Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Ferroptosis
The role of ferroptosis in...
Conclusions and future...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(9):2119-2131. doi:10.7150/ijms.107057 This issue Cite
Review
Novel insights into the role of ferroptosis in temporomandibular joint osteoarthritis and knee osteoarthritis
Yuxin Zhang1,2# ![]() , Dahe Zhang1#, Xiaoyu Liao3, Qingyu Xu1, Lingtong Bu1, Jisi Zheng1, Pei Shen1
, Dahe Zhang1#, Xiaoyu Liao3, Qingyu Xu1, Lingtong Bu1, Jisi Zheng1, Pei Shen1 ![]() , Chi Yang1
, Chi Yang1 ![]()
1. Department of Oral Surgery, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine; College of Stomatology, Shanghai Jiao Tong University; National Center for Stomatology; National Clinical Research Center for Oral Diseases; Shanghai Key Laboratory of Stomatology; Shanghai Research Institute of Stomatology; Research Unit of Oral and Maxillofacial Regenerative Medicine, Chinese Academy of Medical Sciences, Shanghai 200011, China.
2. Shanghai Key Laboratory of Orthopedic Implants, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200011, China.
3. Department of Rehabilitation Medicine, the Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital, Suzhou 215008, China.
#These authors contributed equally to this work.
Received 2024-11-16; Accepted 2025-3-18; Published 2025-4-9
Abstract

Osteoarthritis (OA) is a prevalent degenerative joint disease characterized by pain, limited movement, and joint stiffness, significantly impacting the quality of life and imposing substantial economic burdens. This review paper delves into the novel insights of ferroptosis, an iron-dependent form of cell death associated with lipid peroxidation, in the context of temporomandibular joint osteoarthritis (TMJ OA) and knee osteoarthritis (KOA). We explore the pathogenic characteristics of OA, including synovitis, chondrocyte death, and extracellular matrix (ECM) degradation, and discuss the limitations of current therapeutic interventions. Emerging evidence suggests a significant relationship between ferroptosis and OA, with iron accumulation and lipid peroxidation observed in osteoarthritic cartilage. This review highlights the role of ferroptosis in chondrocyte malfunction and apoptosis, inflammation, and extracellular matrix breakdown, which are central to OA pathogenesis. We also discuss potential therapeutic targets, such as Transient Receptor Potential Vanilloid 1 (TRPV1), Glutathione Peroxidase 4 (GPX4), and Nuclear Factor Erythroid 2-Related Factor 2 (NRF2), which modulate ferroptosis and OA progression. The paper consolidates studies on ferroptosis in OA, offering a comprehensive understanding of its role and the development of innovative therapies targeting this cell death mechanism to improve treatment outcomes for OA patients.
Keywords: ferroptosis, temporomandibular joint osteoarthritis, knee osteoarthritis, mechanism, iron
Introduction
More than 22 percent of adults over the age of 40 suffer from osteoarthritis (OA), a degenerative joint disease which causes pain, limited movement, as well as stiffness in the affected joints [1-3]. Aside from negatively impacting people's quality of life, this ailment also places a heavy financial strain on families and society as a whole [1-3]. The pathogenic characteristics of OA include synovitis, chondrocyte death, deterioration of the extracellular matrix (ECM), subchondral osteosclerosis, and osteophyte formation [4], resulting in elevated incidence and disability rates [5]. Approximately 240 million persons globally are afflicted with symptomatic OA, with a prevalence incidence of 70% to 90% among those over 75 years of age [6,7]. It is estimated that global case numbers for each site of OA will increase by 48.6% to 95.1% between 2020 and 2050 [8], underscoring the pressing need for better therapies [9,10]. Current conservative interventions for OA, such as physical workouts and oral nonsteroidal anti-inflammatory drugs (NSAIDs), seek to mitigate the disease's development during its early to mid-stages [11]. Nonetheless, these therapies possess drawbacks, notably the considerable toxicity linked to NSAIDs, which may result in gastrointestinal complications and reduced renal blood flow [12,13]. In cases when patients are resistant to NSAIDs, intra-articular corticosteroid injections are contemplated; nevertheless, their use is contentious owing to the possibility of hastening cartilage degeneration [14].
In this context, there is increasing interest in elucidating the significance of ferroptosis, a newly founded mechanism of cell death, exhibiting morphological characteristics such as mitochondrial shrinkage and abnormalities in cristae [15]. Ferroptosis has been associated with multiple disciplines, including cardiology [16], hepatology [17], nephrology [18,19], oncology [20], and musculoskeletal research [21]. Recent studies have underscored its significant relationship with OA, demonstrating iron accumulation and lipid peroxidation in osteoarthritic cartilage [22,23]. The possible involvement of ferroptosis in the pathogenesis of OA is complex. Lipid peroxidation mediated by ferroptosis may result in chondrocyte malfunction and apoptosis, hence hastening cartilage deterioration [19]. Elevated inflammatory markers in OA may trigger ferroptosis [24], hence perpetuating a detrimental cycle that intensifies joint inflammation and cartilage degradation [25]. Ferroptosis may further affect the production of inflammatory factors in synovial fibroblasts, hence hastening the breakdown of the ECM of chondrocytes [26]. Considering the pivotal function of ferroptosis in OA, targets such as transient receptor potential vanilloid 1 (TRPV1) [27,28], glutathione peroxidase 4 (GPX4) [29], nuclear factor erythroid 2-related factor 2 (NRF2) [30], acyl-CoA synthetase long-chain family member 4 (ACSL4) [31], and nuclear coactivator adaptor 4 (NCOA4) [32] have surfaced as prospective regulators of ferroptosis and modulators of OA advancement. Ferroptosis is an iron-dependent mechanism of controlled cell death marked by the formation of lipid hydroperoxides [33,34]. The significance of iron buildup in inflamed tissue, resulting in cellular toxicity, in autoimmune and autoinflammatory illnesses associated with arthritis, such as OA, remains ambiguous [35]. The hypoxic environment and the promotion of angiogenesis by hypoxia-inducible factors indicate that oxidative stress may significantly influence synovial tissue [36]. The correlation between inflammation and ferroptosis has been investigated, since ferroptosis is associated with the production of pro-inflammatory chemicals, including IL-1β and IL-18 [37], positioning it as a possible therapeutic target for arthritis-related disorders.
Articular cartilage, mostly consisting of chondrocytes and ECM, is the primary locus of basic aetiology of OA [38]. Pro-inflammatory cytokines activate chondrocytes to release catabolic factors, which impede the production of COL2, proteoglycans, and other synthetic metabolic components, hence facilitating chondrocyte catabolic processes and the degradation of articular cartilage [39,40]. Chronic inflammation, oxidative stress, and ferroptosis are thought to induce chondrocytes to excessively generate catabolic factors, resulting in the degradation of chondrocytes and ECM, hence accelerating the degeneration of articular cartilage in OA [41-46]. Ferroptosis is characterised by increased reactive oxygen species (ROS), lipid peroxidation, iron accumulation, as well as depletion of glutathione (GSH) [47], and it has been seen in chronic bone disorders, such as OA [48-50]. The buildup of ROS resulting in lipid peroxidation and initiation of ferroptosis in chondrocytes aggravates OA. Augmenting the body's antioxidant capacity and removing surplus ROS may constitute an effective treatment strategy for addressing OA. This review seeks to consolidate ferroptosis studies in OA, providing insights into the targeting of its critical elements for innovative OA therapies.
Ferroptosis
Definition and characteristics of ferroptosis
Ferroptosis, introduced by Dixon in 2012, is an iron-dependent form of programmed cell death (PCD) that primarily impacts cellular organelle structures, particularly mitochondrial architecture, and is marked by the buildup of lipid peroxides [51,52]. The morphological characteristics include rupture of mitochondrial cristae, decrease or absence of mitochondria, degradation of the mitochondrial outer membrane, and mitochondrial shrinkage [53,54]. The metabolic features include iron overload, ROS buildup, and the inactivation of the antioxidant enzyme GPX4 [51,55].
Molecular mechanisms of ferroptosis
The role of GPX4 and system Xc-/GSH/GPX4 regulatory axis in ferroptosis
The inactivity of the lipid repair enzyme GPX4 may result in elevated levels of lipid-based ROS, hence triggering ferroptosis [56]. The system Xc-/GSH/GPX4 regulatory axis is regarded as the classical antioxidant regulatory axis of ferroptosis. The system Xc- is an upstream molecule in the axis, functioning as a sodium-independent reverse transporter. The primary function of it is to transport intracellular glutamate and extracellular cystine in a 1:1 ratio [57]. Cystine is transformed into cysteine (Cys) by a redox reaction upon entering the cell, then condensing with glutamic acid (Glu) and glycine (Gly) to produce GSH [58]. The body facilitates the conversion of oxidised glutathione (GSSG) to reduced GSH via GPX4, therefore safeguarding cells against peroxidative damage [59]. Although the GPX4 system is the primary route, it is not the exclusive mechanism; GPX4-independent pathways, including FSP1/CoQ10, DHODH, and GCH1/BH4, all contribute significantly [56,60-62] (Figure 1).
Molecular mechanisms of ferroptosis in chondrocytes.
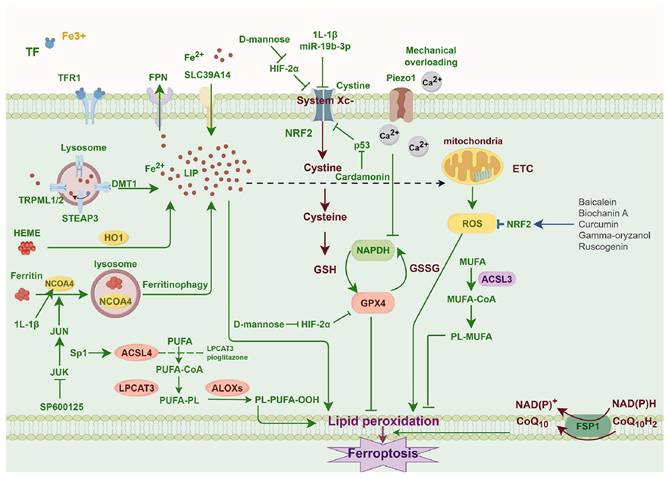
Lipid peroxidation in ferroptosis
Lipid peroxidation principally involving the oxidative degradation of polyunsaturated fatty acids (PUFAs) in membrane phospholipids, in conjunction with reactive oxygen species. It may occur by enzymatic mechanisms involving lipoxygenases (LOXs), cyclooxygenases (COXs), and ACSL4, or through nonenzymatic processes driven by free radicals [63]. One of them, ACSL4, is involved in lipid metabolism and may catalyse the conversion of PUFAs; this process can cause ferroptosis and an overabundance of lipid ROS [64]. Lipid radicals react with oxygen molecules to shape lipid peroxide radicals (LOO•) and lipid hydroperoxides (LOOH), which in turn obtain hydrogen atoms from other PUFAs. This chain reaction continues until oxidative products like hydroxyl radicals (OH•) and hydrogen peroxide (H2O2) are formed. Lipid peroxidation of PUFAs is characterised by a cascade reaction because LOO• may react with PUFAs continually. Peroxidation compounds including lipid peroxide (LPO) and malondialdehyde (MDA) may be produced when PUFAs degrade [65]. In addition to destroying the cell membrane's fluidity and stability, LOOH may enhance the membrane's permeability, which in turn causes lipid membrane rupture and, eventually, ferroptosis [63,66,67].
Iron overload in ferroptosis
A further hallmark of ferroptosis is the accumulation of iron. In physiological settings, iron primarily engages in biological activities like oxygen transport, adenosine triphosphate (ATP) production, and immunological modulation [68,69]. The regulation of iron balance is mostly achieved by the synthesis and release of iron homeostasis regulators by hepatocytes, such as transferrin receptor (TFR), ferritin, and ferroportin (FPN) [68-70]. In pathological settings, iron excess elevates the generation of oxygen free radicals, which subsequently exacerbates the oxidation and destruction of many cellular components [71].
Role of mitochondria in ferroptosis
The current dominant opinion maintains that mitochondria are involved in ferroptosis and create ROS, since mitoROS is primarily produced by oxidative phosphorylation complexes [72]. MitoROS have the potential in causing mitochondrial membrane lipid peroxidation [73]. This is corroborated by the fact that lipid-ROS first localised to the mitochondrial distribution in ferroptosis triggered by cystine deprivation and erastin [70]. Mitochondria not only promote ferroptosis via generation of ROS but also appear to inhibit it through consuming glutamate, counteracting the suppressive influence from elevated extracellular glutamate upon the Xc system [74]. The causal link between mitochondrial morphological alterations and ferroptosis remains ambiguous, despite the tight association of mitochondria with ROS generation and iron metabolism [59,70,75].
The role of ferroptosis in temporomandibular joint osteoarthritis (TMJ OA) and knee osteoarthritis (KOA)
The pathogenic role of ferroptosis in TMJ OA and KOA
OA is a chronic degenerative condition increasingly acknowledged for its intricate pathophysiology, which encompasses genetic, biomechanical, and metabolic components. A notable aspect that has lately garnered attention is ferroptosis, an iron-dependent variant of PCD that differs from apoptosis and other cell death modalities [76]. This review seeks to encapsulate the contemporary comprehension of ferroptosis's function in the aetiology of OA, emphasising its metabolic control and the ramifications for treatment strategies.
Ferroptosis is defined by the buildup of lipid peroxides, causing the breakdown of plasma and organelle membranes, finally lead to cellular death [77]. Redox-active iron significantly accelerates lipid peroxidation events, whereas the glutathione-dependent antioxidant defence system, especially GPX4, is essential in inhibiting lipid peroxidation [78]. The cystine/GSH/GPX4 system is a conventional method for inhibiting iron removal, with system Xc-, being vital for the production of GSH, a key antioxidant defence route [78]. The beginning and advancement of ferroptosis are primarily governed by lipid ROS, ferrous iron, and GPX4 activity, where increased ferrous iron levels and reduced GPX4 activity signify the commencement of ferroptosis [79].
Ferroptosis has been associated with reduced chondrocyte activity in OA, with new studies investigating the pathogenic pathways leading to chondrocyte mortality in this situation [76]. The tumour suppressor gene p53 inhibits the expression of SLC7A11, leading to reduced GSH production and the suppression of GPX4. It eventually induces ferroptosis in many cell lines and maybe in chondrocytes as well [80]. Inflammatory cytokines, including IL-1β/TNF-α, stimulate the expression of transferrin receptors, enhancing iron absorption and buildup, hence facilitating ferroptosis in inflammatory contexts [81]. Disruption of mitochondrial membranes elevates cytoplasmic iron concentrations and impairs oxidative phosphorylation, leading to the production of ROS and exacerbating iron-induced cell death [81]. Research indicates that therapies using the iron chelator deferoxamine (DFX) may alleviate the advancement of OA, underscoring the significance of iron in the condition [82]. Hypoxia-inducible factor 2α (HIF-2α) was shown to significantly influence cartilage formation, the development of OA, and cellular resistance to ferritin [83]. Investigations applying IL-1β to stimulate chondrocytes within the OA environment have shown an up-regulation of HIF-1α and LPO levels, with a down-regulation of GSH levels in chondrocytes subjected to IL-1β stimulation [84]. Patients with OA exhibit elevated iron levels in synovial tissue, and the impairment of iron homeostasis-regulating genes causes cellular iron accumulation, resulting in heightened production of ROS as well as the increased level of collagen II and MMP13, which are established hallmarks of OA [85] (Figure 2).
The mechanism for ferroptosis in OA.
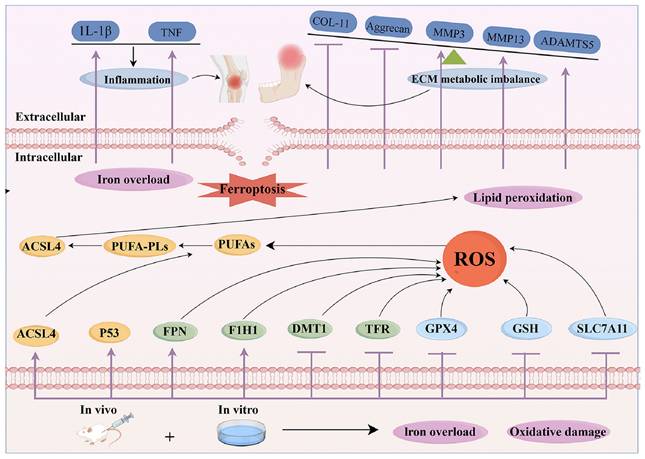
The pathogenic mechanism of OA include damage to periarticular cartilage and subchondral bone, synovitis, and the critical involvement of chondrocytes in this process. In individuals with OA, the iron content in synovial fluid is elevated, and greater serum ferritin levels correlate with increased radiographic severity of the joints compared to healthy controls [86,87]. Chondrocyte ferroptosis facilitates the advancement of OA, as shown by both in vivo and in vitro studies [88]. Crucial enzymes, including ACSL4 and LPCAT3, are essential to the biosynthesis of PUFA-Pls, which serve as substrates for LPO [89,90]. The cumulative consequences of these processes induce lipid peroxidation, eventually causing ferroptosis of chondrocytes in OA (Figure 3).
Ferroptosis in TMJ OA and KOA. ACSL4, acylCoA synthetase long-chain family member 4; DMT1, ferrous ion membrane transport protein DMT1; LIP, intracellular labile iron pool; FPN, ferroportin; LPCAT3, lysophosphatidylcholine acyltransferase 3; GCL, glutamate cysteine ligase; GPX4, glutathione peroxidase 4; GSH, glutathione; GSS, glutathione synthetase; GSSG, oxidized glutathione; PUFA, polyunsaturated fatty acid; PL, phospholipid; STEAP3, metalloreductase STEAP3; SOD, superoxide dismutase; Tf, transferrin; TFR1, transferrin receptor protein 1.
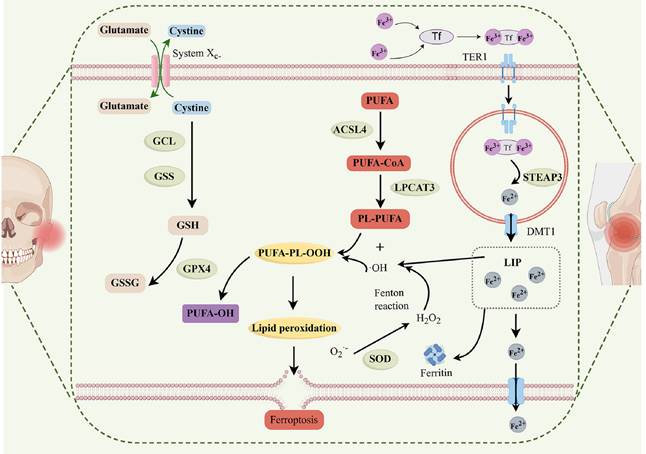
Using high-resolution mass spectrometry, Zou et al. completed a thorough analysis of the protein expression patterns in synovial fluid of patients with temporomandibular joint (TMJ) anterior disc displacement (ADD) [91]. The research revealed notable elevations in proteins including FGL2, THBS4, and TNC in ADD without OA, signifying active ECM and collagen metabolism, whereas FGFR1, FBLN2, and GRB2 were heightened in ADD with OA, implying a role in ferroptosis. Moreover, proteins such as P4HB and CBLN4 exhibited persistent overexpression throughout the development of the illness. These results elucidate the molecular processes of TMJ OA and provide possible treatment targets [91]. Ferroptosis is essential in chondrocytes and is more susceptible to cellular damage due to heightened buildup. This vulnerability is linked to inflammatory mediators like IL-1β and IL-6 that were shown to trigger ferroptosis in OA. The exact mechanism of ferroptosis in OA, coupled with discovery of possible targets in the ferroptosis pathway, is essential to improve therapeutic approaches for persons suffering from OA.
The ferroptosis-based strategies in TMJ OA and KOA
OA is marked by cartilage deterioration, synovial inflammation, and alterations in subchondral bone. Despite comprehensive research, effective therapies for OA remain constrained, especially in preventing or reversing disease progression. Recent studies have emphasised the significance of ferroptosis, a kind of PCD characterised by iron-dependent lipid peroxidation, in the aetiology of OA. This review encapsulates the current comprehension of ferroptosis in OA and examines prospective treatment approaches aimed at this pathway (Figure 4).
The ferroptosis-based strategies in TMJ OA and KOA.
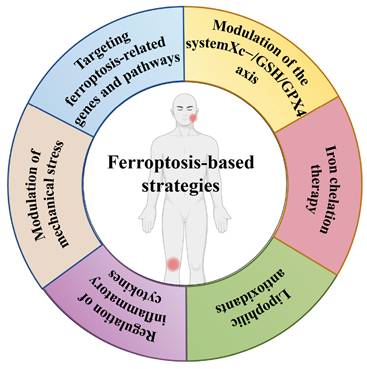
Ferroptosis and chondrocyte death
Chondrocytes, the intrinsic cells of cartilage, are essential for preserving cartilage homeostasis. In OA, chondrocytes experience substantial alterations, including senescence, apoptosis, and necrosis. Ferroptosis has become a notable factor in chondrocyte apoptosis in OA. Recent studies indicate that ferroptosis indicators, including GPX4 and SLC7A11, are reduced in osteoarthritic chondrocytes, although lipid peroxidation and iron buildup are elevated [92,93]. These alterations indicate that ferroptosis plays a significant role in chondrocyte apoptosis and cartilage deterioration in OA.
Ferroptosis-related genes and pathways
Ferroptosis regulation in osteoarthritic chondrocytes has been linked to various genes and mechanisms. The system Xc-/GSH/GPX4 regulation axis, crucial for sustaining cellular redox equilibrium, is impaired in OA. The downregulation of SLC7A11, a component of system Xc-, results in diminished GSH production and GPX4 activity, hence increasing chondrocyte vulnerability to ferroptosis [92,93]. Moreover, miRNAs like miR-19b-3p have shown the ability to induce ferroptosis in osteoarthritic chondrocytes by targeting SLC7A11 [92]. Pang et al. examine the function of GPX4 and ferroptosis in the advancement of TMJ OA [94]. The authors show, via the examination of patient samples and animal models, that ferroptosis is triggered in TMJ OA tissues and synovial fluid, as indicated by elevated levels of ferroptosis markers and diminished antioxidant system function. GPX4 expression is significantly reduced in TMJ OA, facilitating the induction of ferroptosis [94]. The research indicates that the inhibition of ferroptosis, either by GPX4 overexpression or the use of ferroptosis inhibitors, markedly improves the pathological alterations in TMJ OA in rat models. Overexpression of GPX4 enhances cell viability by suppressing ferroptosis and increasing the activity of the antioxidant system. The results suggest that ferroptosis and GPX4 targeting might be an innovative treatment approach for the therapy of TMJ OA [94]. Additionally, inflammatory cytokines, including IL-1β, have been shown to induce ferroptosis in chondrocytes. Treatment with IL-1β enhances iron absorption, ROS generation, and lipid peroxidation, concurrently reducing GPX4 expression [95]. These alterations result in chondrocyte apoptosis and cartilage deterioration. The regulation of ferroptosis is influenced by several variables, including EAAT1, a membrane protein, and HSPA5, a member of the heat shock protein A family. Inhibiting IL-1β-induced chondrocyte ferroptosis, overexpressing HSPA5 activates the glutathione system, and upregulating EAAT1 suppresses ferroptosis and slows the course of OA, according to previous research [96,97].
The effects of ferroptosis in the fibrocartilage of the TMJ and the hyaline cartilage of the knee exhibit both similarities and potential differences, which can be attributed to the distinct tissue characteristics and microenvironments of these two joint types. Firstly, ferroptosis, an iron-dependent form of PCD characterized by the accumulation of lipid peroxides, plays a pathogenic role in both TMJ OA and KOA. In both conditions, ferroptosis contributes to chondrocyte death and cartilage degeneration. This is evidenced by reduced expression of ferroptosis-inhibiting genes such as GPX4 and SLC7A11, along with increased lipid peroxidation and iron accumulation in osteoarthritic chondrocytes from both the TMJ and the knee. However, there are some potential differences in the effects of ferroptosis between these two types of cartilage. Fibrocartilage, which is the primary tissue type in the TMJ, has a unique composition and structure compared to hyaline cartilage found in the knee. Fibrocartilage contains a higher proportion of type I collagen and is more fibrous, which may influence its susceptibility to ferroptosis and the subsequent disease progression. Additionally, the mechanical stress experienced by the TMJ and the knee differs, with the TMJ being subjected to complex movements and loads during jaw function, while the knee experiences primarily compressive and shear forces. These differences in mechanical stress may modulate the ferroptosis pathway in distinct ways, affecting chondrocyte metabolism and survival. Furthermore, the inflammatory milieu in TMJ OA and KOA may also contribute to differential effects of ferroptosis. Inflammatory cytokines, such as IL-1β, have been shown to induce ferroptosis in chondrocytes. However, the specific inflammatory profiles and cytokine levels may vary between the TMJ and the knee, leading to distinct outcomes in terms of ferroptosis-mediated chondrocyte death and cartilage damage. Collectively, while ferroptosis plays a significant role in the pathogenesis of both TMJ OA and KOA, the specific effects may differ due to the distinct tissue characteristics, mechanical stress, and inflammatory environments of TMJ and knee. Further research is needed to elucidate these differences and to develop targeted therapeutic strategies that address the unique aspects of ferroptosis in each joint type.
Therapeutic strategies targeting ferroptosis in OA
Modulation of the system Xc-/GSH/GPX4 axis
Numerous treatment approaches have focused on modifying the system Xc-/GSH/GPX4 axis because of its significance in controlling ferroptosis. By stimulating the system Xc-/GSH/GPX4 axis, for instance, it has been shown that traditional Chinese medicines (TCMs) prevent ferroptosis in OA chondrocytes. A substance called icariside II, which comes from epimedium, has been shown to repress ferroptosis-related gene expression in chondrocytes, activate the GSH/GPX4 axis, and reduce the production of iron transporter proteins [98]. Similarly, by upregulating GPX4 and SLC7A11 expression in chondrocytes, baicalein, an extract from scutellaria baicalensis, has been shown to prevent ferroptosis and postpone the development of OA [99]. A further research examines the contribution of chondrocyte ferroptosis to the development of TMJ OA and the possibility of liproxstatin-1 (Lip-1) as a treatment to reduce cartilage deterioration [100]. Utilizing a variety of animal models of TMJ OA caused by unilateral anterior crossbite (UAC), occlusion disorder (OD), monosodium iodoacetate (MIA), and IL-1β, the study shows that ferroptosis, which is typified by elevated p53 and ACSL4 and decreased GPX4, is a major contributor to TMJ OA.
A potential treatment strategy for TMJ OA could be the injection of the ferroptosis inhibitor Lip-1, which successfully reduced chondrocyte ferroptosis, restored mitochondrial function, decreased ROS and lipid peroxidation, and improved the integrity of the cartilage matrix [100]. In their study, Guo et al. detail the steps used to create and characterize PCA NPs, which are nanoparticles made from the naturally occurring phenolic acid p-coumaric acid, with the aim of treating TMJ OA [101]. In vitro investigations showed that PCA NPs were more effective than hyaluronic acid at reducing inflammation and oxidative damage. Injecting PCA NPs into the joints of TMJ OA-affected rats slowed the disease's course over the long term by improving cell proliferation and matrix formation, decreasing inflammation and oxidative stress, and chondrocyte ferroptosis [101]. This study's results demonstrate that PCA NPs have therapeutic potential by addressing several TMJ OA pathological pathways. By influencing important markers associated with ferroptosis, including GPX4, ACSL4, and SLC7A11, PCA NPs were discovered to suppress chondrocyte ferroptosis, a newly defined cell death pathway involved in the advancement of OA. This comprehensive approach positions PCA NPs as a viable choice for the formulation of novel therapeutic techniques for the treatment of TMJ OA [101]. He et al. examine the preventive benefits of Biochanin A (BCA) against KOA produced by iron overload [102]. In vivo and in vitro tests revealed that BCA markedly decreased iron accumulation in the knee joints of mice with iron overload, therefore mitigating the severity of KOA. Moreover, BCA was shown to impede chondrocyte ferroptosis via modulating iron homeostasis, diminishing ROS buildup, and activating the Nrf2/system XC-/GPX4 signaling pathway, which combined facilitated its chondroprotective effects [102]. The study emphasizes the possible therapeutic benefits of BCA in treating KOA related to iron excess. BCA exhibits a unique method of action by directly reducing intracellular iron levels and alleviating oxidative stress, perhaps paving the way for new therapeutic options for this illness. The study's results elucidate the molecular pathways that underlie BCA's preventive actions against iron overload-induced KOA.
Iron chelation therapy
Iron accumulation is a prevalent characteristic in OA and facilitates ferroptosis-mediated chondrocyte apoptosis. Consequently, iron chelation treatment has been suggested as a viable therapeutic approach. Deferoxamine (DFO), an iron chelator, were shown the ability to suppress ferroptosis among osteoarthritic chondrocytes by diminishing iron buildup and lipid peroxidation [103]. Intra-articular administration of DFO has been shown to impede the development of OA in murine models [103]. These results indicate that iron chelation therapy might be a feasible treatment for OA by addressing ferroptosis.
Lipophilic antioxidants
Lipophilic antioxidants, including ferrostatin-1 (Fer-1), have shown the ability to impede ferroptosis by neutralizing lipid peroxides. Fer-1 has been shown to counteract ferroptosis-induced chondrocyte mortality and cartilage deterioration in OA models [104,105]. Guan et al. examine the influence of the gut microbiota metabolite capsiate on the inhibition of ferroptosis-associated OA progression [106]. Through the analysis of clinical data and the execution of in vivo and in vitro tests, the researchers discovered that capsiate levels were markedly reduced in OA patients relative to healthy controls. They found that capsiate suppressed the production of HIF-1α and activated SLC2A1, thereby diminishing ferroptosis and alleviating OA in murine models [106]. The research clarifies the molecular mechanism by which capsiate has a preventive effect against OA. Utilizing bioinformatics analysis, gene knockout tests, and molecular modeling, the researchers demonstrated that SLC2A1 modulated HIF-1α expression and oxidative stress levels, which are essential for ferroptosis [106]. The results indicate that capsiate, a product of gut microbiota, may serve as a possible therapeutic approach for OA by targeting the gut-joint axis. The results indicate that lipophilic antioxidants might be beneficial in the treatment of OA by safeguarding chondrocytes against ferroptosis.
Regulation of inflammatory cytokines
Inflammatory cytokines, including IL-1β, are pivotal in triggering ferroptosis in osteoarthritic chondrocytes. Consequently, targeting these cytokines may provide a useful treatment approach. Puerarin, a substance extracted from pueraria lobata, has shown the capacity to enhance chondrocyte viability and reduce inflammatory cytokine levels in chondrocytes exposed with IL-1β [107]. Gamma-oryzanol, a molecule derived from rice bran oil, has been shown to inhibit ferroptosis and cartilage degradation by engaging with the Keap1 and Nrf2 binding sites in IL-1β-treated chondrocytes [108].
Modulation of mechanical stress
Mechanical stress is a significant component that induces ferroptosis in osteoarthritic chondrocytes. Moderate mechanical stress could activate Nrf2/GPX4/HO-1 axis and inhibit NF-κB pathway, therefore reducing chondrocyte ferroptosis [109]. Excessive mechanical stress may trigger catabolic processe in chondrocytes to facilitate ferroptosis. Consequently, regulating mechanical stress might serve as an effective therapeutic approach for addressing OA by targeting ferroptosis. Through the modulation of ferroptosis in fibroblast-like synoviocytes (FLSs), Hu et al. examine the therapeutic effects of Lipoxin A4 (LXA4) on the development of KOA [110]. The authors show that LXA4 activates the expression of GPX4, lysophosphatidic acid receptor-3 (LPAR3), and estrogen receptor beta (ESR2) in FLSs, which inhibits ferroptosis and reduces inflammation and chondrocyte matrix breakdown. The work shows that LXA4 acts via the ESR2/LPAR3/Nrf2 axis to provide its anti-inflammatory and anti-ferroptotic actions in vitro cell co-culture and in vivo rat models, offering a novel therapeutic target for the intervention of KOA [110]. The research examines the impact of exercise on KOA therapy, demonstrating that moderate-intensity treadmill exercise enhances LXA4 synthesis, which subsequently suppresses synovial ferroptosis and mitigates KOA pathogenesis. Utilizing PHTPP, a selective ESR2 antagonist, the authors demonstrate that the therapeutic benefits of exercise are largely reliant on the ESR2-mediated signaling system, therefore providing novel insights into the processes governing exercise treatment for KOA [110].
Targeting ferroptosis-related genes and pathways
Moreover, focusing on ferroptosis-related genes and pathways may be beneficial in the treatment of OA. Inhibition of miR-19b-3p expression, which facilitates ferroptosis by targeting SLC7A11, has shown benefit effects on chondrocytes against ferroptosis in OA [92]. Likewise, the activation of TRPV1, which suppresses the ferroptosis pathway in chondrocytes, has been shown to slow the development of OA [111,112]. This research conducted by Bao et al. examined the influence of METTL3 on the advancement of KOA via regulating ferroptosis in chondrocytes [113]. The study demonstrated that METTL3 promotes ferroptosis in chondrocytes via m6A methylation of high mobility group box 1 (HMGB1), intensifying pain and cartilage deterioration in KOA model. The suppression of METTL3 mitigated these adverse effects, suggesting that the METTL3/HMGB1 axis is integral to the pathophysiology of KOA [113]. The research revealed that METTL3 depletion suppressed inflammatory responses, ECM disintegration, and ferroptosis in chondrocytes, resulting in enhanced cartilage regeneration and decreased knee discomfort in KOA mice. The results indicate that focusing on the METTL3/HMGB1 pathway might provide a new therapeutic approach for treating KOA [113]. The results indicate that targeting genes and pathways associated with ferroptosis could provide new options for the treatment of OA. He et al. examine the therapeutic benefits and underlying mechanisms of vitamin K2 (VK2) on OA [114]. The study shows that VK2 substantially mitigates OA by inhibiting chondrocyte ferroptosis and the destruction of the ECM via in vivo and in vitro trials. VK2 has protective effects via dual control of GPX4, which suppresses ferroptosis and modifies ECM breakdown through the MAPK/NF-κB axis [114]. The results indicate that VK2 enhances bone mass and cartilage thickness in OA rats, alleviates pain, and lowers the OARSI score. VK2 therapy enhances GPX4 expression, resulting in reduced Fe2+ levels, ROS, lipid-ROS generation, and mitochondrial membrane potential, hence preventing chondrocyte ferroptosis. Moreover, VK2 suppresses the MAPK/NF-κB axis, resulting in decreased production of MMP3 and MMP13, while enhancing the synthesis of type II collagen and proteoglycans, so collectively mitigating ECM breakdown [114].
Challenges
Notwithstanding the encouraging therapeutic prospects of targeting ferroptosis in OA, several difficulties persist. The precise function of ferroptosis in the pathophysiology of OA is inadequately understood. Additional study is required to clarify the molecular pathways that drive ferroptosis-related chondrocyte death and cartilage deterioration. The effectiveness and safety of existing ferroptosis-targeting medicines must be confirmed via clinical studies. The diversity among OA patients may restrict the efficacy of single-target treatments. Combination medicines that target various pathways may enhance the efficacy of OA treatment. Future research must concentrate on discovering new ferroptosis-related biomarkers and therapeutic targets, creating more effective and safer ferroptosis inhibitors, and assessing the effectiveness of combination therapy in clinical trials. Furthermore, comprehending the interplay between ferroptosis and other cell death mechanisms, including apoptosis and necrosis, might provide novel insights into the etiology of OA and inform the formulation of more holistic treatment approaches. Ferroptosis has emerged as an important factor in chondrocyte death and cartilage deterioration in OA. Focusing on ferroptosis-related pathways, including the system Xc-/GSH/GPX4 axis, iron metabolism, and inflammatory cytokines, may provide novel therapeutic strategies for managing OA. Further study is needed to comprehensively demonstrate the role of ferroptosis in the etiology of OA and to devise effective and safe treatment options. Ongoing improvements in ferroptosis research are anticipated to provide novel therapeutics targeting this pathway, therefore enhancing the quality of life for people with OA.
Collectively, the role of ferroptosis in OA is multifaceted and has been extensively explored in recent studies. Ferroptosis, an iron-dependent form of cell death characterized by excessive lipid peroxidation, is distinct from apoptosis and necrosis. It plays a significant role in the pathogenesis of OA, particularly in the context of chondrocyte death and cartilage degradation. Ferroptosis contributes to OA through several mechanisms: 1) Iron overload and lipid peroxidation: elevated iron levels in OA tissues lead to increased lipid peroxidation, which damages cell membranes and induces cell death. This process is exacerbated by the downregulation of protective enzymes like GPX4 and the disruption of the system Xc-/GSH/GPX4 axis. 2) Inflammatory cytokines: inflammatory cytokines such as IL-1β and TNF-α upregulate transferrin receptors, leading to increased iron uptake and accumulation, thereby promoting ferroptosis. These cytokines also downregulate GPX4, further sensitizing chondrocytes to ferroptosis. 3) Mechanical stress: excessive mechanical stress can induce ferroptosis in chondrocytes by disrupting iron homeostasis and increasing oxidative stress. Moderate mechanical stress, however, can activate protective pathways like the Nrf2/GPX4/HO-1 axis, thereby reducing ferroptosis. 4) Genetic and epigenetic regulation: certain genes and pathways, such as miR-19b-3p and the METTL3/HMGB1 axis, have been identified as key regulators of ferroptosis in OA. For example, METTL3 promotes ferroptosis through m6A methylation of HMGB1, leading to cartilage degradation and pain in OA. Causes of ferroptosis in OA includes: 1) Iron accumulation: elevated levels of iron in synovial fluid and cartilage tissues are a hallmark of OA. This iron overload can be due to disrupted iron homeostasis, increased iron absorption via transferrin receptors, or decreased iron export via ferroportin. 2) Oxidative stress: increased production of ROS and lipid peroxides due to impaired antioxidant defenses (e.g., reduced GPX4 activity) leads to oxidative damage and cell death. 3) Inflammatory mediators: inflammatory cytokines not only induce iron accumulation but also directly activate ferroptosis pathways. For instance, IL-1β can increase iron uptake and decrease GPX4 expression, thereby promoting ferroptosis. 4) Genetic and molecular pathways: dysregulation of genes involved in iron metabolism, lipid peroxidation, and antioxidant defense (e.g., SLC7A11, GPX4, Nrf2) contributes to the susceptibility of chondrocytes to ferroptosis. Ferroptosis plays a crucial role in OA by contributing to chondrocyte death, cartilage degradation, and joint inflammation. Its initiation is driven by iron overload, oxidative stress, inflammatory cytokines, and disrupted genetic pathways. Understanding these mechanisms provides valuable insights for developing targeted therapies to mitigate OA progression.
Conclusions and future perspectives
OA is a common degenerative joint disease marked by the gradual degradation of cartilage, synovitis, and remodeling of subchondral bone. Effective therapies for OA are limited, sometimes necessitating joint replacement in late stages. Comprehending the etiology of OA is essential for formulating more efficacious therapy approaches. Recent study has emphasized the significance of ferroptosis, an iron-dependent mode of cell death induced by lipid peroxidation, in the development of OA. Research has focused on three primary domains related to ferroptosis in OA: iron homeostasis, lipid metabolism, and the antioxidant defense mechanism. Chondrocytes in OA are less viable because disturbances in iron homeostasis, such changes in the expression of proteins involved in iron metabolism, cause an increase in iron input and a decrease in iron efflux. An important factor in ferroptosis is lipid peroxidation, especially of polyunsaturated fatty acids, in which LOX and ACSL4 play essential roles. In OA, ferroptosis may be inhibited by antioxidant systems such as the NRF2 signaling pathway and the system Xc-/GSH/GPX4 system. Also involved in OA are many other forms of PCD, including autophagy, pyroptosis, apoptosis, cuproptosis, and necroptosis. Inflammatory substances, which may be produced by these processes, might worsen the symptoms of OA. The development of OA is regulated by molecules including IER1, NF-κB, Jak, and mTOR.
Subsequent research may concentrate on the following aspects: 1) Expansion of ferroptosis research: additional research into the involvement of ferroptosis in OA is essential, especially with critical regulators such as FSP1, DHODH, and GCH1, which remain unexamined in the context of OA. Research should include other cell types implicated in OA, including osteoblasts, macrophage, and synovial cells, rather than just concentrating on chondrocytes. 2) Iron homeostasis and lipid metabolism: a comprehensive knowledge of iron homeostasis and lipid metabolism in OA is essential. Investigating how variables such as IL-1β and H2O2 influence the level of iron related proteins might provide insights into illness development. Inhibiting LOX and ACSL4 activity may effectively prevent chondrocyte ferroptosis and decelerate OA development. 3) Antioxidant systems and ROS: the function of antioxidant systems in mitigating ferroptosis in OA warrants further investigation. Pharmaceuticals that stimulate these pathways may alleviate chondrocyte ferroptosis and slow the course of OA. ROS are pivotal molecules in OA, governing not only ferroptosis but also mitophagy and many kinds of autophagy. Examining ROS interaction in OA microautophagy and its modulation of ferroptosis is a viable research avenue. 4) PCD and signaling pathways: additional investigation is required to clarify the intricate interactions among several forms of PCD in OA. Comprehending the regulatory roles of molecules like as NF-κB, Jak, and mTOR in these processes may provide novel therapeutic targets. Investigating therapies that focus on PCD and signaling pathways associated with OA progression may provide more efficacious and less invasive options compared to existing treatments. 5) Therapeutic development: compounds capable of inhibiting PCD, namely ferroptosis, warrant exploration as prospective therapeutic agents for OA. The precise network processes of crosstalk governing OA ferroptosis and other types of autophagy need comprehensive investigation to pinpoint novel treatment targets. 6) Global study collaboration: additional confirmatory studies and study from a wider array of nations are important to verify results and enhance the comprehension of ferroptosis in OA. Collaborative initiatives should be promoted to bridge research gaps and expedite the development of novel therapies for OA. Collectively, ferroptosis and other types of PCD are pivotal in the course of OA. Additional investigation into these mechanisms, their control, and prospective therapeutic targets is crucial for formulating more effective therapeutics for OA. Through comprehensive experimental and clinical research, PCD may be used as a target for the development of therapeutic medications that tackle the underlying causes of OA without adverse effects or consequences.
Acknowledgements
Funding
This study was supported by National Key R&D Program of China (2023YFC2509100), Science and Technology Commission of Shanghai Municipality (23Y31900400), National Natural Science Foundation of China (82370984 & 82370980), Natural Science Foundation of Shanghai (22ZR1437600), Shanghai's Top Priority Research Center (2022ZZ01017), and Dominant disease biological sample project of the Ninth People's Hospital Affiliated to Shanghai Jiao Tong University School of Medicine (YBKA202201).
Author contributions
YxZ: Writing-original draft, Visualization, Investigation, Funding acquisition; DhZ: Formal analysis, Validation, Investigation; XyL, QyX and LtB: Investigation and Data curation; JsZ: Resources and Supervision. PS: Writing-review & editing, Supervision, Funding acquisition. CY: Writing-review & editing, Supervision, Resources, Funding acquisition, Conceptualization. All authors gave final approval and agreed to be accountable for all aspects of the work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yao Q, Wu X, Tao C, Gong W, Chen M, Qu M. et al. Osteoarthritis: pathogenic signaling pathways and therapeutic targets. Sig Transduct Target Ther. 2023;8(1):56
2. Quicke JG, Conaghan PG, Corp N, Peat G. Osteoarthritis year in review 2021: epidemiology & therapy. Osteoarthritis and Cartilage. 2022;30(2):196-206
3. Lo J, Chan L, Flynn S. A systematic review of the incidence, prevalence, costs, and activity and work limitations of amputation, osteoarthritis, rheumatoid arthritis, back pain, multiple sclerosis, spinal cord injury, stroke, and traumatic brain injury in the United States: a 2019 update. Archives of Physical Medicine and Rehabilitation. 2021;102(1):115-31
4. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB. et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2(1):1-18
5. Glyn-Jones S, Palmer AJR, Agricola R, Price AJ, Vincent TL, Weinans H. et al. Osteoarthritis. The Lancet. 2015;386(9991):376-87
6. Lane NE, Corr M. Anti-NGF treatments for pain — two steps forward, one step back? Nat Rev Rheumatol. 2017;13(2):76-8
7. Allen KD, Thoma LM, Golightly YM. Epidemiology of osteoarthritis. Osteoarthritis and Cartilage. 2022;30(2):184-95
8. Steinmetz JD, Culbreth GT, Haile LM, Rafferty Q, Lo J, Fukutaki KG. et al. Global, regional, and national burden of osteoarthritis, 1990-2020 and projections to 2050: a systematic analysis for the Global Burden of Disease Study 2021. The Lancet Rheumatology. 2023;5(9):e508-22
9. Kloppenburg M, Berenbaum F. Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthritis and Cartilage. 2020;28(3):242-8
10. Peat G, Thomas MJ. Osteoarthritis year in review 2020: epidemiology & therapy. Osteoarthritis and Cartilage. 2021;29(2):180-9
11. Bannuru RR, Osani MC, Vaysbrot EE, Arden NK, Bennell K, Bierma-Zeinstra SMA. et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthritis and Cartilage. 2019;27(11):1578-89
12. Katz JN, Arant KR, Loeser RF. Diagnosis and Treatment of Hip and Knee Osteoarthritis. JAMA. 2021;325(6):568
13. Li X, Dai B, Guo J, Zheng L, Guo Q, Peng J. et al. Nanoparticle-Cartilage Interaction: Pathology-Based Intra-articular Drug Delivery for Osteoarthritis Therapy. Nano-Micro Lett. 2021;13(1):149
14. Zeng C, Lane NE, Hunter DJ, Wei J, Choi HK, McAlindon TE. et al. Intra-articular corticosteroids and the risk of knee osteoarthritis progression: results from the Osteoarthritis Initiative. Osteoarthritis and Cartilage. 2019;27(6):855-62
15. Li J, Cao F, Yin H liang, Huang Z jian, Lin Z tao, Mao N. et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88
16. Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. 2019;116(7):2672-80
17. Wang H, An P, Xie E, Wu Q, Fang X, Gao H. et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449-65
18. Tonnus W, Meyer C, Steinebach C, Belavgeni A, von Mässenhausen A, Gonzalez NZ. et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun. 2021;12(1):4402
19. Ru Q, Li Y, Xie W, Ding Y, Chen L, Xu G. et al. Fighting age-related orthopedic diseases: focusing on ferroptosis. Bone Res. 2023;11(1):12
20. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381-96
21. Cao S, Wei Y, Yue Y, Liu P, Zeng H. Global research landscape on the crosstalk between ferroptosis and musculoskeletal diseases: a bibliometric and visualized analysis. Heliyon. 2023;9(12):e23113
22. Sun K, Guo Z, Hou L, Xu J, Du T, Xu T. et al. Iron homeostasis in arthropathies: From pathogenesis to therapeutic potential. Ageing Research Reviews. 2021;72:101481
23. Zhang X, Hou L, Guo Z, Wang G, Xu J, Zheng Z. et al. Lipid peroxidation in osteoarthritis: focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov. 2023;9(1):320
24. Yao X, Sun K, Yu S, Luo J, Guo J, Lin J. et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. Journal of Orthopaedic Translation. 2021;27:33-43
25. Chen BY, Pathak JL, Lin HY, Guo WQ, Chen WJ, Luo G. et al. Inflammation triggers chondrocyte ferroptosis in TMJOA via HIF-1α/TFRC. J Dent Res. 2024;103(7):712-22
26. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966-75
27. Lv Z, Han J, Li J, Guo H, Fei Y, Sun Z. et al. Single cell RNA-seq analysis identifies ferroptotic chondrocyte cluster and reveals TRPV1 as an anti-ferroptotic target in osteoarthritis. eBioMedicine. 2022;84:104258
28. Li W, Lv Z, Wang P, Xie Y, Sun W, Guo H. et al. Near Infrared Responsive Gold Nanorods Attenuate Osteoarthritis Progression by Targeting TRPV1. Advanced Science. 2024;11(16):2307683
29. Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao J. et al. Contribution of ferroptosis and GPX4's dual functions to osteoarthritis progression. eBioMedicine. 2022;76:103847
30. Zhou Y, Jia Z, Wang J, Huang S, Yang S, Xiao S. et al. Curcumin reverses erastin-induced chondrocyte ferroptosis by upregulating Nrf2. Heliyon. 2023;9(10):e20163
31. He W, Lin X, Chen K. Specificity protein 1-mediated ACSL4 transcription promoted the osteoarthritis progression through suppressing the ferroptosis of chondrocytes. J Orthop Surg Res. 2023;18(1):188
32. Sun K, Hou L, Guo Z, Wang G, Guo J, Xu J. et al. JNK-JUN-NCOA4 axis contributes to chondrocyte ferroptosis and aggravates osteoarthritis via ferritinophagy. Free Radical Biology and Medicine. 2023;200:87-101
33. Han C, Liu Y, Dai R, Ismail N, Su W, Li B. Ferroptosis and Its Potential Role in Human Diseases. Front Pharmacol. 2020;11:239
34. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401-21
35. Jing X, Lin J, Du T, Jiang Z, Li T, Wang G. et al. Iron Overload Is Associated With Accelerated Progression of Osteoarthritis: The Role of DMT1 Mediated Iron Homeostasis. Front Cell Dev Biol. 2021;8:594509
36. Biniecka M, Connolly M, Gao W, Ng CT, Balogh E, Gogarty M. et al. Redox-Mediated Angiogenesis in the Hypoxic Joint of Inflammatory Arthritis. Cell. 2022;8(1):2401-21
37. Yan H fa, Zou T, Tuo Q zhang, Xu S, Li H, Belaidi AA. et al. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflamm Res. 2023;72(2):281-99
38. Rim YA, Nam Y, Ju JH. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. IJMS. 2020;21(7):2358
39. Hosseinzadeh A, Kamrava SK, Joghataei MT, Darabi R, Shakeri-Zadeh A, Shahriari M. et al. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. Journal of Pineal Research. 2016;61(4):411-25
40. Robinson WH, Lepus CM, Wang Q, Raghu H, Mao R, Lindstrom TM. et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(10):580-92
41. Hui W, Young DA, Rowan AD, Xu X, Cawston TE, Proctor CJ. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann Rheum Dis. 2016;75(2):449-58
42. Wang FS, Kuo CW, Ko JY, Chen YS, Wang SY, Ke HJ. et al. Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy. Antioxidants. 2020;9(9):810
43. Hodgkinson T, Kelly DC, Curtin CM, O'Brien FJ. Mechanosignalling in cartilage: an emerging target for the treatment of osteoarthritis. Nat Rev Rheumatol. 2022;18(2):67-84
44. Kawata M, Teramura T, Ordoukhanian P, Head SR, Natarajan P, Sundaresan A. et al. Krüppel-like factor-4 and Krüppel-like factor-2 are important regulators of joint tissue cells and protect against tissue destruction and inflammation in osteoarthritis. Ann Rheum Dis. 2022;81(8):1179-88
45. Zhou X, Yang Y, Qiu X, Deng H, Cao H, Liao T. et al. Antioxidant taurine inhibits chondrocyte ferroptosis through upregulation of OGT/Gpx4 signaling in osteoarthritis induced by anterior cruciate ligament transection. Journal of Advanced Research. 2025 S2090-1232(25)00029-3
46. Cao S, Wei Y, Yue Y, Chen Y, Qian J, Wang D. et al. Rosiglitazone retards the progression of iron overload-induced osteoarthritis by impeding chondrocyte ferroptosis. iScience. 2024;27(9):110526
47. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273-85
48. Liu D, Cai ZJ, Yang YT, Lu WH, Pan LY, Xiao WF. et al. Mitochondrial quality control in cartilage damage and osteoarthritis: new insights and potential therapeutic targets. Osteoarthritis and Cartilage. 2022;30(3):395-405
49. Liu Y, Xu T, Ma Z, Zhang C, Xu M, Li Q. et al. Cartilage protective and anti-edema effects of JTF in osteoarthritis via inhibiting NCOA4-HMGB1-driven ferroptosis and aquaporin dysregulation. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2024;129:155593
50. Yan C, Zhang J, An F, Wang J, Shi Y, Yuan L. et al. Research Progress of Ferroptosis Regulatory Network and Bone Remodeling in Osteoporosis. Front Public Health. 2022;10:910675
51. Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W. et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Bull Exp Biol Med. 2018;32(5):662-8
52. Tong L, Yu H, Huang X, Shen J, Xiao G, Chen L. et al. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 2022;10(1):60
53. Guan Z, Jin X, Guan Z, Liu S, Tao K, Luo L. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Oxidative Medicine and Cellular Longevity. 2017;171(2):273-85
54. Gao G, Li J, Zhang Y, Chang YZ. Cellular Iron Metabolism and Regulation. Adv Exp Med Biol. 2019;1173:21-32
55. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Cell Res. 2019;133:144-52
56. Abramoff B, Caldera FE. Osteoarthritis. Medical Clinics of North America. 2020;104(2):293-311
57. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261(5):2256-63
58. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I. et al. The oxidative stress-inducible cystine/glutamate antiporter, system x c - : cystine supplier and beyond. Gene. 2012;42(1):231-46
59. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS. et al. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Medicine and Cellular Longevity. 2019;2019:1-13
60. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-92
61. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. 2021;220(9):e202105043
62. Yang H, Zhong C, Tan X, Chen G, He Y, Liu S. et al. Transcriptional Responses of Copper-Transport-Related Genes ctr1, ctr2 and atox1 and Their Roles in the Regulation of Cu Homeostasis in Yellow Catfish Pelteobagrus fulvidraco. IJMS. 2022;23(20):12243
63. Gudjoncik A, Guenancia C, Zeller M, Cottin Y, Vergely C, Rochette L. et al. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. IJMS. 2022;24(1):449
64. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-8
65. Abusarah J, Bentz M, Benabdoune H, Rondon PE, Shi Q, Fernandes JC. et al. An overview of the role of lipid peroxidation-derived 4-hydroxynonenal in osteoarthritis. Inflamm Res. 2017;66(8):637-51
66. Gochenauer GE, Robinson MB. Progress in Understanding Ferroptosis and Challenges in Its Targeting for Therapeutic Benefit. Cell. 2019;256(1):1-13
67. Kuang F, Liu J, Tang D, Kang R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front Cell Dev Biol. 2020;8:586578
68. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C. et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Neurotox Res. 2019;26(11):2284-99
69. Kawakami Y, Bhullar NK. Potential Implications of Interactions between Fe and S on Cereal Fe Biofortification. Inflamm Res. 2022;79(8):146460
70. Krümmel B, Plötz T, Jörns A, Lenzen S, Mehmeti I. Role of Mitochondria in Ferroptosis. Nat Chem Biol. 2019;73(2):354-363.e3
71. Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T. et al. The iron-regulatory hormone hepcidin: A possible therapeutic target? Neurotox Res. 2013;15(1):e0201369
72. Li J, Cao F, Yin H liang, Huang Z jian, Lin Z tao, Mao N. et al. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Nat Chem Biol. 2015;6:472-85
73. Barrera G, Gentile F, Pizzimenti S, Canuto R, Daga M, Arcaro A. et al. Mitochondrial Dysfunction in Cancer and Neurodegenerative Diseases: Spotlight on Fatty Acid Oxidation and Lipoperoxidation Products. Antioxidants. 2016;5(1):7
74. Lin X, Ping J, Wen Y, Wu Y. The Mechanism of Ferroptosis and Applications in Tumor Treatment. Front Pharmacol. 2020;11:1061
75. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W. et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biology. 2017;12:558-70
76. Djulbegovic MB, Uversky VN. Ferroptosis - An iron- and disorder-dependent programmed cell death. International Journal of Biological Macromolecules. 2019;135:1052-69
77. Dar NJ, John U, Bano N, Khan S, Bhat SA. Oxytosis/Ferroptosis in Neurodegeneration: the Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4). Mol Neurobiol. 2024;61(3):1507-26
78. Xia C, Xing X, Zhang W, Wang Y, Jin X, Wang Y. et al. Cysteine and homocysteine can be exploited by GPX4 in ferroptosis inhibition independent of GSH synthesis. Redox Biology. 2024;69:102999
79. Jafari-Gharabaghlou D, Pilehvar-Soltanahmadi Y, Dadashpour M, Mota A, Vafajouy-Jamshidi S, Faramarzi L. et al. Combination of metformin and phenformin synergistically inhibits proliferation and hTERT expression in human breast cancer cells. Iranian Journal of Basic Medical Sciences. 2018;21(Online First):1167-73
80. Shao M, Jiang Q, Shen C, Liu Z, Qiu L. Sinapine induced ferroptosis in non-small cell lung cancer cells by upregulating transferrin/transferrin receptor and downregulating SLC7A11. Gene. 2022;827:146460
81. Yang F, Jiang X, Cao L, Gu Q, Teng X, He L. Diverse Sesquiterpenoids from the Roots of Croton crassifolius and Their Inhibitory Effects on Ferroptosis. Chemistry & Biodiversity. 2022;19(4):e202101028
82. Guo Z, Lin J, Sun K, Guo J, Yao X, Wang G. et al. Deferoxamine Alleviates Osteoarthritis by Inhibiting Chondrocyte Ferroptosis and Activating the Nrf2 Pathway. Front Pharmacol. 2022;13:791376
83. Zhang ZJ, Hou YK, Chen MW, Yu XZ, Chen SY, Yue YR. et al. A pH-responsive metal-organic framework for the co-delivery of HIF-2α siRNA and curcumin for enhanced therapy of osteoarthritis. J Nanobiotechnol. 2023;21(1):18
84. Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51(2):249-57
85. Simão M, Gavaia PJ, Camacho A, Porto G, Pinto IJ, Ea H. et al. Intracellular iron uptake is favored in Hfe-KO mouse primary chondrocytes mimicking an osteoarthritis-related phenotype. BioFactors. 2019;45(4):583-97
86. Kennish L, Attur M, Oh C, Krasnokutsky S, Samuels J, Greenberg JD. et al. Age-dependent ferritin elevations and HFE C282Y mutation as risk factors for symptomatic knee osteoarthritis in males: a longitudinal cohort study. BMC Musculoskelet Disord. 2014;15(1):8
87. Sheng W, Liao S, Wang D, Liu P, Zeng H. The role of ferroptosis in osteoarthritis: Progress and prospects. Biochemical and biophysical research communications. 2024;733:150683
88. Zhou X, Zheng Y, Sun W, Zhang Z, Liu J, Yang W. et al. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell proliferation. 2021;54:e13134
89. Xu Y, Yang Z, Dai T, Xue X, Xia D, Feng Z. et al. Characteristics and time points to inhibit ferroptosis in human osteoarthritis. Sci Rep. 2023;13(1):21592
90. Klinger W, Lupp A, Karge E, Baumbach H, Eichhorn F, Feix A. et al. Estradiol, testosterone, dehydroepiandrosterone and androstenedione: novel derivatives and enantiomers. Interactions with rat liver microsomal cytochrome P450 and antioxidant/radical scavenger activities in vitro. Toxicology Letters. 2002;128(1-3):129-44
91. Zou L, Yang K, Yu Y, Wang C, Zhao J, Lu C. et al. Analysis of joint protein expression profile in anterior disc displacement of TMJ with or without OA. Oral Diseases. 2024;30(7):4463-82
92. Kong R, Ji L, Pang Y, Zhao D, Gao J. Exosomes from osteoarthritic fibroblast-like synoviocytes promote cartilage ferroptosis and damage via delivering microRNA-19b-3p to target SLC7A11 in osteoarthritis. Front Immunol. 2023;14:1181156
93. Wang S, Li W, Zhang P, Wang Z, Ma X, Liu C. et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. Front Cell Dev Biol. 2022;41:63-75
94. Pang C, Zhang H, Liu Y, Tang N, Tian K, Mu Y. et al. Glutathione peroxidase 4 restrains temporomandibular joint osteoarthritis progression by inhibiting ferroptosis. J Cellular Molecular Medi. 2024;28(9):e18377
95. Fan F, Yang C, Piao E, Shi J, Zhang J. Mechanisms of chondrocyte regulated cell death in osteoarthritis: Focus on ROS-triggered ferroptosis, parthanatos, and oxeiptosis. Biochem Biophys Res Commun. 2024;705:149733
96. Wen Z, Xia G, Liang C, Wang X, Huang J, Zhang L. et al. Selective Clearance of Senescent Chondrocytes in Osteoarthritis by Targeting Excitatory Amino Acid Transporter Protein 1 to Induce Ferroptosis. Antioxidants & Redox Signaling. 2023;39(4-6):262-77
97. Lv M, Cai Y, Hou W, Peng K, Xu K, Lu C. et al. The RNA-binding protein SND1 promotes the degradation of GPX4 by destabilizing the HSPA5 mRNA and suppressing HSPA5 expression, promoting ferroptosis in osteoarthritis chondrocytes. Inflamm Res. 2022;71(4):461-72
98. Yu R, Zhou Y, Shi S, Wang X, Huang S, Ren Y. Icariside II induces ferroptosis in renal cell carcinoma cells by regulating the miR-324-3p/GPX4 axis. Phytomedicine. 2022;102:154182
99. Wan Y, shen K, Yu H, Fan W. Baicalein limits osteoarthritis development by inhibiting chondrocyte ferroptosis. Free Radical Biology and Medicine. 2023;196:108-20
100. Cheng B, Zhang J, Shen Q, Sun Z, Luo Y, Hu Y. Liproxstatin-1 alleviates cartilage degradation by inhibiting chondrocyte ferroptosis in the temporomandibular joint. Biol Cell. 2024;116(1):e202300042
101. Guo J, Su K, Wang L, Feng B, You X, Deng M. et al. Poly(p-coumaric acid) nanoparticles alleviate temporomandibular joint osteoarthritis by inhibiting chondrocyte ferroptosis. Bioactive Materials. 2024;40:212-26
102. He Q, Yang J, Pan Z, Zhang G, Chen B, Li S. et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System xc-/GPX4 axis. Biomedicine & Pharmacotherapy. 2023;157:113915
103. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA. et al. Ferroptosis: mechanisms and links with diseases. Signal transduction and targeted therapy. 2021;6:49
104. Xu W, Zhang B, Xi C, Qin Y, Lin X, Wang B. et al. Ferroptosis plays a role in human chondrocyte of osteoarthritis induced by IL-1β in vitro. CARTILAGE. 2023;14(4):455-66
105. Wang X, Liu Z, Peng P, Gong Z, Huang J, Peng H. Astaxanthin attenuates osteoarthritis progression via inhibiting ferroptosis and regulating mitochondrial function in chondrocytes. Chemico-biological interactions. 2022;366:110148
106. Guan Z, Jin X, Guan Z, Liu S, Tao K, Luo L. The gut microbiota metabolite capsiate regulate SLC2A1 expression by targeting HIF-1α to inhibit knee osteoarthritis-induced ferroptosis. Aging Cell. 2023;22(6):e13807
107. Deng W, Zhang W, He Q. Study on the mechanism of puerarin against osteoarthritis from ferroptosis based on network pharmacology and bioinformatics. Naunyn-Schmiedeberg's Arch Pharmacol. 2024;397(2):959-68
108. Dai ZH, Zhou CC, Yu CY, Qian CJ, Jin SQ, Du SQ. et al. Gamma-oryzanol alleviates osteoarthritis development by targeting Keap1-Nrf2 binding to interfere with chondrocyte ferroptosis. Naunyn-Schmiedeberg's Arch Pharmacol. 2024;397(2):959-68
109. Han J, Zhan L nan, Huang Y, Guo S, Zhou X, Kapilevich L. et al. Moderate mechanical stress suppresses chondrocyte ferroptosis in osteoarthritis by regulating NF-κB p65/GPX4 signaling pathway. Sci Rep. 2024;14(1):5078
110. Hu Z, Chen L, Zhao J, Zhang W, Jin Z, Sun Y. et al. Lipoxin A4 ameliorates knee osteoarthritis progression in rats by antagonizing ferroptosis through activation of the ESR2/LPAR3/Nrf2 axis in synovial fibroblast-like synoviocytes. Redox Biology. 2024;73:103143
111. Lv Z, Xu X, Sun Z, Yang YX, Guo H, Li J. et al. TRPV1 alleviates osteoarthritis by inhibiting M1 macrophage polarization via Ca(2+)/CaMKII/Nrf2 signaling pathway. Cell death & disease. 2021;12:504
112. Lv Z, Wang P, Li W, Xie Y, Sun W, Jin X. et al. Bifunctional TRPV1 Targeted Magnetothermal Switch to Attenuate Osteoarthritis Progression. Research. 2024;7:research.0316
113. Bao T, Liao T, Cai X, Lu B, Dai G, Pei S. et al. METTL3 mediated ferroptosis in chondrocytes and promoted pain in KOA via HMGB1 m6A modification. Cell Biology International. 2024;48(11):1755-65
114. He Q, Lin Y, Chen B, Chen C, Zeng J, Dou X. et al. Vitamin K2 ameliorates osteoarthritis by suppressing ferroptosis and extracellular matrix degradation through activation GPX4's dual functions. Biomedicine & Pharmacotherapy. 2024;175:116697
Author contact
![]() Corresponding authors: Yuxin Zhang, Email: yxzhang0129edu.cn; Pei Shen, Email: shenpei1215com; Chi Yang, Email: yangchi1963com.
Corresponding authors: Yuxin Zhang, Email: yxzhang0129edu.cn; Pei Shen, Email: shenpei1215com; Chi Yang, Email: yangchi1963com.