 | 0.594±0.024 | 0.554±0.034 | 0.342±0.051* | 0.197±0.027** | 0.178±0.048** |
mRNA and protein expressions of hMSH2, ERCC1, XRCC1, and hOGG1 were determined in untransformed controls; 5th, 15th, and 35th passage-transformed cells; and tumorigenic cells. mRNA and protein expressions were measured by real-time PCR and Western blot analysis, respectively. mRNA and Protein expression levels were calculated relative to β-actin. *P<0.05 and **P<0.01 compared with untransformed controls from the corresponding group.
mRNA and protein expression of hMSH2, ERCC1, XRCC1, and hOGG1 during cadmium-induced malignant transformation of 16HBE cells. (A) mRNA and (B) protein expression of hMSH2, ERCC1, XRCC1, and hOGG1 was determined in untransformed controls; 5th, 15th, and 35th passage-transformed cells; and tumorigenic cells from nude mice with real-time PCR and Western blot analysis, respectively. mRNA and protein expression levels were calculated relative to β-actin.

Cadmium-induced genetic alterations
Genomic instability
To investigate cadmium-induced genomic instability, a RAPD-PCR assay was performed using genomic DNA from long-duration cadmium-exposed cells and passage-matched, untreated controls. Figure 7A shows representative photographs of RAPD fingerprints. Cadmium-induced changes detected in RAPD fingerprints generated by primer OPC07 (50-ATTCTGGTTT-30) indicate the affected genomic region RAPD fingerprints generated by primer OPC12 (50-TGTCATCCCC-30), showing a similar pattern indicating unaffected genomic regions (Figure 7B). A loss of 1,250 base pairs (bp) was seen in 35th passage-transformed cells. A 500 bp loss was seen in 35th passage-transformed and tumorigenic cells compared with controls (Figure 7A). Similarly, gains of 1,100, 950, and 700 bp were seen at 35th passage-transformed cells. Gains of 650 bp were seen in tumorigenic cells compared with DNA fingerprints of controls (Figure 7A). Genomic instability shown by maximum changes in RAPD products was observed in 35th passage-transformed cells. No change was common to passage-transformed and tumorigenic cells.
Representative RAPD-PCR fingerprints generated from genomic DNA during cadmium-induced malignant transformation of 16HBE cells. (A) Fingerprints generated by primer OPC07(50-ATTCTGGTTT-30) showing affected genomic regions in cadmium-induced malignant transformation of 16HBE cells. Arrows indicate changes in RAPD-PCR-amplified loci in cadmium-induced cells compared with passage-matched control cells. (B) Fingerprints for passage-matched controls for cadmium-induced cells tested generated by primer OPC12 (50-TGTCATCCCC-30) showing unaffected genomic regions.

Sequence analysis of DNA repair genes
To investigate cadmium-induced genomic instability, the DNA polymorphic characteristics of hMSH2, ERCC1, XRCC1, and hOGG1 were detected by sequencing amplified PCR and primers in 16HBE cells and tumorigenic cells induced by CdCl2. The objective fragments of the exons in these four DNA repair genes were obtained with PCR amplification. After the application was identified by gel electrophoresis, the amplified DNA strips were purified. Then the exons were detected by DNA sequence. We observed no mutations at exon6 in hMSH2, exon7 in hMSH2, exon3 in ERCC1, exon6 in XRCC1, or exon9 in XRCC1 identified in 16HBE cells and tumorigenic cells of nude mice induced by CdCl2. In the CdCl2-induced tumorigenic cells, there were thymine deletions at the first, second, and seventh sites of exon8 in hMSH2; adenine deletions at the 20th and 182th sites of exon9 in hMSH2; adenine insertions at the 241th site of exon12 in hMSH2; adenine deletions at the first site of exon4 in ERCC1; and adenine in the 162th site of exon7 in hOGG1 (Figure 8). All mutations of exons in these DNA repair genes were frame shift mutations.
Sequence analysis of exons in hMSH2, ERCC1, XRCC1, and hOGG1. Sequence of exons in hMSH2, ERCC1, XRCC1, and hOGG1. Sequence analysis of exons in hMSH2, ERCC1, XRCC1, and hOGG1. Sequence of exons in hMSH2, ERCC1, XRCC1, and hOGG1 were detected by sequencing amplified PCR and primers in untransformed control and tumorigenic cells induced by cadmium chloride. (A) Sequence of exon8 in hMSH2; (B) Sequence of exon9 in hMSH2; (C) Sequence of exon12 in hMSH2; (D) Sequence of exon14 in ERCC1; (E) Sequence of exon17 in hOGG1. The mutation “↓” are marked.

DISCUSSION
Cadmium is an important heavy metal widely used in nickel-cadmium batteries, metal plating, pigments, plastics, and alloys [25]. This metal stimulates free-radical production, resulting in oxidative deterioration of lipids, proteins, and DNA, and initiating various pathological conditions in humans and animals [26]. Several reports have shown that cadmium can induce apoptosis and DNA damage [21,27-29]. However, the underlying mechanism remains to be elucidated, and few reports have shown the effects and toxic mechanism of cadmium on the respiratory system. In this study, we used the malignant transformation of the 16HBE line to show that submicromolar concentrations of cadmium result in apoptotic resistance and aberrant expression of some genes related to cell cycle, apoptosis, DNA damage, and DNA repair. An increase in cell apoptosis and a decrease in DNA repair capacity following cadmium exposure together may lead to an accumulation of mutations in DNA repair genes, induction of genomic instability, and neoplastic transformation of cadmium-exposed cells. Therefore, this study provides a potential mechanism for cadmium-induced carcinogenesis.
Increased cell proliferation and cell apoptosis are characteristic features of transformed cells and most types of cancer [30]. In this study, we observed a significant increase in the G0/G1 phase of 35th passage-transformed and tumorigenic cells. The observed increase in the G0/G1 phase of cadmium-exposed cells could be explained by the mitogenic effect of cadmium. The increased cell growth and expression of the cell proliferation marker gene PCNA and the cell cycle gene CyclinD1 in cells treated with cadmium suggest that cadmium has mitogenic potential. Although there are several reports on increased cell proliferation from cadmium exposure [31,32], there are very few reports on increased expression of PCNA and CyclinD1 in cadmium-exposed cells[33,34].
Therefore, this study further provides evidence at the molecular level for the mitogenic potential of cadmium at submicromolar concentrations. Cadmium-induced apoptosis has been described in various cells, including renal cells[35,36,37] and, so far, no uniform molecular mechanism has been proposed. We also observed a marked increase in the expression of the pro-apoptotic gene Bax and decreased expression of the anti-apoptotic gene Bcl-2 in cadmium-exposed cells compared with unexposed cells. The increase in pro-apoptotic gene expression and decrease in anti-apoptotic gene expression could explain the observed increased apoptosis. Similar findings in other cells have been reported [38,39]. Therefore, these previous reports and data from this study suggest that apoptotic resistance could be a mechanism for cadmium-induced malignant transformation.
DNA damage, repair, replication, tolerance, and escape are associated with gene mutation and malignant transformation. In this study, a significant increase in DNA damage rate, tail lengths, and expression of the DNA damage checkpoint genes ATM, ATR, Chk1m, Chk2, and Cdc25A in 35th passage- transformed cells and tumorigenic cells was observed. There is much debate as to whether cadmium induces mutations directly by interacting with DNA as an adduct or indirectly by inhibiting cellular antioxidants and DNA repair systems [40]. DNA repair genes play a crucial role in the maintenance of genome stability, and impairment of these genes has been found to promote carcinogenesis [41,42]. The removal or repair of DNA damage is important plays a key role in protecting cells from the insults of carcinogens. Both the low expression and mutation of DNA repair genes reduce DNA repair capacity, which in turn increases the probability of mutation and, consequently, of cancer initiation. Thus far, more than 130 DNA repair genes have been found, and they belong to 5 DNA repair systems: base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), directness repair, and double-stranded DNA break repair. Each participate in one or more DNA repair pathways.
Among the 130 DNA repair genes, hMSH2, hMLH1, ERCC1, ERCC2, XRCC1, hOGG1, and MGMT are the key genes. They represent different DNA repair pathways and capabilities. Abnormalities in these genes cause cell transformation and carcinogenesis. Our results show that the mRNA and protein expression of the DNA repair genes hMSH2, ERCC1, XRCC1, and hOGG1 was progressively reduced and DNA damage increased with the degree of cadmium-induced malignant transformation in 16HBE cells. This suggests that the BER, NER, and MMR pathways are inhibited, and DNA repair capacity is lowered during transformation.
Genetic changes have an important role in environmental chemical-induced carcinogenesis [43]. Some studies have shown that the mutations of the exons in hMLH1, hMSH2, ERCC1, XRCC1, and hOGG1 impair DNA repair function, which increases DNA damage and even carcinogenesis in some diseases [44,45,46]. In this study, the RAPD-PCR analysis revealed changes in 35th passage-transformed cells and tumorigenic cells, and sequence analysis showed shift mutation of exons in hMSH2, ERCC1, XRCC1, and hOGG1. The gains and losses of DNA amplification products in the RAPD-PCR fingerprints generated from the DNA of cadmium-treated samples compared with the RAPD-PCR fingerprints from the DNA of untreated control samples revealed the mutagenic effects of cadmium. Previous reports also provide evidence of promutagenic DNA damage resulting from cadmium exposure[47]. A recent study showed that even submicromolar concentrations of cadmium can induce mutations in tetranucleotide repeats in human fibrosarcoma cells[19]. Therefore, these previous reports and the data presented here for RAPD-PCR analysis showing genomic instability and sequence analysis showing mutations in cadmium-induced malignant transformation of 16HBE cells clearly suggest that submicromolar concentrations of cadmium are environmentally relevant and mutagenic.
In conclusion, this study clearly indicates that cadmium increases cell apoptosis and DNA damage mediated by aberrant expression of related genes and decreases the DNA repair capacity through mutation of the exons in hMSH2, ERCC1, XRCC1, and hOGG1. The observed decrease in DNA repair capacity coupled with changes in the cell cycle distribution and apoptosis after cadmium exposure would create a microenvironment for genomic instability, leading to malignant transformation. This study therefore provides insight into the mechanism underlying cadmium-induced carcinogenesis in 16HBE cells. Further research into the mechanism for cadmium-induced cancers of the respiratory system is needed.
FUNDING INFORMATION
This work was supported by the National Natural Science Foundation of China (81202236 Zhou, Z.H; 81072322 to Lei, Y.X) and the Science and Technology Planning Project of Guangdong Municipality, China (2013J410037).
Acknowledgements
The authors thank Dr. Yuanhui Huang for his helpful suggestions and advice on this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107-120
2. Satarug S, Baker JR, Urbenjapol S, Haswell-Elkins M, Reilly PEB, Williams DJ, Moore MR. A global perspective on cadmium pollution and toxicity in non-occupationally exposed population. Toxicol Lett. 2003;137:65-83
3. Swaddiwudhipong W, Mahasakpan P, Funkhiew T, Limpatanachote P. Changes in cadmium exposure among persons living in cadmium-contaminated areas in northwestern Thailand: a five-year follow-up. J Med Assoc Thai. 2010;93:1217-1222
4. Koyu A, Gokcimen A, Ozguner F, Bayram DS, Kocak A. Evaluation of the effects of cadmium on rat liver. Mol Cell Biochem. 2006;20:1-5
5. Nordberg G, Jin T, Bernard A, Fierens S, Buchet JP, Ye T, Kong Q, Wang H. Low bone density and renal dysfunction following environmental cadmium exposure in China. Ambio. 2002;31:478-481
6. Schöpfer J, Drasch G, Schrauzer GN. Selenium and cadmium levels and ratios in prostates, livers, and kidneys of nonsmokers and smokers. Biol Trace Elem Res. 2010;134:180-187
7. Saidi SA, Azaza MS Windmolders P, van Pelt J, El-Feki A. Cytotoxicity assessment and antioxidant enzyme related to heavy metals found in tuna by-products meal: An in vitro study in human and rat liver cell lines. Exp Toxicol Pathol. 2013 doi: 10.1016/j.etp.2013.03.001
8. IARC. Beryllium, cadmium, mercury and exposures in the glass manufacturing industry. Lyon, France: International Agency for Research on Cancer. 1993:119-238
9. Sens D.A, Park S, Gurel V, Sens MA, Garrett SH, Somji S. Inorganic cadmium- and arsenite-induced malignant transformation of human bladder urothelial cells. Toxicol Sci. 2004;79:56-63
10. Achanzar WE, Diwan BA, Liu J, Quader ST, Webber MM, Waalkes MP. Cadmium-induced malignant transformation of human prostate epithelial cells. Cancer Res. 2001;61:455-458
11. Terracio L, Nachtigal M. Oncogenicity of rat prostate cells transformed in vitro with cadmium chloride. Arch Toxicol. 1988;61:450-456
12. Harstad EB, Klaassen CD. Tumor necrosis factor-alpha-null mice are not resistant to cadmium chloride-induced hepatotoxicity. Toxicol Appl. 2002;179(3):155-62
13. Tzirogiannis KN, Panoutsopoulos GI, Demonakou MD. et al. Time-course of cadmium-induced acute hepatotoxicity in the rat liver: the role of apoptosis. Arch Toxicol. 2003;77(12):694-701
14. Hossain S, Liu HN, Nguyen M. et al. Cadmium exposure induces mitochondria-dependent apoptosis in oligodendrocytes. Neurotoxicology. 2009;30(4):544-54
15. Krumschnabel G, Ebner HL, Hess MW. et al. Apoptosis and necroptosis are induced in rainbow trout cell lines exposed to cadmium. Aquat Toxicol. 2010;99(1):73-85
16. Yuan Ya, Bian J C, Liu XZ. et al. Oxidative Stress and Apoptotic Changes of Rat Cerebral Cortical Neurons Exposed to Cadmium in Vitro. Biomed Environ Sci. 2012;25(2):172-181
17. Bjerregaard H. Effects of cadmium on differentiation and cell cycle progression in cultured Xenopus kidney distal epithelial (A6) cells. Altern Lab Anim. 2007;35:343-348
18. Cao F, Zhou T, Simpson D, Zhou Y, Boyer J, Chen B, Jin T, Cordeiro-Stone M, Kaufmann W. p53-Dependent but ATM-independent inhibition of DNA synthesis and G2 arrest in cadmium-treated human fibroblasts. Toxicol Appl Pharmacol. 2007;218:174-185
19. Slebos RJ, Li M, Evjen AN, Coffa J, Shyr Y, Yarbrough WG. Mutagenic effect of cadmium on the tetranucleotide repeats in human cells. Mutat Res. 2006;602:92-99
20. Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, Kunkel TA, Resnick MA, Gordenin DA. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet. 2003;34:326-329
21. Kamaleshwar PS, Ragini K, Christina P, Desiree J, James WD. Long duration exposure to cadmium leads to increased cell survival,decreased DNA repair capacity, and genomic instability in mouse testicular Leydig cells. Cancer letter. 2009;279:84-92
22. Lei YX, Wei L, Wang M, Wu GR, Li M. Malignant transformation and abnormal expression of eukaryotic initiation factor during human bronchial epithelial cells induced by cadmium chloride. Biomed Environ Sci. 2008;21:332-338
23. Andrighetti-Fröhner CR, Kratz JM, Antonio RV, Creczynski-Pasa TB, Barardi CR, Simões CM. In vitro testing for genotoxicity of violacein assessed by comet and micronucleus assays. Mutat Res. 2006;603:97-103
24. Singh KP, Roy D. Allelic loss and mutations in a new ETRG-1 gene are early events in diethylstilbestrol-induced renal carcinogenesis in Syrain hamsters. Gene. 2008;408:18-26
25. Agency for Toxic Substances, Disease Registry (ATSDR). Toxicological profile for cadmium, draft for public comment (update). Public Health Service, US Department of Health and Human Services. 1997
26. Manca D, Richard AC, Trottier B, Chevallier G. Studies on lipid peroxidation in rat tissues following administration of low and moderate doses of cadmium chloride. Toxicology. 1991;67:303-323
27. Migliarini B, Campisi AM, Maradonna F, Truzzi C, Annibaldi A. et al. Effects of cadmium exposure on testis apoptosis in the marine teleost Gobius niger. Gen Comp Endocrinol. 2005;142:241-247
28. Xu C, Johnson JE, Singh PK, Jones MM, Yan H. et al. In vivo studies of cadmium-induced apoptosis in testicular tissue of the rat and its modulation by a chelating agent. Toxicology. 1996;107:1-8
29. Blottner S, Frolich K, Roelants H, Streich J, Tataruch F. Influence of environmental cadmium on testicular proliferation in roe deer. Reprod Toxicol. 1999;13:261-267
30. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
31. Misra UK, Gawdi G, Akabani G, Pizzo SV. Cadmium-induced DNA synthesis and cell proliferation in macrophages: the role of intracellular calcium and signal transduction mechanisms. Cell Signal. 2002;14:327-340
32. Brama M, Gnessi L, Basciani S, Cerulli N, Politi L, Spera G, Mariani S, Cherubini S, Abusco A.S, Scandurra R, Migliaccio S. Cadmium induces mitogenic signaling in breast cancer cell by ER alpha-dependent mechanism. Mol Cell Endocrinol. 2007;264:102- 108
33. Fang MZ, Mar W, Cho MH. Cadmium affects genes involved in growth regulation during two-stage transformation of Balb/3T3 cells. Toxicology. 2002;177:253-265
34. Arriazu R, Pozuelo JM, Martin R, Rodriguez R, Santamaria L. Quantitative and immunohistochemical evaluation of PCNA, androgen receptors, apoptosis, and Glutathione-S-Transferase P1 on preneoplastic changes induced by cadmium and zinc chloride in the rat ventral prostate. Prostate. 2005;63:347-357
35. Thevenod F, Friedmann JM, Katsen AD, Hauser IA. Upregulation of multidrug resistance P-glycoprotein via nuclear factor-_b activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. The Journal of biological chemistry. 2000;275:1887-1896
36. Nordberg M, Jin T, Nordberg GF. Cadmium, metallothionein and renal tubular toxicity. IARC scientific publications. 1992;118:293-297
37. Xie J, Shaikh ZA. Cadmium-induced apoptosis in rat kidney epithelial cells involves decrease in nuclear factor-kappa B activity. Toxicological sciences. 2006;91:299-308
38. Hossain S, Liu HN, Nguyen M. et al. Cadmium exposure induces mitochondria-dependent apoptosis in oligodendrocytes. Neurotoxicology. 2009;30(4):544-54
39. Krumschnabel G, Ebner HL, Hess MW. et al. Apoptosis and necroptosis are induced in rainbow trout cell lines exposed to cadmium. Aquat Toxicol. 2010;99(1):73-85
40. Bialkowski K, Bialkowska A, Kasprzak KS. Cadmium (II), unlike nickel (II), inhibits 8-oxo-dGTPase activity and increases 8-oxo-dG level in DNA of the rat testis, a target organ for cadmium (II) carcinogenesis. Carcinogenesis. 1999;20:1621-1624
41. Singh KP, Kumari R, Pevey C, Jackson D, DuMond JW. Long duration exposure to cadmium leads to increased cell survival, decreased DNA repair capacity, and genomic instability in mouse testicular Leydig cells. Cancer Letters. 2009;279:84-92
42. Sieber OM, Heinimann K, Tomlinson IP. Genome instability - the engine of tumorigenesis? Nat Rev Cancer. 2003;3:701-708
43. Singh KP, DuMond JWJr. Genetic and epigenetic changes induced by chronic low dose exposure to arsenic of mouse testicular Leydig cells. Int J Oncol. 2007;30:253-260
44. Wenqian W, Fangqi L, Lei L. et al. Distinct mutations in MLH1 and MSH2 genes in Hereditary Non-polyposis Colorectal Cancer (HNPCC) families from China. BMB Report. 2011;44(5):317
45. Sheng JQ, Chan TL, Chan YW, Huang JS, Chen JG, Zhang MZ, Guo XL, Mu H, Chan AS, Li SR, Yuen ST, Leung SY. Microsatellite instability and novel mismatch repair gene mutations in northern Chinese population with Hereditary non-polyposis colorectal cancer. Chinese J Digest Dis. 2006;7:197-205
46. Lin Y, Wilson JH. Nucleotide excision repair, mismatch repair, and R-loops modulate convergent transcription-induced cell death and repeat instability. PLoS One. 2012;7(10):e46807
47. Mikhailova MV, Littlefield NA, Hass BS, Poirier LA, Chou MW. Cadmium-induced 8-hydroxydeoxyguanosine formation DNA strand breaks and antioxidant enzyme activities in lymphoblastoid cells. Cancer Lett. 1997;115:141-148
Author contact
![]() Corresponding author: Yixiong Lei, School of public health, Guangzhou Medical University, 195 Dongfengxi Road, Guangzhou 510182, People's Republic of China. Fax: +86-20-81340196. E-mail: gz-leizengcom
Corresponding author: Yixiong Lei, School of public health, Guangzhou Medical University, 195 Dongfengxi Road, Guangzhou 510182, People's Republic of China. Fax: +86-20-81340196. E-mail: gz-leizengcom