Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2008; 5(4):169-180. doi:10.7150/ijms.5.169 This issue Cite
Research Paper
Infliximab and Etanercept Are Equally Effective in Reducing Enterocyte APOPTOSIS in Experimental Colitis
Walter Fries1, Carmelo Muja2, Carmela Crisafulli2, Giuseppe Costantino1, Giuseppe Longo1, Salvatore Cuzzocrea2,3, Emanuela Mazzon2,3 ![]()
1. Dipartimento di Medicina Interna e Terapia Medica, Sezione di Farmacologia, Università di Messina, Messina, Italy.
2. Dipartimento Clinico-Sperimentale di Medicina e Farmacologia, Sezione di Farmacologia, Università di Messina, Messina, Italy.
3. IRCCS Centro Neurolesi “Bonino-Pulejo”, Messina, Italy.
Received 2008-3-30; Accepted 2008-7-1; Published 2008-7-3
Abstract
Loss of epithelial barrier integrity is considered an early step in the pathogenesis of Crohn's disease (CD), and the rate of enterocyte apoptosis is one of the determinants of the intestinal barrier function. Tumor necrosis factor-α (TNF-α), one of the major proinflammatory mediators in CD, is one of the extrinsic signals which initiate apoptosis of enterocytes. The aim of this study was to investigate the early effects of experimental colitis on enterocyte apoptosis, and the effects of two anti-TNF treatments, infliximab (IFX) and etanercept (ETC). In addition, the importance of receptor I for TNF was tested in TNFR-1-/- mice.
Circulating TNF-α levels were effectively reduced by IFX and ETC (p<0.01, both) at 3 and 6 h. Apoptosis of the ileal enterocytes, assessed by TUNEL staining, staining for Fas-ligand, and bax, increased at 3 and 6h. These alterations were prevented by both anti-TNF strategies, and in TNFR-1-/- animals. The anti-apoptotic protein Bcl-2 was expressed in the ileal epithelium under control conditions, but was suppressed in DNB-colitis. Expression of Bcl-2 was maintained in both anti-TNF treatments and TNFR-1-/- mice.
DNB colitis induced a very early, rapid increase of enterocyte apoptosis. Both anti-TNF strategies, IFX and ETC, were equally effective in suppressing enterocyte apoptosis, most likely by inactivation of circulating TNF-α.
Keywords: TNF-α, Enterocyte, Apoptosis, Experimental Colitis
INTRODUCTION
A defect of the gastrointestinal barrier function can be considered an important stage in the pathogenesis of Crohn's disease (CD), leading to increased penetration of luminal antigens in the gut wall, thus initiating a self-perpetuating immune response.
Whereas most research in the past few years has focused on the apical junctional complex between enterocytes [for review see ref 1-3], little attention has been given to the programmed cell death of enterocytes, i.e. apoptosis. Under normal conditions, epithelial cell shedding does not contribute to an alteration of the intestinal barrier [4], but an increase of the apoptotic rate may generate leaks contributing to a decrease in barrier function. It has been shown that tumor necrosis factor-α (TNF-α)-induced apoptosis contributes significantly to the loss of ions and water, and to the passage of small antigens [5].
In CD, an imbalance of apoptosis is reported, characterized by prolonged survival of lamina propria proinflammatory cells [6,7] together with a shortened survival of the epithelial cell lining [8]. With the recent introduction of biologic therapies, it has been shown that Infliximab, a chimeric mouse/human IgG1 antibody against TNF-α, normalizes apoptotic rates by increasing T-cell death [9] and decreasing epithelial cell apoptosis [5,10]. However, most research has concentrated on the regulatory effect of Infliximab (IFX), or its fully human analog adalimumab (ADA), on apoptosis of proinflammatory cells, such as monocytic cell lines and lamina propria T-cells, and this effect has been explained by the direct interaction of these antibodies with membrane-bound TNF. Etanercept (ETC), a synthetic fusion protein of a recombinant human TNF receptor subunit with a human IgG1, failed to show clinical efficacy [11] and appeared to be less effective at inducing apoptosis of peripheral blood monocytes and of lamina propria derived T-cells [12,13], although some activity in cell-dependent cytotoxicity and antibody-dependent cell-mediated cytoxicity has been reported recently [14]. Finally, some doubt about the importance of the pro-apotptotic effect in the mediation of clinical efficacy has been added recently after positive clinical studies with certolizumab, a pegylated humanized Fab' fragment of an IgG4 [15].
Since no information on the epithelial effects of the anti-TNF strategy is yet available, we designed the present study to investigate the very early effects of experimental colitis on ileal enterocyte apoptosis, induced by dinitrobenzenesulphonic acid (DNB), a model which is very close to the classical Th-1 model employing trinitrobenzenesulphonic acid (TNB). In this model, we have previously shown that both IFX and ETC, prevent alterations of enterocyte tight junctions [16]. In the present setting, we studied the effects of experimental colitis on ileal enterocyte apoptosis by means of TUNEL staining, immunohistochemistry for Fas-Ligand, the expression and immunostaining of bax and the anti-apoptotic protein bcl-2, and the effects of TNF-blockade by IFX and ETC. In an additional experiment, the results were compared to the effects of DNB colitis in knockout (k.o.) mice for the receptor I of TNF-α (TNFR-1-/-).
MATERIALS AND METHODS
All experiments were carried out in accordance with the national law on animal protection. Experimental protocols were approved by the Ethics Committee of the University of Messina. Male CD mice (Harlan, Italy) were housed for two weeks under standard conditions, with free access to tap water and standard lab chow. Colitis was induced by intrarectal administration of 5 mg of dinitrobenzenesulphonic acid (DNB) dissolved in 50% ethanol under light ether anesthesia. Control colitis was induced by intrarectal instillation of 50% ethanol only. Twenty-five mice per group were sacrificed at 3 and 6 hours after colitis induction. Blood was drawn by intracardiac puncture under ether anaesthesia, the abdomen was opened by a midline incision and the entire colon was removed, opened and macroscopic damage score determined [17]. Subsequently, the distal colon and terminal ileum were cut in longitudinal slices and fixed in buffered formalin.
In a second experiment, mice were treated one hour before colitis induction with Infliximab (IFX; Schering-Plough, Milan; Italy; 5 mg/kg i.p.) or with Etanercept (ETC; Wyeth, Milan; Italy; 5 mg/kg s.c.). In parallel, mice subjected to anti-TNF treatment with sham colitis and colitis with sham treatment served as controls with an identical protocol as above. Both treatments have been shown to effectively antagonize TNF effects in vivo in mice [18,19].
In a third experimental setting, in mice lacking the TNF membrane receptor I (TNFR-1-/-, Jackson Laboratory, Bar Harbor, MN; USA) colitis was induced as above and animals were sacrificed at 3 and 6 h.
Serum TNF-α determination
Blood was spun and serum stored at -80°C, until analysis. Serum concentrations of TNF-α were determined by ELISA (Euroclone, Devon; UK).
Histologic evaluation
After fixation for 1 week at room temperature in buffered formaldehyde solution (10% in phosphate buffered saline), samples were dehydrated in graded ethanol and embedded in paraplast (Sherwood Medical, Mahwah, NJ; USA). Thereafter, 7 μm sections were deparaffinized with xylene, stained hematoxylin/eosin, observed with a Axostar Plus equipped with AxioCam MRc (Zeiss, Milan, Italy) and studied using an Imaging computer program (AxioVision, Zeiss, Milan, Italy).
Immunohistochemical assessment of apoptosis
After deparaffinization, slices were treated with protease (type XIV, Sigma) (2 mg/ml) for 10 min at 37°C. Endogenous peroxidase was quenched with 0.3% (v/v) hydrogen peroxide in 60% (v/v) methanol for 30 min. Non-specific adsorption was minimized by incubating the section in 2% (v/v) normal goat serum in PBS for 20 min. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with biotin and avidin (DBA, Milan, Italy), respectively. Sections were incubated overnight with 1) polyclonal rabbit anti-Bcl-2 antibody (1:200 in PBS, w/v), 2) anti-bax (1:200, w/v) (anti-Bcl-2 and –bax antibodies, Santa Cruz Biotechnology, Inc., Santa Cruz, CA; USA), or 3) anti-Fas-L (1:50, w/v)(Novocastra Lab Ltd, Newcastle upon Tyne, UK). Sections were washed with PBS, and incubated with secondary antibody obtained from Jackson Immuno Research, Laboratories, INC (Jackson San Francisco, CA, USA).,Specific labeling was detected with a biotin-conjugated goat anti-rabbit IgG and avidin-biotin peroxidase complex (DBA, Milan, Italy). The counter stain was carried out with nuclear fast red (red background). All sections were observed using light microscopy (Axostar Plus equipped with AxioCam MRc, Zeiss, Milan, Italy) and studied using an Imaging computer program (AxioVision, Zeiss, Milan, Italy).
Terminal Deoxynucleotidyltransferase-Mediated UTP End Labelling (TUNEL) Assay
TUNEL assay was conducted by using a TUNEL detection kit according to the manufacturer's instruction (Apotag, HRP kit, Chemicon International, Milan, Italy). Briefly, sections were incubated with 20 μg/ml proteinase K for 15 min at room temperature and then washed with PBS. Endogenous peroxidase was inactivated by 3% H2O2 for 30 min at room temperature and then washed with PBS. Sections were incubated with Working Strength TdT Enzyme in a humid atmosphere at 37°C for 60 min, and then washed with stop wash buffer. The sections were incubated at room temperature for 30 min with anti-digoxigenin-peroxidase and the signals were visualized with diaminobenzidine. Sections were counterstained with methyl-green (green background) (Vector Lab, Milan; Italy).
Total protein extraction and Western blot analysis
Tissue samples from the terminal ileum were homogenized with an Ultra-turrax T8 homogenizer in a buffer containing 20mM HEPES pH 7.9, 1.5mM MgCl2, 400 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM ethyleneglycoltetraacetic acid (EGTA), 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulphonyl fluoride (PMSF), 1.5 μg/ml trypsin inhibitor, 3 μg/ml pepstatin, 2 μg/ml leupeptin, 40 μM benzidamin, 1% NP-40, 20% glycerol. The homogenates were centrifuged (13000 rpm, 15 min, 4°C); the surnatant was collected to evaluate contents.
Protein concentration was determined with the Bio-Rad protein assay kit. Proteins were mixed with gel loading buffer; (50 mM Tris, 10% (w/v) sodium dodecyl sulphate (SDS), 10% (w/v) glycerol, 10% (v/v) 2-mercaptoethanol, 2 mg/ml bromophenol), boiled for 5 min and centrifuged at 10000 rpm for a few seconds. Protein concentration was determined and equivalent amounts (50 μg) of each sample electrophoresed in a 12% (w/v) discontinuous polyacrylamide minigel. Proteins were separated electrophoretically and transferred to nitrocellulose membranes. For immunoblotting, membranes were blocked with 5% non-fat dry milk in Tris-buffered saline (TBS) for 1 h and then incubated with primary antibodies against Bcl-2 (1:100), bax (1:100), and β-actin (1:5000) (Santa Cruz Biotechnology, CA; USA) over-night at 4°C. The membranes were washed three times for 10 min in TBS with 0.1% Tween 20 and incubated with AffiniPure Goat Anti-Rabbit IgG coupled to peroxidase (1:5000). The immune complexes were visualized using the SuperSignal West Pico chemiluminescence Substrate (PIERCE, USA).
Data presentation and statistics
Data of circulating TNF concentrations, macroscopic damage score, and densitometric units (Western-blot) are given as mean values ± SEM; comparison was made with the Mann-Whitney test and Bonferroni's correction; a p value <0.025 was considered significant.
RESULTS
Macroscopic damage score (Table 1) and serum TNF-α (Table 2) are effectively reduced by anti-TNF treatment
DNBS colitis led to high circulating TNF-α levels at 3 and 6 hours post-induction, which were effectively reduced by IFX at 3h and 6 h (p<0.01, both) compared with untreated colitis, and only at 6 h by ETC (p<0.01). Macroscopic damage score was similar in all groups at 3h, but significantly (p<0.05) reduced in both treatment groups at 6 h compared to untreated colitis. At both time-points k.o. mice for TNFR-1 had a similar damage score to untreated animals.
macroscopic damage score 3 h and 6 h after colitis induction in untreated colitis (DNBS), colitis treated with IFX 5 mg/kg i.p. (DNBS + IFX), in colitis treated with ETC 5 mg/kg s.c., and in TNFR1 k.o. mice (TNFR-/-); * p<0.05 vs corresponding untreated colitis; unpaired t-test.
| Macroscopic damage score | ||
|---|---|---|
| 3 hours | 6 hours | |
| DNBS (n = 25) | 1.4 ± 0.2 | 2.2 ± 0.3 |
| DNBS + IFX (n = 25) | 1.2 ± 0.2 | 1.1 ± 0.4 * |
| DNBS + ETC (n = 25) | 1.9 ± 0.4 | 0.9 ± 0.4 * |
| TNFR-/- (n = 25) | 0.8 ± 0.1 | 1.4 ± 0.4 |
serum TNF-alpha concentrations at 3 h and 6 h in untreated colitis (DNBS), colitis treated with IFX 5 mg/kg i.p. (DNBS + IFX), and in colitis treated with ETC 5 mg/kg s.c.; * p<0.01 vs corresponding untreated colitis; unpaired t-test.
| Serum TNF-alpha; pg/ml | ||
|---|---|---|
| 3 hours | 6 hours | |
| DNBS (n = 25) | 149 ± 8 | 100 ± 2 |
| DNBS + IFX (n = 25) | 42 ± 2 * | 67 ± 6 * |
| DNBS + ETC (n = 25) | 110 ± 9 | 39 ± 9 * |
Enterocyte apoptosis in experimental colitis and effects of TNF- inhibition and lack of TNFR-1
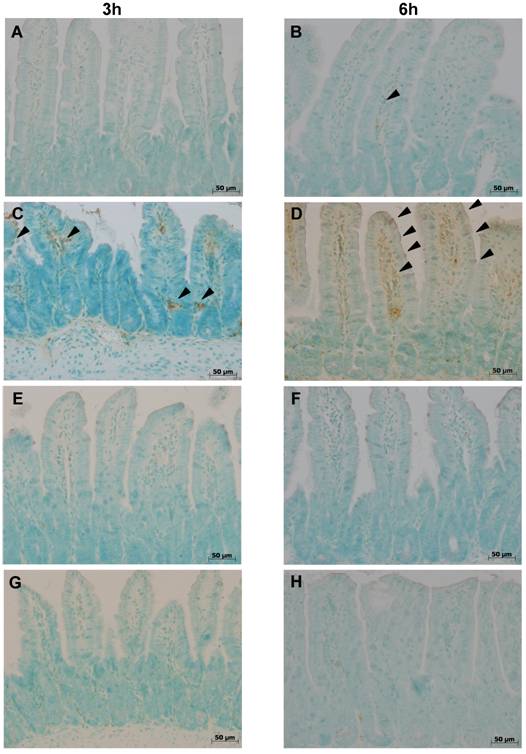
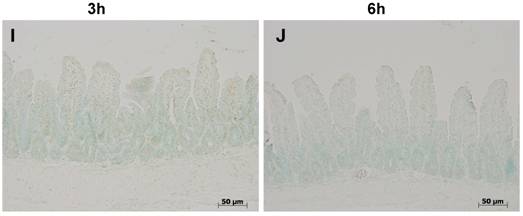
Isolated positivity was observed at any time-point in control ilea on TUNEL staining (fig.1), nor in sham colitis treated with ETC or IFX. Three hours after colitis induction a few cells distributed along the entire length of the villi stained positive (fig. 1 C). At 6 h, the number of apoptotic cells had increased (fig. 1 D). No TUNEL-positive cells were observed at either time-points with either treatment, ETC or IFX (fig. 1 E-H), nor in TNFR-1-/- mice (fig. 1 I,J).
Tunel staining at 3 h (left column) at 6 h (right column) in control ilea (A,B) showing no positivity except for isolated enterocytes at both time-points; positivity (arrowheads) appeared 3 h (B) after colitis induction with DNB/ethanol along the villus axis and increased at 6 h (C); positivity of Tunel staining was completely prevented by treatment with ETC (5 mg/kg s.c.) (E,F) and with IFX (G,H), and in TNFR-1-/- mice (I,J).
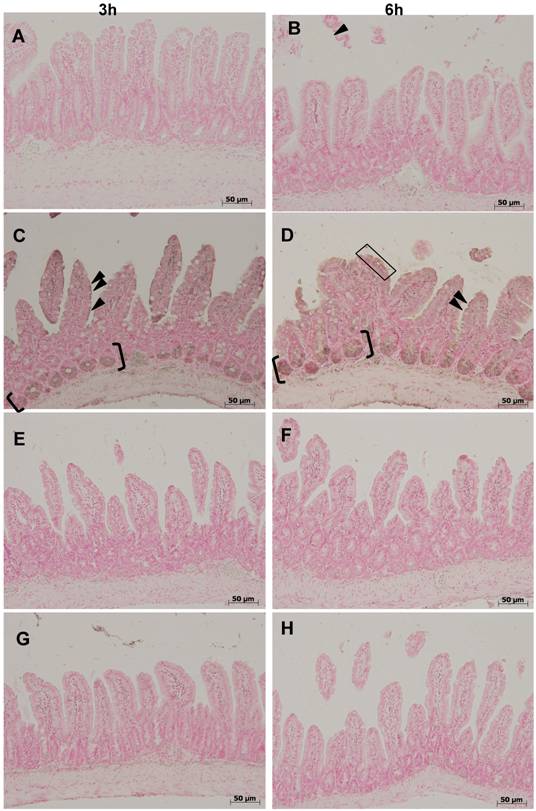
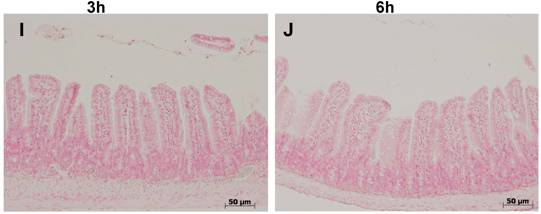
Fas-L (fig 2) was not detectable under control conditions in the ileal mucosa. Three and 6 hours after colitis induction, enterocyte nuclei and cytoplasm stained positive in the two upper thirds of the villi, whereas the lower third remained negative (fig. 2 B,C). Expression of Fas-L was absent at both time-points in colitis treated with ETC or IFX (fig. 2 E-H), and in TNFR-1-/- mice (fig. 2 I,J).
Immunostaining for Fas-L showed its absence under control conditions in the ileal mucosa (A,B). Three (C) and 6 hours (D) after colitis induction with DNB/ethanol, enterocyte nuclei and cytoplasm stained positive in the two upper thirds along the villi (arrowheads) (the rectangle indicate an area where almost all entercytes stained positive for Fas-L), whereas the lower third remained negative. Both treatments, ETC (5 mg/kg s.c.) (E,F) and IFX (5 mg/kg i.p.) (G,H), prevented positivity for Fas-L, as well as the absence of the receptor 1 for TNF-α (I,J).


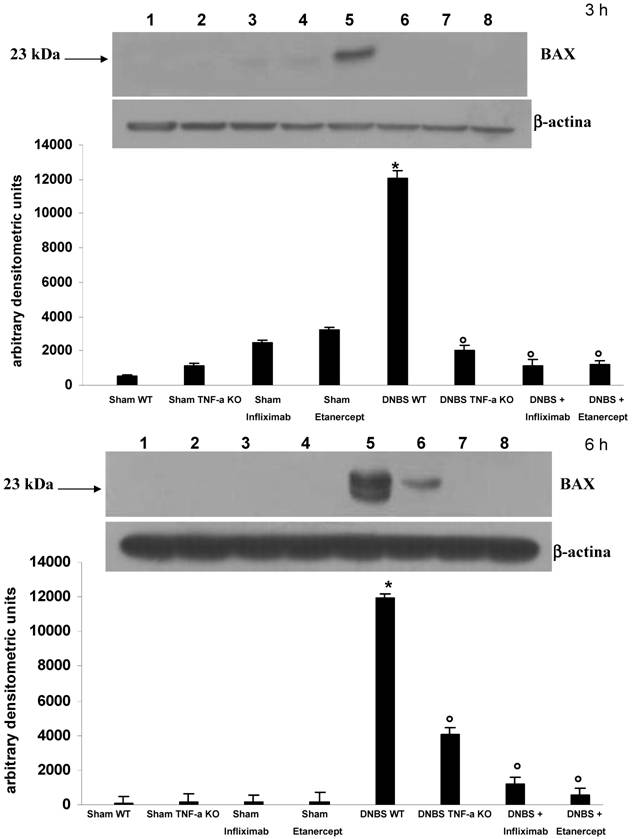
Like Fas-L, bax was also not detectable in controls (fig 3 A,B), but was increased in animals with colitis (fig. 3 C,D), especially in the crypt compartment. Treatment with ETC (fig 3 E,F) and with IFX (fig. 3 G,H) and the absence of the receptor-1 for TNF-α (fig. 3 I,J) prevented the expression of bax in the ileal enterocytes. On Western-blot analysis (fig. 4) bax was expressed strongly in the ileal mucosa of animals with colitis at 3 and 6h. This increased expression was effectively prevented by both IFX and ETC. In TNFR-1-/- mice with colitis, some expression of bax in the subepithelial layer was present.
Immunostaining for bax showed its absence under control conditions in the ileal mucosa (A,B). Three (C) and 6 hours (D) after colitis induction with DNB/ethanol, enterocyte nuclei and cytoplasm stained positive in the two upper thirds along the villi (arrowheads) and the crypt compartment (a short segment is highlighted within brackets). Both treatments, ETC (5 mg/kg s.c.) (E,F) and IFX (5 mg/kg i.p.) (G,H), prevented positivity for bax, as well as the absence of the receptor 1 for TNF-α (I,J).
Western-blot analysis of ileal concentrations of bax three hours (a; upper panel) and 6 hours (b; lower panel) in control animals with sham colitis (sham WT), in control TNFR-1-/- animals and sham colitis (sham TNF-α KO), in animals treated with ETC or IFX alone without colitis, in animals with DNBS/ethanol colitis and sham treatment (DNBS WT), in TNFR-1-/- animals with colitis (DNBS TNF-α KO), and in animals with colitis and ETC or IFX treatment; protein expression of bax was significantly increased in DNBS colitis compared with all other conditions. At 6 h in TNFR-1-/- animals with colitis traces of bax were detectable. These trace amounts were due to subepithelial bax expression.
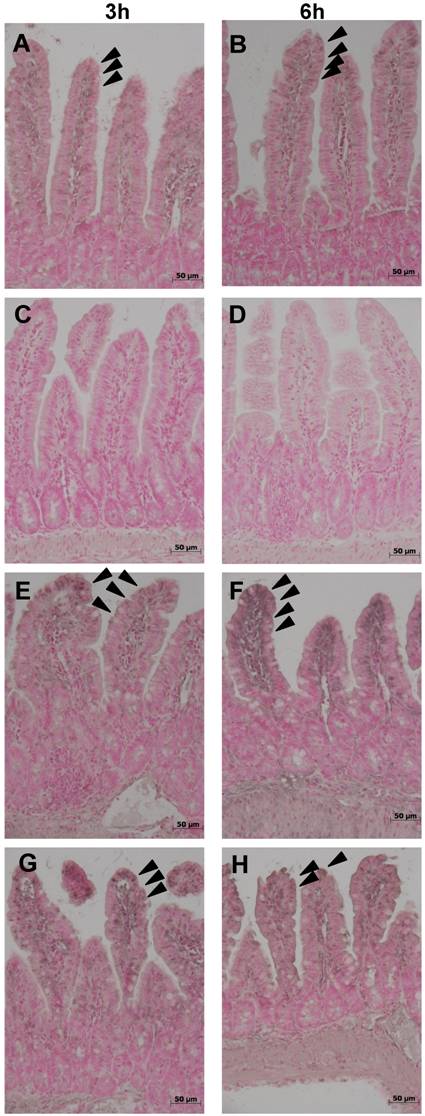
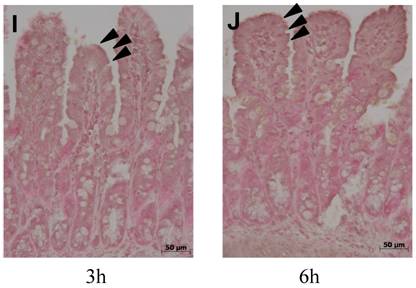
Expression of the anti-apoptotic protein bcl-2 (fig. 5) was evident under control conditions in the upper ¾ of ileal villi (fig. 5 A,B). Expression of bcl-2 was completely abolished in mice with experimental colitis, at both time-points (fig. 5 C,D). Both treatments, ETC and IFX, prevented the loss of bcl-2 at 3 and 6 h (fig. 5 E-H). Similarly, in TNFR-1-/- mice, bcl-2 was preserved at both time-points (fig. 5 I,J).
Expression of the anti-apoptotic protein Bcl-2 was evident under control conditions in the upper ¾ of ileal villi (arrowheads) (A,B), but was completely abolished by experimental colitis at both time-points (C,D). Both treatments, ETC (5 mg/kg s.c.) (E,F) and IFX (5 mg/kg i.p.) (G,H), prevented the loss of bcl-2; immunohistologic expression of the anti-apoptotic protein Bcl-2 was not altered in TNFR-1-/- mice after induction of colitis with DNB/ethanol at 3 (I) and 6 hours (J).
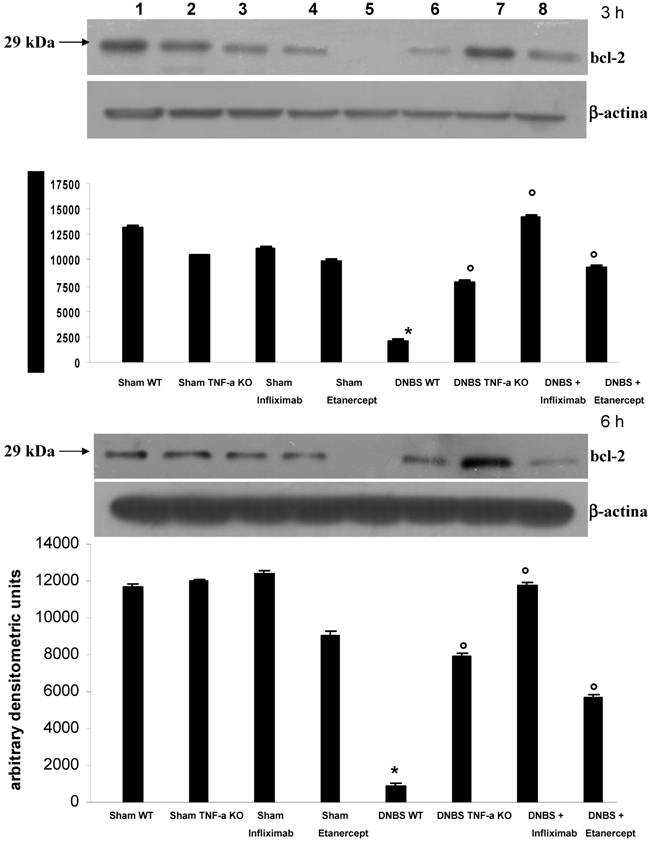
On Western-blot analysis, protein concentration of bcl-2 (fig. 6) was significantly reduced (p<0.01 or less) in mice with colitis compared to untreated sham colitis or sham colitis treated with IFX or ETC, but was preserved in mice with colitis, by either treatment, IFX and ETC, at 3 and 6 hours.
Western-blot analysis of ileal concentrations of Bcl-2 three hours (a; upper panel) and 6 hours (b; lower panel) in control animals with sham colitis (sham WT), in control TNFR-1-/- animals and sham colitis (sham TNF-α KO), in animals treated with ETC or IFX alone without colitis, in animals with DNBS/ethanol colitis and sham treatment (DNBS WT), in TNFR-1-/- animals with colitis (DNBS TNF-α KO), and in animals with colitis and ETC or IFX treatment; protein expression of Bcl-2 was significantly reduced in DNBS colitis compared with all other conditions.
DISCUSSION
In the present experiment, the short term effects of DNB colitis on apoptosis of the ileal enterocytes have been shown for the first time in a whole organism, thus underlining the evidence that this model may be used to study inflammation-associated effects on programmed cell death of enterocytes. Additionally, we have demonstrated that both anti-TNF treatments, ETC and IFX, were equally effective in abolishing colitis-induced enterocyte apoptosis, and we confirmed that the proapoptotic effect is mediated through receptor 1 for TNF- α.
At present, few studies have addressed the effect of TNF-α in vivo on epithelial cell apoptosis [20] and its role in chronic inflammatory bowel diseases (IBD), especially in non-inflamed gut segments. Most research has focused on actively inflamed mucosa in experimental models [21] as well as in human disease [5,10,22]. In a very recent paper, an upregulation of TNF-related apoptosis-inducing ligand (TRAIL), a membrane-bound protein with a very close homology to TNF-α and FasL, has been found on enterocytes of actively inflamed mucosa of patients with IBD[23]. TRAIL thus represents an additional pro-apoptotic mechanism in IBD. In the present paper, for the first time we show that epithelial cell apoptosis increased distantly from actively inflamed mucosa in this model of experimental colitis. Similar effects on enterocyte apoptosis have been described in models of experimental liver failure associated with high circulating TNF-α levels [24].
The interrelation between apoptosis and loss of barrier function has been addressed in some previous studies [25] concluding that the physiologic event does not lead to permeability defects, whereas apoptosis induced by exposure to cytokines such as TNF-α was found recently to disrupt barrier function in terms of epithelial conductivity [5]. From studies in several cell lines it appears that IFN-γ or TNF-α sensitize cells to Fas-mediated apoptosis [26,27].
In our study, enterocyte apoptosis was completely prevented by both anti-TNF treatments, and in TNFR-1-/- mice, together with a preserved expression of anti-apoptotic protein Bcl-2. Our findings agree both with a recent paper by Zeissig and coll [10] who investigated the effects of IFX treatment in vivo on colonic mucosa in CD patients showing a significant reduction of epithelial cell apoptosis, and with Marini et al [21] in Samp/YitFc mice, a murine model with spontaneous ileitis resembling human CD very closely, where the administration of a chimeric rat/mouse anti-TNF-α antibody reduced epithelial apoptosis in the inflamed ileum.
The nature of available anti-TNF strategies, ETC, onercept, IFX, and ADA is different. ETC and onercept are genetically engineered fusion proteins consisting of two molecules of the natural p75 or p55 TNF receptors and the Fc domain of human IgG1, whereas IFX and ADA are IgG1 antibodies against TNF-α, the former a chimeric mouse/human IgG1 antibody, the latter a fully human IgG1-antibody. The affinity of all these molecules is extremely high for circulating TNF-α; however, membrane-bound TNF is not, or less, recognized by ETC [28], whereas IFX and ADA also bind to this uncleaved form. This different TNF binding pattern may explain the distinct therapeutic effects of these anti–TNF drugs. Indeed, studies in Crohn's disease patients were disappointing with ETC [11], whereas the efficacy of IFX and ADA have been well documented.
A very important aspect is the ability of IFX, as well as of ADA, not only to bind both soluble and membrane-bound TNF but also to induce apoptosis of TNF-expressing cells. It has been shown that lamina propria T-cells survive too long in Crohn's disease [29]. This increased survival of pro-inflammatory cells is associated with the perpetuation of inflammatory stimuli and an increased production of proinflammatory cytokines, such as TNF-α. IFX induces apoptosis of peripheral blood monocytes from healthy volunteers and patients with CD dose-dependently by activation of caspase-8, 9 and 3 in a Fas-independent manner [30]. This effect has also been shown for ADA but not for ETC [13]. Subsequently, it was shown that IFX [9], but not ETC [12], induces apoptosis of lamina propria T cells in vivo and in a CD3/CD28-activated T-cell line. The pro-apoptotic effect of IFX was also shown by DiSabatino et al. [31] in vitro on lamina propria T cells and peripheral blood T cells from CD patients.
Beyond this important difference between ETC and IFX, other discrepancies have been described, e.g. the response of peripheral T cells, in terms of cytokine production (TNF-α, interleukin-4, and interferon-γ) to non-specific and antigen specific stimulation in patients with AS, is downregulated during IFX treatment but increased by ETC [32,33]. In vitro, the opposite effect was not confirmed on intestinal T cells from CD patients, but there was a fourfold reduction of efficacy of ETC compared to IFX [14, 34].
As shown in our present study, ETC and IFX were equally effective in abolishing the colitis-associated increase of ileal enterocytes and in the preservation of bcl-2 expression, suggesting an exclusive effect of circulating and not membrane-bound TNF-α. Moreover, the same results were achieved using k.o. mice for TNFR-1, confirming the mediation of propapototic stimuli through this receptor [35]. Interestingly, knock out mice for TNFR-1 showed a higher mortality in TNBS induced colitis [36], whereas those who lacked TNFR-2 developed a less severe disease despite higher serum TNF-α levels. This may be explained by secondary defence mechanisms mediated by TNFR-1.
In conclusion, induction of enterocyte apoptosis represents a very early response of the ileum mucosa to distal colitis induced by DNBS, emphasizing the highly dynamic aspect of programmed cell death. This effect was most likely mediated by circulating TNF-α since its inhibition or the lack of TNFR-1 completely prevented epithelial apoptosis. The upregulation of enterocyte apoptosis may represent a mucosal defence mechanism but, shooting down intestinal barrier function, the influx of antigens may be enhanced, inducing local inflammatory response in genetic susceptible organisms.
Our results show that intestinal segments distant from inflammation are highly sensitive, in terms of apoptosis and TJ protein expression/redistribution. In this context, it seems more likely that barrier dysfunction is a secondary event to inflammation.
Acknowledgements
This work was supported by ministerial grants (PRA 2003, Cofin 2004063577_003) (WF).
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Clayburgh DR, Shen L, Turner JR. A porous defence: the leaky epithelial barrier in ontestinal disease. Lab Invest. 2004;84:282-291
2. Bruewer M, Samarin S, Nusrat A. Inflammatory Bowel Disease and the apical junctional complex. Ann NY Acad Sci. 2006;1072:242-252
3. Arrieta MC, Bistritz L, Meddings JB. Alterations in intestinal permeability. Gut. 2006;55:1512-1520
4. Watson AJM, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, McKenna M, Rose T, Montrose MH. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129:902-912
5. Schulzke JD, Bojarski C, Zeissig S, Heller F, Bitter AH, Fromm M. Disrupted barrier function through epithelial cell apoptosis. Ann NY Acad Sci. 2006;1072:288-299
6. Peppelenbosch MP, van Deventer SJH. T-cell apoptosis and inflammatory bowel disease. Gut. 2004;53:1556-1558
7. Di Sabatino A, Corazza GR. Surviving too long in Crohn's disease. Gut. 2001;49:6-8
8. Di Sabatino A, Ciccocioppo R, Luinetti O, Ricevuti L, Morera R, Cifone MG, Solcia E, Corazza GR. Increased enterocyte apoptosis in inflamed areas of Crohn's disease. Dis Colon Rectum. 2003;46:1498-1507
9. Ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJH. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's disease. Gut. 2002;50:206-211
10. Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Downregulatuion of epithelial apoptosis and barrier repair in active Crohn's disease by tumor necrosis factor α antibody treatment. Gut. 2004;53:1295-1302
11. Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, Tremaine WJ, Johnson T, Diehl NN, Zinsmeister AR. Etanercept for active Crohn's disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121:1088-1094
12. Van den Brande JMH, Braat H, van den Brink GR, Versteeg HH, Bauer C.A, Hoedemaeker I, van Monfrans C, Hommes DW, Peppelenbosch MP, van Deventer SJH. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn's disease. Gastroenterology. 2003;124:1774-1785
13. Shen C, van Assche G, Colpaert S, Maerten P, Geboes K, Rutgeerts P, Ceuppens JL. Adalimumab induces apoptosis of human monocytes: a comparative study with infliximab and etanercept. Aliment Pharmacol Ther. 2005;21:251-258
14. Nesbitt A, Fossati G, Bergin M, Stephens P, Stephens S, Foulkes R, Brown D, Robinson M, Bourne T. Mechanism of action of Certolizumab Pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor α agents. Inflamm Bowel Dis. 2007;13:1323-1332
15. Sandborn WJ, Feagan BG, Stoinov S, Honiball PJ, Rutgeerts P, Mason D, Bloomfield R, Schreiber S. Certolizumab Pegol for the Treatment of Crohn's Disease. N Engl J Med. 2007;357:228-238
16. Fries W, Muja C, Crisafulli C, Cuzzocrea S, Mazzon E. Dynamics of enterocyte tight junctions - effect of experimental colitis and two different anti-TNF strategies. Am J Physiol Gastrointest Liver Physiol. 2008;294:G938-G947
17. Wallace JL, Keenan CM, Gale D, Shoupe TS. Exacerbation of experimental colitis by non-steroidal anti-inflammatory drugs is not related to elevated leukotriene B4 synthesis. Gastroenterology. 1992;102:18-27
18. Siegel SA, Shealy DJ, Nakada MT, Le J, Woulfe DS, Probert L, Kollias G, Ghrayeb J, Vilcek J, Daddona PE. The mouse/human chimeric monoclonal antibody cA2 neutralizes TNF in vitro and protects transgenic mice from cachexia and TNF lethality in vivo. Cytokine. 1995;7:15-25
19. Genovese T, Mazzon E, Crisafulli C, di Paola R, Muia C, Bramanti P, Cuzzocrea S. Immunomodulatory effects of etanercept in an experimental model of spinal cord injury. J Pharmacol Exp Ther. 2006;316:1006-1016
20. Garside P, Bunce C, Tomlinson RC, Nichols BL, Mowat AM. Analysis of enteropathy induced by tumour necrosis factor α. Cytokine. 1993;5:24-30
21. Marini M, Bamias G, River-Nieves J, Moskaluk CA, Hoang SB, Ross WG, Pizarro TT, Cominelli F. TNF-α neutralization ameliorates the severity of murine Crohn's-like ileitis by abrogation of intestinal epithelial cell apoptosis. PNAS. 2003;100:8366-8371
22. Iwamoto M, Koji I, Makiyama K, Kobayashi N, Nakane PK. Apoptosis of crypt epithelial cells in ulcerative colitis. J Pathol. 1996;180:152-159
23. Begue B, Wajant H, Bambou JC, Dubuquoy L, Siegmund D, Beaulieu JF, Canioni D, Berrebi D, Brousse N, Desreumaux P, Schmitz J, Lentze MJ, Goulet O, Cerf-Bensussan N, Ruemmele FM. Implication of TNF-related apoptosis inducing ligand in inflammatory intestinal epithelial lesions. Gastroenterology. 2006;130:1962-1974
24. Song HL, Lu S, Liu P. Tumor necrosis factor-alpha induces apoptosis of enterocytes in mice with acute hepatic failure. World J Gastroenterol. 2005;11:3701-3709
25. Abreu MT, Palladino AA, Ernold ET, Kwon RS, McRoberts JAM. Modulation of barrier function during Fas-mediated apoptosis in human intestinal epithelial cells. Gastroenterology. 2000;119:1524-1536
26. O'Connell J, Bennett MW, Nally K, O'Sullivan GC, Collins JK, Shanahan F. Interferon-γ sensitizes colonic epithelial cell lines to physiological and therapeutic inducers of colonocyte apoptosis. J Cell Physiol. 2000;185:331-338
27. Ruemmele FM, Russo P, Beaulieu J, Dionne S, Levy E, Lentze MJ, Seidman EG. Susceptibility to Fas-induced apoptosis in human nontumoral enterocytes: role of costimulatory factors. J Cell Physiol. 1999;181:45-54
28. Scallon BF, Moore MA, Trinh H, Knight DM, Ghrayeb J. Chimeric anti-TNF-alpha monoclonal antibody cA2 binds recombinant transmembrane TNF-alpha and activates immune effector functions. Cytokine. 1995;7:251-259
29. Ina K, Itoh J, Fukushima K, Kusugami K, Yamaguchi T, Kyokane K, Imada A, Binion DG, Musso A, West GA, Dobrea GM, McCormick TS, Lapetina EG, Levine AD, Ottaway CA, Fiocchi C. Resistance of Crohn's disease T cells to multiple apoptotic signals is associated with a Bcl-2/Bax mucosal imbalance. J Immunol. 1999;163:1081-1090
30. Lűgering A, Schmidt M, Lűgering N, Pauels HG, Domschke W, Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn's disease by using a caspase dependent pathway. Gastroenterology. 2001;121:1145-1157
31. Di Sabatino A, Ciccocioppo R, Cinque B, Millimaggi D, Morera R, Ricevuti L, Cifone MG, Corazza GR. Defective mucosal T cell death is sustainably reverted by infliximab in a caspase dependent pathway in Crohn's disease. Gut. 2004;53:70-77
32. Zou J, Rudwaleit M, Brandt J, Thiel A, Braun J, Sieper J. Down-regulation of the non-specific and antigen-specific T cell cytokine response in ankylosing spondylitis during treatment with infliximab. Arthritis Rheum. 2003;48:780-790
33. Zou J, Rudwaleit M, Brandt J, Thiel A, Braun J, Sieper J. Up regulation of the production of tumour necrosis factor alpha and interferon gamma by T cells in ankylosing spondylitis during treatment with etanercept. Arthritis Rheum. 2003;62:561-564
34. Agnholt J, Dahlerup JF, Kaltoft K. The effect of etanercept and infliximab on the production of tumour necrosis factor alpha, interferon gamma and GM-CSF in in vivo activated intestinal T lymphocyte cultures. Cytokine. 2003;23:76-85
35. Piguet PF, Vesin C, Guo J, Donati Y, Barazzone C. TNF-induced enterocyte apoptosis in mice is mediated by the TNF- receptor 1 and does not require p53. Eur J Immunol. 1998;28:3499-2505
36. Ebach DR, Newberry R, Stenson WF. Differential role of tumor necrosis factor receptors in TNBS colitis. Inflamm Bowel Dis. 2005;11:533-540
Author contact
![]() Correspondence to: Walter Fries, MD, Dipartimento di Medicina Interna e Terapia Medica, Policlinico Universitario, Pad C – III piano, Via Consolare Valeria, 1, 98100 Messina – Italy. Tel.: ++39/090/2212373; fax: ++39/090/2935162; e-mail: fwalterit
Correspondence to: Walter Fries, MD, Dipartimento di Medicina Interna e Terapia Medica, Policlinico Universitario, Pad C – III piano, Via Consolare Valeria, 1, 98100 Messina – Italy. Tel.: ++39/090/2212373; fax: ++39/090/2935162; e-mail: fwalterit