Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(3):630-640. doi:10.7150/ijms.102266 This issue Cite
Research Paper
NACC1 accelerates the progression of AML by regulating the ADAM9/PI3K/AKT axis
Ying Zhang1, Liang Zhong2, Peng Wan1, Yi Zhao1, Meng Wang1, Hongyan Zhang1, Yang Liao1, Ying Deng1, Beizhong Liu1,2,3 ![]()
1. Central Laboratory of Yongchuan Hospital, Chongqing Medical University, Chongqing 402160, China.
2. Key Laboratory of Laboratory Medical Diagnostics, Ministry of Education, Department of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, China.
3. Clinical Laboratory of The Affiliated Rehabilitation Hospital, Chongqing Medical University, Chongqing 400050, China.
Received 2024-8-12; Accepted 2024-12-12; Published 2025-1-6
Abstract

Nucleus accumbens-associated protein 1 (NACC1) regulates various types of biological processes. It is a transcription factor associated with cancer. NACC1 is overexpressed in many human malignancies and can regulate the progression, metastasis, and drug resistance of cancer cells. However, its precise role in acute myeloid leukemia (AML) remains unknown. This study aimed to unravel the basic mechanism of NACC1 in AML. Our findings demonstrated that NACC1 is immensely expressed in AML cells. Lentiviral vector-mediated knockdown of NACC1 inhibited the PI3K/AKT signaling pathway. Simultaneously, NACC1 knockdown promoted apoptosis, suppressed the proliferative capacity of AML cells, and resulted in cell cycle arrest during the G0/G1 phase. Additionally, A disintegrin and metalloproteinase 9 (ADAM9) was markedly expressed in AML cells. NACC1 regulated ADAM9 expression. ADAM9 expression was also downregulated after NACC1 knockdown. Concurrently, ADAM9 knockdown affected the activity of AML cells by decelerating the growth rate, promoting apoptosis, and blocking cell cycle progression. In addition, the AKT activator SC79 restored the inhibited cell proliferation after NACC1 knockdown and ADAM9 knockdown. In conclusion, our study suggested that the NACC1/ADAM9/PI3K/AKT axis is crucial for sustaining the survival of AML cells, indicating that NACC1 may be a viable target for treating AML.
Keywords: Acute myeloid leukemia, NACC1, ADAM9, PI3K/AKT pathway, Proliferation, Cell apoptosis
Introduction
Acute myeloid leukemia (AML) affects myeloid hematopoietic stem cells. The clonal proliferation of abnormally differentiated mother cells from the bone marrow line is a hallmark of AML [1, 2]. AML is a prominent type of leukemia in adult patients, and the prevalence gradually increases with age [3]. The majority of patients do not experience substantial remission after initial treatment and develop refractory AML [4]. The mortality rate of the disease is higher among elderly patients [5, 6]. AML therapy has significantly advanced over the past years; however, patients with AML are at a higher risk of relapsing and are prone to medication resistance [7]. Therefore, to explore new treatments for AML, more studies are needed on the molecular target of AML.
BTB is also frequently referred to as the POZ domain. It regulates many cellular functions and is a major motif involved in protein-protein interactions [8-11]. BTB/POZ family proteins are involved in biological processes. They play a key role in transcriptional regulation [12]. Previous studies have demonstrated its significance in the growth of many cancers and diseases [13-15]. Multiple proteins containing this domain have been linked to human cancer, including B-cell lymphoma 6 (BCL-6) [16], promyelocytic leukemia zinc finger (PLZF) [17, 18], and zinc finger and BTB domain protein 1 (ZBTB1) [19].
Nucleus accumbens-associated protein 1 (NACC1), containing a BTB/POZ domain, is a transcription factor associated with cancer [20]. Initially, NACC1 was discovered in the nucleus accumbens of cocaine-treated rats. The nucleus accumbens is a key region of the brain that is strongly linked to addictive behaviors [21]. Moreover, NACC1 is closely linked to the pluripotency of embryonic stem cells, mediating various biological functions [22-25]. Meanwhile, abnormal expression of NACC1 is linked to the treatment resistance of cancer cells [26, 27]. Abnormal expression of NACC1 was shown to be linked to the poor prognostic features of many types of malignant tumors, such as ovarian cancer [28, 29], colorectal cancer [30], melanoma [31, 32], hepatocellular carcinoma [33] and other diseases. However, the expression level of NACC1 and its biological roles and pathogenic mechanism in AML is still unknown. Thus, it is needed to investigate NACC1 as a target for treating AML.
A disintegrin and metalloproteinases (ADAMs), a multi-domain family of proteins, are regarded as a prospective target for treating cancer [34]. This protein family possesses multiple biological functions [35]. As a member of the ADAMs family, ADAM9 is deemed a potential target for treating cancer [36]. Many cancers, including ovarian cancer [37], lung cancer [38, 39], colorectal cancer [40], and stomach cancer [41], have ADAM9 overexpression. ADAM9 promotes cancer cells to migrate and grow, thereby contributing to tumor development and progression [42, 43]. Our study proved that NACC1 regulates the expression of ADAM9. As a downstream effector of NACC1, ADAM9 is a potential target for AML treatment.
This research aimed to delve into the role of NACC1 and ADAM9 in AML development and found that NACC1 promotes the progression of AML by modulating ADAM9 expression and activating the PI3K/AKT axis.
Materials and Methods
Cell culture
This study used AML cell lines (HL-60, NB4, U937, KG1a, and THP1), all derived from the American Type Culture Collection (ATCC, USA). The cell lines were cultured in RPMI-1640 medium (#C11875500BT, Gibco, USA) containing 10% FBS (#900-108, Gemini, USA) and 1% penicillin-streptomycin (#C0222, Beyotime, China). The culture was kept at 5% CO2 and 37°C.
Separation of peripheral blood mononuclear cells (PBMCs)
Peripheral blood samples from healthy donors were collected and placed in EDTA-coated tubes. A medium for mononuclear cell separation (#LDS1075, TBD, Tianjin, China) was employed to separate PBMCs and preserve them at a temperature of -80°C.
qRT-PCR
After extracting total RNA from AML cells using TRIzol reagent (#9108, Takara, Japan), the extracted RNA was reverse transcribed into cDNA using the PrimeScript™ RT reagent kit (#RR600, Takara, Japan). The SYBR® Premix Ex Taq™ II kit (#RR820A, Takara) and the CFX Connect™ RT-qPCR system (Bio-Rad, USA) were used to conduct real-time quantitative polymerase chain reaction (RT-qPCR).
All primers were synthesized by Sangon Biotech (Shanghai, China). NACC1 primers: (F: 5′-CTCTCCCGGCTGAACTTATCAAC-3′, R: 5′-GTACACGTTGGTGCCTGTCAC-3′.). ADAM9 primers: (F: 5′-TCCATTGCTCTTAGCGACTGT-3′, R: 5′-GGGGTTCAATCCCATAACTCG-3′.). β-actin primers: (F: 5′-TGACGTGGACATCCGCAAAG-3′, R: 5′-CTGGAAGGTGGACAGCGAGG-3′).
Western blotting
After collecting pretreated cells, they underwent three rounds of washing in phosphate-buffered saline (PBS), which was cooled beforehand. Then, total protein was extracted using ice-cold RIPA buffer (#P0013B, Beyotime, China) with PMSF (#ST505, Beyotime, China). The BCA protein concentration detection kit (#P0010, Beyotime, China) was employed to measure protein concentrations. After separating proteins via 10% SDS-PAGE gel electrophoresis, they were transferred to PVDF membranes (#IPVH00010, Millipore, USA) and then blocked with 5% skimmed milk for 2 hours. Subsequently, the membrane was covered with specific primary antibodies and placed in a refrigerator at 4 degrees for a minimum of 16 hours. The following primary antibodies were employed in this study: Cleaved PARP (#5625T, Cell Signaling Technology, USA), Cleaved-Caspase3 (#9664T, Cell Signaling Technology, USA), Cleaved-Caspase9 (#7237T, Cell Signaling Technology, USA), p-PI3K (#4228T, Cell Signaling Technology, USA), p-AKT (#4060T, Cell Signaling Technology, USA), Bcl-2 (#381702, ZENBIO, China), Bax (#R22708, ZENBIO, China), c-Myc (#343250, ZENBIO, China), p21 (#HA500156, HUABIO, China), p27kip1(#ET1608-61, HUABIO, China), AKT1/2/3 (#ET1609-51, HUABIO, China), PI3K (#ET1608-70, HUABIO, China), Cyclin D1 (#WL01435a, Wanleibio, China), Cyclin E (#WL01072, Wanleibio, China), NACC1 (#sc-376216, Santa Cruz Biotechnology, USA), ADAM9 (#ab218242, Abcam, UK), and β-actin (#BA2305, Boster, USA). Finally, a secondary antibody (Biosharp, China) was added to the membrane for an hour. The secondary antibody was incubated at a 1:4000 dilution ratio. The protein signal was evaluated using an ECL kit (EMD Millipore, USA).
Cell infection
AML cell lines with steady NACC1 and ADAM9 knockdown were produced using recombinant lentivirus (GenePharma, Shanghai, China). Following 48 h of lentiviral infection, the fluorescence level of GFP in cells was assessed. Subsequently, AML cells were subjected to a two-week screening process using 2 µg/ml of puromycin (#P8230, Solarbio, Beijing, China), and then, the cells were harvested for subsequent analyses.
CCK8
We used the CCK8 kit (#CA1210, Solarbio, Beijing, China) to determine cell viability. 5×103 cells were put in each well of the 96-well plates. At 0/24/48/72/96 hours, 10 μL of the CCK8 reagent was added. An enzyme-labeled instrument was utilized to obtain optical density (OD) values at 450 nm. Data were sorted and analyzed. In another experiment, cells were treated with 10 μM SC79 (#HY-18749, MCE, USA) for 48 hours and 72 hours, and cell viability was measured as described above.
Flow cytometry assay
Collected cells (1×106) were kept at 4°C overnight with 75% ethanol after three rounds of washing with cold PBS. After centrifugation the supernatant was discarded, and the cells were exposed to RNase A for 25 minutes at 37°C in darkness. Thereafter, propidium iodide (PI, Millipore) was added to the mixture. The cell cycle phase was determined utilizing FACSCaliburTM Flow cytometry (BD Biosciences) at 4°C in a dark environment.
To determine the apoptosis rate, cells (1×106) were collected and subjected to three rounds of washing and resuspending with 1 ml of PBS. Thereafter, the Annexin V APC-A and DAPI kits were used to stain cells. Finally, the CytoFLEX flow cytometer (Beckman, USA) was used to examine the stained cells. Data processing was conducted with CytExpert software (Beckman Coulter, USA).
RNA sequencing
AML cells from control and NACC1 knockdown groups were harvested. Three times independent replication experiments were conducted in each group. Following two successive washes with sterile PBS, the total RNA for each group was extracted using TRIzol (Takara, Japan) kit. The RNA-Seq library construction and subsequent data processing were conducted by LC-Bio Technology Co. Differential expression analysis of genes was conducted between two different groups using DESeq2 software (and between two samples using edgeR). The genes with the parameter of p-value less than 0.05 and absolute fold change ≥ 2 were considered differentially expressed genes.
Bioinformatics analysis
The expression of NACC1 (Dataset#227651_at) and ADAM9 (Dataset#1555326_a_at) in hematopoietic stem cells (HSCs) and normal AML patients were analyzed by investigating the BloodPool: AML samples and normal cells dataset of BloodSpot data. The downloaded data were imported into GraphPad Prism 8 for statistical analysis.
Statistical analysis
Data were obtained from 3 individual experiments. Data are displayed as mean ± standard deviation (SD). GraphPad Prism 8 was used to conduct a detailed statistical evaluation of data, including but not limited to unpaired student's t-tests and one-way ANOVA. Two samples were compared using the student's t-tests, and several samples were compared using the one-way analysis of variance. Accordingly, P<0.05 was used to define statistical significance. *p <0.05, **p < 0.01, ***p <0.001, and ****p < 0.0001, and ns indicated no significant difference.
Results
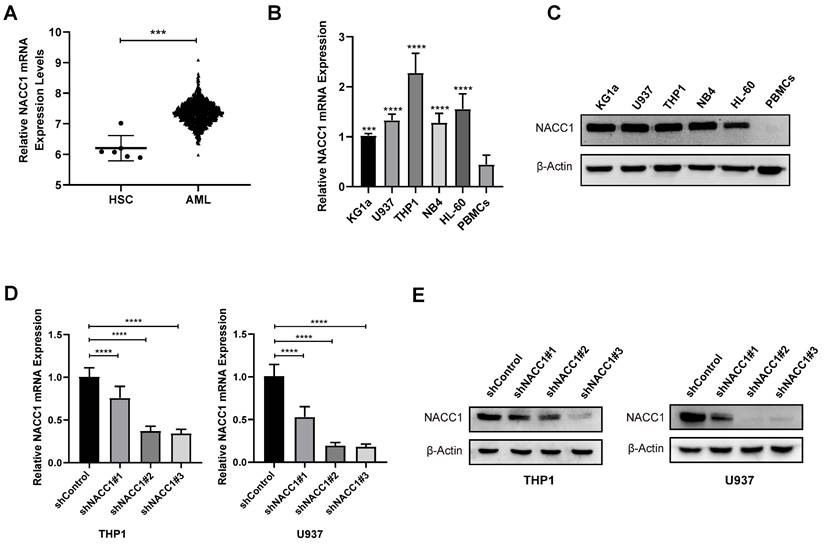
NACC1 expression and effectiveness of knockdown in AML cells
The Bloodspot database was employed to determine the levels of NACC1 mRNA in patients with AML. We found that NACC1 expression in patients with AML is much higher than that of normal human hematopoietic stem cells (HSCs) (Fig. 1A). Subsequently, we measured NACC1 expression in various AML cell lines, including HL-60, THP1, NB4, U937, and KG1a by qRT-PCR and immunoblotting. We observed a significantly higher NACC1 expression in AML cells than in PBMCs (Fig. 1B and C). Based on these data, we selected THP1 and U937 cells with high expression of NACC1 for subsequent experimental analyses. We successfully knocked down NACC1 in THP1 and U937 cells using lentivirus and proved the effectiveness of knockdown at mRNA (Fig. 1D) and protein levels (Fig. 1E). Subsequently, we selected shRNA#3 with optimal knockdown efficacy for subsequent experiments.
NACC1 expression and effectiveness of knockdown in AML cells. The BloodSpot database provided RNA-seq data. (A). Using qRT-PCR, the mRNA levels of NACC1 in PBMCs and AML cell lines (KG1a, U937, NB4, HL-60, and THP1) were determined (B). The protein levels of NACC1 in AML cells and PBMCs were measured using immunoblotting (C). THP1 and U937 cells were treated with different NACC1 shRNAs. The effectiveness of knockdown was evaluated using qRT-PCR (D) and immunoblotting (E).
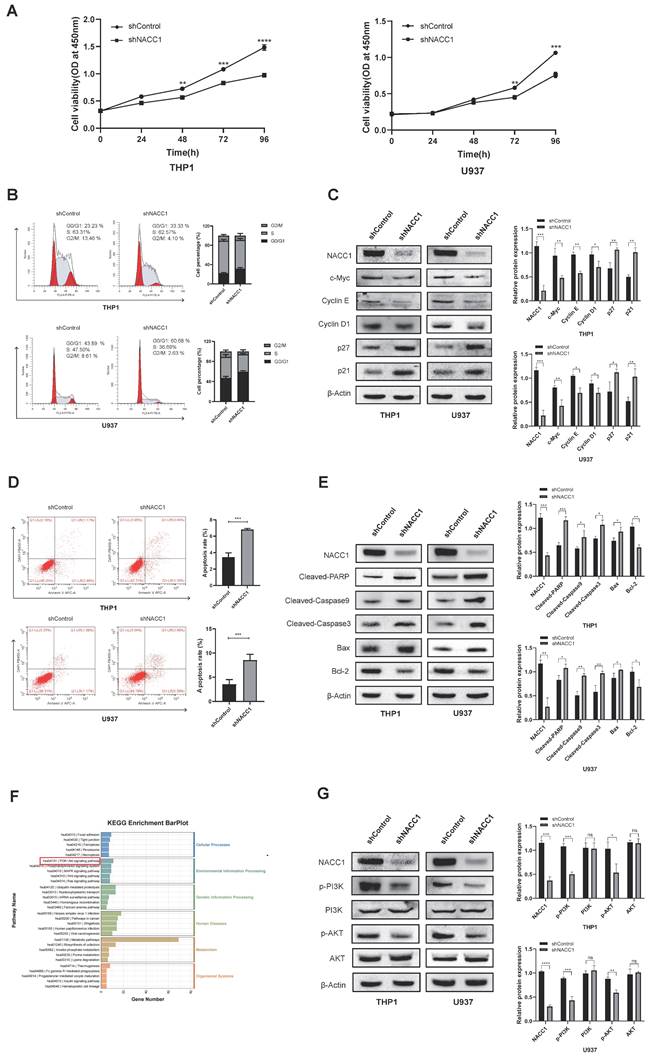
NACC1 knockdown inhibited proliferation, led to G0/G1 phase arrest, and accelerated apoptosis via the PI3K/AKT axis
To explore the exact function that NACC1 plays in AML cells, we initially employed the CCK8 assay to ascertain cell growth rates. The findings demonstrated that NACC1 knockdown markedly decelerated the proliferation of U937 and THP1 cells (Fig. 2A). We utilized flow cytometry (FCM) to assess variation in cell cycle progression. Our findings indicated that NACC1 knockdown enhanced the proportions of the G0/G1 phase in THP1 and U937 cells (Fig. 2B). Then, we measured the G0/G1 phase-related molecules and observed their changes. Immunoblotting indicated that the expression of c-Myc, Cyclin E, and Cyclin D1 reduced after NACC1 knockdown, while the levels of p21/p27 significantly elevated (Fig. 2C). Next, we used flow cytometry to investigate cells' apoptosis rate after NACC1 knockdown. Based on the results, NACC1 knockdown enhanced the apoptosis rate of AML cells (Fig. 2D). Meanwhile, Western blotting showed that the trial group with NACC1 knockdown had considerably lower expression levels of Bcl-2 compared to controls. In contrast, apoptosis-promoting molecules Bax, Cleaved-PARP, Cleaved-caspase9, and Cleaved-caspase3 all had higher expression levels in the NACC1 knockdown group (Fig. 2E). Subsequently, we conducted RNA sequencing in the NACC1 knockdown group and control group of U937 cells. Then, we conducted KEGG enrichment analysis using the sequencing data (Fig. 2F). NACC1 is abnormally expressed in cancer cells and activates the AKT-related signaling pathway, thereby exerting its biological function [44]. Therefore, it is inferred that the predominant biological role of NACC1 in AML cells is achieved by activating the PI3K/AKT axis. Immunoblotting indicated that PI3K and AKT phosphorylation levels were markedly downregulated after NACC1 knockdown, which inhibited the PI3K/AKT signaling pathway (Fig. 2G). Collectively, our results indicated that the role of NACC1 in AML is modulated through the PI3K/AKT axis.
NACC1 knockdown inhibited proliferation, led to G0/G1 phase arrest, and accelerated apoptosis via the PI3K/AKT axis. CCK8 test indicated that NACC1 knockdown suppressed the proliferation of U937 and THP1 cell lines (A). Using flow cytometry, the cell cycle distribution of THP1 and U937 cells was identified (B). Cell cycle-related molecules were detected using immunoblotting (C). The apoptotic rates of THP1 and U937 cells were assessed using flow cytometry (D). Immunoblotting of proteins linked to apoptosis (E). KEGG enrichment analysis was conducted for the significant gene signatures after NACC1 knockdown in U937 cells (F). Immunoblotting was utilized to assess the levels of proteins associated with the PI3K/AKT axis (G).
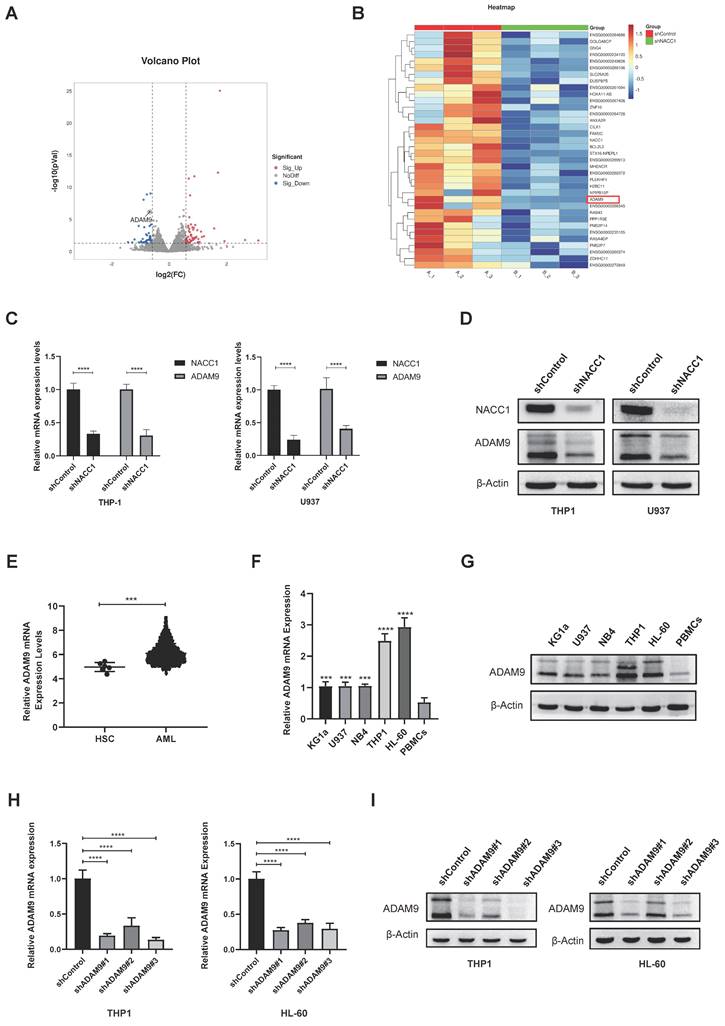
Suppression of NACC1 expression downregulated ADAM9 expression
To unravel the downstream regulatory mechanisms underpinning the effect of NACC1 on AML cells, we conducted a detailed analysis of differentially expressed genes obtained through RNA sequencing. Sequencing analysis demonstrated that NACC1 knockdown remarkably decreased the expression levels of ADAM9 (Fig. 3A and B). Immunoblotting and qRT-PCR demonstrated that ADAM9 expression was remarkably downregulated in THP1 and U937 cell lines after NACC1 silencing (Fig. 3C and D). We hypothesized that reduced NACC1 expression substantially decreased ADAM9 expression, thereby affecting the biological function of AML. To verify this theory, we first confirmed that ADAM9 was expressed at a high level in AML cells. By analyzing the Bloodspot database, we observed that normal AML patients had considerable upregulation of ADAM9 (Fig. 3E). Our findings indicated that ADAM9 expression was higher in AML cells than in PBMCs (Fig. 3F and G). Subsequently, two cell lines THP1 and HL-60 with high ADAM9 expression were selected for the lentiviral knockdown experiment. Based on the data of qRT-PCR and immunoblotting, shRNA#3 with a better knockdown efficiency was selected for subsequent experiments (Fig. 3H and I). These results indicated that low NACC1 expression downregulated ADAM9 expression and ADAM9 was highly expressed in AML.
Suppression of NACC1 expression downregulated ADAM9 expression. The volcano plot depicts some of the genes expressed in the control group and NACC1 knockdown group of U937 cells. Genes with negligible changes are shown by black dots, downregulated genes are shown by blue dots, and upregulated genes are shown by red dots. |log2FC|>=1 p<0.05 (A) Heat maps revealed differential expression genes in RNA sequences between the comparison group and the NACC1 suppression group (B). qRT-PCR (C) and immunoblotting (D) were performed to investigate the effectiveness of NACC1 knockdown in THP1 and U937 cell lines and measure ADAM9 expression after NACC1 silencing. Data used in this chart are from the BloodSpot database (E). ADAM9 expression in PBMCs and 5 AML cell lines was measured by qRT-PCR (F) and immunoblotting (G). qRT-PCR (H) and immunoblotting (I) were conducted to assess the efficiency of ADAM9 knockdown in THP1 and HL-60 cells.
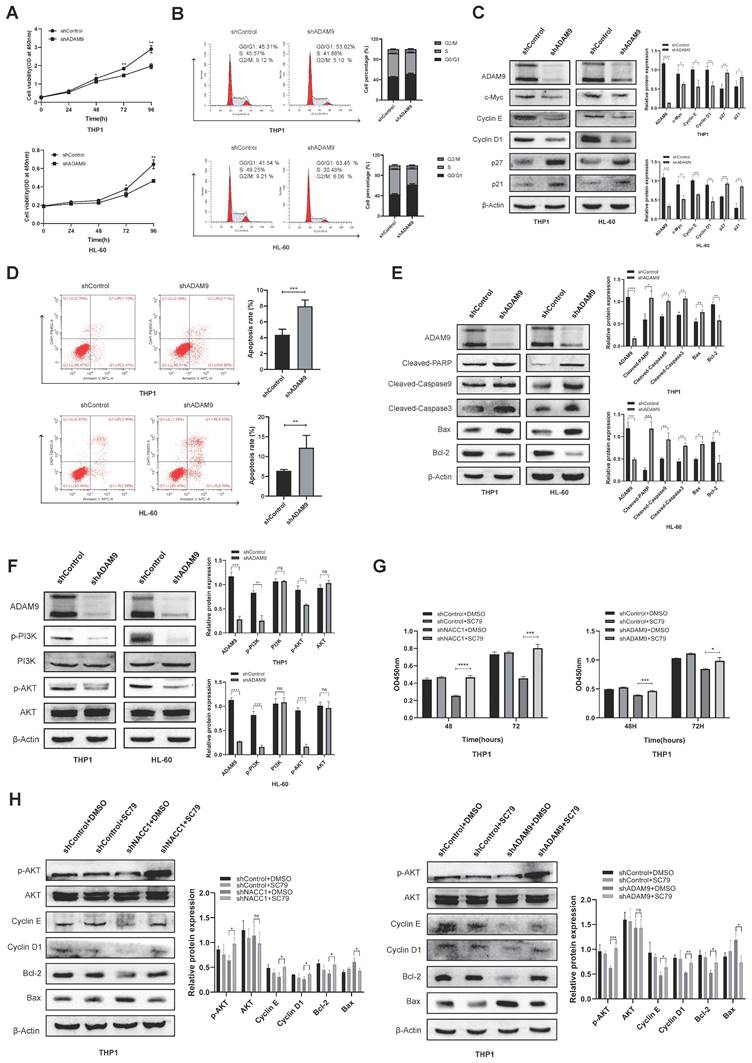
The effect of ADAM9 downregulation on AML cell activity was mediated by the PI3K/AKT pathway
Initially, we used the CCK8 assay to investigate how ADAM9 knockdown affects THP1 and HL-60 cell growth. The experimental findings indicated significant suppression of growth in both cell lines (Fig. 4A). Using flow cytometry, we discovered that ADAM9 knockdown induced the G0/G1 phase arrest (Fig. 4B). We determined the expression levels of pertinent cyclins via immunoblotting, and it was found that the protein levels of p21/p27 increased, whereas those of c-Myc, Cyclin D1 and E decreased (Fig. 4C). We examined the activity of AML cells by FCM, revealing that the apoptotic ratio of these cells was enhanced after ADAM9 silencing (Fig. 4D). Western blotting indicated that ADAM9 repression enhanced the expression of pro-apoptotic proteins, including Cleaved-PARP, Cleaved-caspase9, Cleaved-caspase3, and Bax and reduced that of Bcl-2 (Fig. 4E). Then, we explored the mechanism of ADAM9 in AML and investigated the effect of ADAM9 on the PI3K/AKT axis by knocking down ADAM9. The results showed that PI3K phosphorylation and AKT phosphorylation were downregulated, indicating that ADAM9 knockdown suppressed the activity of the PI3K/AKT axis (Fig. 4F). We treated NACC1 knockdown THP1 cells and ADAM9 knockdown THP1 cells with the AKT activator SC79 to determine whether the carcinogenic effects of NACC1 and ADAM9 are mediated by the PI3K/AKT pathway. The findings demonstrated that the addition of SC79 markedly enhanced the proliferation of cells with NACC1 and ADAM9 knockdown (Fig. 4G). In the knockdown cell lines, SC79 enhanced the expression level of p-AKT protein and altered the expression of apoptosis-related proteins Bcl-2 and Bax, as well as Cyclin E and Cyclin D1 (Fig. 4H). In conclusion, ADAM9 knockdown can promote AML cell apoptosis, decelerate AML cell growth, and change cell cycle distribution via the PI3K/AKT pathway.
The effect of ADAM9 downregulation on AML cell activity was mediated via the PI3K/AKT axis. The growth of THP-1 and HL-60 cells was suppressed by silencing ADAM9 (A). The change in the cellular cycle was determined by FCM (B). The expression levels of proteins associated with cell cycle were assessed via WB (C). The apoptosis rates of THP1 and HL-60 cell lines were evaluated via FCM (D). The expression levels of apoptosis-related proteins were measured using immunoblotting (E). Using immunoblotting, the expression levels of proteins associated with the PI3K/AKT axis were measured (F). CCK8 assay detected the proliferation of THP1 cells treated with NACC1/ADAM9 knockdown and/or SC79 (10µM) after processing (G). Effects of AKT activator SC79 on p-Akt, cycle-related proteins, and apoptosis-related proteins (H).
Discussion
The etiology of AML remains widely unknown, and the prevalence of AML has been rising in recent years [45, 46]. AML mostly affects older people, with a median diagnosis age of almost 70 years [47]. While optimizing drug dosage and combinations of new drugs during induction therapy can prolong the survival of patients with newly diagnosed AML [48-50], this strategy is less effective in older patients, and the recurrence rate remains high [51]. The precise etiology of AML remains elusive in the majority of patients, and to date, there is insufficient compelling evidence to endorse any single method as the gold standard for care [52, 53]. Accordingly, it is of great importance to delve into promising therapeutic targets. Our study clarified the mechanism by which NACC1 and ADAM9 are involved in the pathogenesis of AML. The experiment employed THP1 and U937 cells from AML-M5, with HL-60 cells from AML-M2. NACC1 and ADAM9 are markedly aberrant expressed in these three cell lines, which are often used in AML research. Meanwhile, our results corroborate the pivotal role of the NACC1/ADAM9/PI3K/AKT axis in the proliferation, apoptosis, and cell cycle of AML cells. Therefore, it provides a new idea for finding new therapeutic and diagnostic targets for AML.
NACC1 is overexpressed in various malignancies, correlating positively with tumor progression and recurrence, suggesting a poor prognosis [54]. Gao et al. compared differential genes in ovarian cancer cells after NACC1 overexpression and knockdown. They found that NACC1 was strongly associated with multiple genes and pathways associated with cancer, indicating that it is crucial for the growth of tumors [55]. Furthermore, high expression of NACC1 has been correlated with low overall survival rates of patients suffering from cervical cancer [56]. Our study confirmed that NACC1 is overexpressed in AML cell lines. NACC1 knockdown suppressed the proliferation ability of AML cells, increased the apoptosis rate, and led to cell cycle arrest. Previous reports have elucidated a close association between the abnormal expression of NACC1 in tumor cells and ATK-related signaling pathways [44, 57]. Accumulating data suggest that the PI3K/AKT/mTOR axis is critical for the development and progression of AML [58, 59]. Our KEGG enrichment analysis results indicated that NACC1 is closely associated with the PI3K/AKT signaling pathway. Our results suggested that NACC1 knockdown suppressed the PI3K/AKT pathway. Signaling pathways such as MAPK, Wnt, and Ras, which were enriched by KEGG results, regulate various biological effects, such as cell proliferation and differentiation, and play an important role in AML. Many enzymes involved in these pathways are considered to be potential targets for treating cancer [60, 61]. Thus, these results provide invaluable clues for further exploration of the mechanism of NACC1 in future experiments on AML. Briefly, our results indicated that NACC1 promotes the development of AML. Thus, NACC1 is predicted to be a useful target for the treatment of AML. It may be useful as a tumor marker to assess the prognosis of AML patients.
According to reports, the ADAMs protein family constitutes a promising family of therapeutic targets in cancer [62]. Previous studies have identified that ADAMs family members are expressed in various hematological malignancies [63]. ADAM9 is overexpressed in several tumors and affects tumor progression through different mechanisms [64]. Several studies have confirmed that abnormal ADAM9 expression can efficiently stimulate the growth and invasion of malignant cells [65, 66]. Furthermore, ADAM9 is a transcription factor that encourages angiogenesis in esophageal cancer [67]. The expression and functional role of ADAM9 in AML remain unclarified. In our study, we discovered that ADAM9 is abnormally expressed in AML cells and affects cell survival by triggering the PI3K/AKT axis. Meanwhile, we found that NACC1 knockdown in AML cells downregulates ADAM9 expression. Although the precise mechanism is yet to be fully understood, these results demonstrate the importance of ADAM9 in the treatment of AML.
Taken together, our findings demonstrated that NACC1 and ADAM9 contribute to the development of AML. Nonetheless, there are some shortcomings to this study. The lack of clinical specimens did not allow the assessment of NACC1 and ADAM9 expression in primary isolates. Future endeavors can help overcome these limitations and investigate the specific regulatory role of NACC1 in ADAM9.
Conclusion
In conclusion, this study revealed that NACC1 and ADAM9 were abnormally overexpressed in AML cell lines. The abnormal expression of NACC1 modulated the expression of ADAM9, subsequently activating the PI3K/AKT axis, enhancing the proliferation of AML cells, accelerating cell cycle progression, and inhibiting apoptosis.
Abbreviations
AML: acute myeloid leukemia; NACC1: Nucleus accumbens-associated protein 1; ADAM9: A disintegrin and metalloproteinase 9; BTB Domain: Broad Complex, Tramtrack and Bric a Brac Domain; qRT-PCR: quantitative real-time PCR; PBS: Phosphate Buffered Saline; CCK8: Cell counting kit-8; OD: optical density; FCM: Flow cytometry; FBS: fetal bovine serum; P27: p27kip1; PBMCs: peripheral blood mononuclear cells; p-PI3K: phosphorylated PI3K; p-AKT: phosphorylated AKT.
Acknowledgements
Funding
The research was funded by the Key Technology Innovation Special of Key Industries of the Chongqing Science and Technology Bureau (Grant Number: csct2022ycjh-bgzxm0034), the Postgraduate Innovation Fund of Yongchuan Hospital Affiliated to Chongqing Medical University (Grant Number: YJSCX202305).
Author contributions
Ying Zhang: Formal analysis, Methodology, Conceptualization, Data curation, Funding acquisition, Investigation, Writing - original draft.
Liang Zhong: Conceptualization, Project administration, Resources, Supervision.
Peng Wan: Investigation, Methodology.
Yi Zhao: Investigation, Methodology.
Meng Wang: Software, Methodology.
Hongyan Zhang: Investigation, Methodology.
Ying Deng: Software.
Yang Liao: Validation.
Beizhong Liu: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing - review & editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. The Lancet. 2018;392:593-606
2. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136-52
3. Sanz MA, Iacoboni G, Montesinos P, Venditti A. Emerging strategies for the treatment of older patients with acute myeloid leukemia. Ann Hematol. 2016;95:1583-93
4. Mohamed Jiffry MZ, Kloss R, Ahmed-Khan M, Carmona-Pires F, Okam N, Weeraddana P. et al. A review of treatment options employed in relapsed/refractory AML. Hematology. 2023;28:2196482
5. Zeidan AM, Podoltsev NA, Wang X, Zhang C, Bewersdorf JP, Shallis RM. et al. Patterns of care and clinical outcomes with cytarabine-anthracycline induction chemotherapy for AML patients in the United States. Blood Adv. 2020;4:1615-23
6. Choi JH, Shukla M, Abdul-Hay M. Acute Myeloid Leukemia Treatment in the Elderly: A Comprehensive Review of the Present and Future. Acta Haematol. 2023;146:431-57
7. Ramos NR, Mo CC, Karp JE, Hourigan CS. Current Approaches in the Treatment of Relapsed and Refractory Acute Myeloid Leukemia. J Clin Med. 2015;4:665-95
8. Zollman S, Godt D, Privé GG, Couderc JL, Laski FA. The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proc Natl Acad Sci U S A. 1994;91:10717-21
9. Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M. et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370-4
10. Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T. et al. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311-6
11. Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Privé GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82
12. Chaharbakhshi E, Jemc JC. Broad-complex, tramtrack, and bric-à-brac (BTB) proteins: Critical regulators of development. Genesis. 2016;54:505-18
13. Bonchuk A, Balagurov K, Georgiev P. BTB domains: A structural view of evolution, multimerization, and protein-protein interactions. Bioessays. 2023;45:e2200179
14. Zhang H, Ouyang C. BTB protein family and human breast cancer: signaling pathways and clinical progress. J Cancer Res Clin Oncol. 2023;149:16213-29
15. Mafra D, Alvarenga L, Cardozo L, Stockler-Pinto MB, Nakao LS, Stenvinkel P. et al. Inhibiting BTB domain and CNC homolog 1 (Bach1) as an alternative to increase Nrf2 activation in chronic diseases. Biochim Biophys Acta Gen Subj. 2022;1866:130129
16. Polo JM, Dell'Oso T, Ranuncolo SM, Cerchietti L, Beck D, Da Silva GF. et al. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat Med. 2004;10:1329-35
17. Chen Z, Brand NJ, Chen A, Chen SJ, Tong JH, Wang ZY. et al. Fusion between a novel Krüppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. Embo j. 1993;12:1161-7
18. Yeyati PL, Shaknovich R, Boterashvili S, Li J, Ball HJ, Waxman S. et al. Leukemia translocation protein PLZF inhibits cell growth and expression of cyclin A. Oncogene. 1999;18:925-34
19. Williams RT, Guarecuco R, Gates LA, Barrows D, Passarelli MC, Carey B. et al. ZBTB1 Regulates Asparagine Synthesis and Leukemia Cell Response to L-Asparaginase. Cell Metab. 2020;31:852-61.e6
20. Nakayama K, Nakayama N, Davidson B, Sheu JJ, Jinawath N, Santillan A. et al. A BTB/POZ protein, NAC-1, is related to tumor recurrence and is essential for tumor growth and survival. Proc Natl Acad Sci U S A. 2006;103:18739-44
21. Cha XY, Pierce RC, Kalivas PW, Mackler SA. NAC-1, a rat brain mRNA, is increased in the nucleus accumbens three weeks after chronic cocaine self-administration. J Neurosci. 1997;17:6864-71
22. Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW. et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364-8
23. Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049-61
24. Schoch K, Meng L, Szelinger S, Bearden DR, Stray-Pedersen A, Busk OL. et al. A Recurrent De Novo Variant in NACC1 Causes a Syndrome Characterized by Infantile Epilepsy, Cataracts, and Profound Developmental Delay. Am J Hum Genet. 2017;100:343-51
25. Yap KL, Fraley SI, Thiaville MM, Jinawath N, Nakayama K, Wang J. et al. NAC1 is an actin-binding protein that is essential for effective cytokinesis in cancer cells. Cancer Res. 2012;72:4085-96
26. Tang XH, Li H, Zheng XS, Lu MS, An Y, Zhang XL. CRM197 reverses paclitaxel resistance by inhibiting the NAC-1/Gadd45 pathway in paclitaxel-resistant ovarian cancer cells. Cancer Med. 2019;8:6426-36
27. Wang X, Ji C, Zhang H, Shan Y, Ren Y, Hu Y. et al. Identification of a small-molecule compound that inhibits homodimerization of oncogenic NAC1 protein and sensitizes cancer cells to anticancer agents. J Biol Chem. 2019;294:10006-17
28. Nakayama K, Rahman MT, Rahman M, Yeasmin S, Ishikawa M, Katagiri A. et al. Biological role and prognostic significance of NAC1 in ovarian cancer. Gynecol Oncol. 2010;119:469-78
29. Nakayama K, Rahman M, Rahman MT, Nakamura K, Sato E, Katagiri H. et al. Nucleus accumbens-1/GADD45GIP1 axis mediates cisplatin resistance through cellular senescence in ovarian cancer. Oncol Lett. 2017;13:4713-9
30. Ju T, Jin H, Ying R, Xie Q, Zhou C, Gao D. Overexpression of NAC1 confers drug resistance via HOXA9 in colorectal carcinoma cells. Mol Med Rep. 2017;16:3194-200
31. Tsunoda K, Oikawa H, Tada H, Tatemichi Y, Muraoka S, Miura S. et al. Nucleus accumbens-associated 1 contributes to cortactin deacetylation and augments the migration of melanoma cells. J Invest Dermatol. 2011;131:1710-9
32. Jiao H, Jiang S, Wang H, Li Y, Zhang W. Upregulation of LINC00963 facilitates melanoma progression through miR-608/NACC1 pathway and predicts poor prognosis. Biochem Biophys Res Commun. 2018;504:34-9
33. Yin L, Sun T, Liu R. NACC-1 regulates hepatocellular carcinoma cell malignancy and is targeted by miR-760. Acta Biochim Biophys Sin (Shanghai). 2020;52:302-9
34. Duffy MJ, Mullooly M, O'Donovan N, Sukor S, Crown J, Pierce A. et al. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin Proteomics. 2011;8:9
35. Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258-89
36. Peduto L. ADAM9 as a potential target molecule in cancer. Curr Pharm Des. 2009;15:2282-7
37. Guo T, Yuan D, Lin M, Zhu D, Xu N, Wang J. Aberrant expression of ADAM9 in ovarian cancer and its clinical significance. J Clin Lab Anal. 2020;34:e23136
38. Lin CY, Chen HJ, Huang CC, Lai LC, Lu TP, Tseng GC. et al. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res. 2014;74:5229-43
39. Lin CY, Cho CF, Bai ST, Liu JP, Kuo TT, Wang LJ. et al. ADAM9 promotes lung cancer progression through vascular remodeling by VEGFA, ANGPT2, and PLAT. Sci Rep. 2017;7:15108
40. Chandrasekera P, Perfetto M, Lu C, Zhuo M, Bahudhanapati H, Li J. et al. Metalloprotease ADAM9 cleaves ephrin-B ligands and differentially regulates Wnt and mTOR signaling downstream of Akt kinase in colorectal cancer cells. J Biol Chem. 2022;298:102225
41. Kim JM, Jeung HC, Rha SY, Yu EJ, Kim TS, Shin YK. et al. The effect of disintegrin-metalloproteinase ADAM9 in gastric cancer progression. Mol Cancer Ther. 2014;13:3074-85
42. Mammadova-Bach E, Zigrino P, Brucker C, Bourdon C, Freund M, De Arcangelis A. et al. Platelet integrin α6β1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight. 2016;1:e88245
43. Grützmann R, Lüttges J, Sipos B, Ammerpohl O, Dobrowolski F, Alldinger I. et al. ADAM9 expression in pancreatic cancer is associated with tumour type and is a prognostic factor in ductal adenocarcinoma. Br J Cancer. 2004;90:1053-8
44. Cao Z, Chen H, Mei X, Li X. Silencing of NACC1 inhibits the proliferation, migration and invasion of nasopharyngeal carcinoma cells via regulating the AKT/mTOR signaling pathway. Oncol Lett. 2021;22:828
45. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
46. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74:12-49
47. Versluis J, Hazenberg CL, Passweg JR, van Putten WL, Maertens J, Biemond BJ. et al. Post-remission treatment with allogeneic stem cell transplantation in patients aged 60 years and older with acute myeloid leukaemia: a time-dependent analysis. Lancet Haematol. 2015;2:e427-36
48. Burnett AK, Russell NH, Hills RK, Kell J, Cavenagh J, Kjeldsen L. et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood. 2015;125:3878-85
49. Burnett AK, Russell NH, Hills RK, Hunter AE, Kjeldsen L, Yin J. et al. Optimization of chemotherapy for younger patients with acute myeloid leukemia: results of the medical research council AML15 trial. J Clin Oncol. 2013;31:3360-8
50. Willemze R, Suciu S, Meloni G, Labar B, Marie JP, Halkes CJ. et al. High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia: results of the EORTC-GIMEMA AML-12 trial. J Clin Oncol. 2014;32:219-28
51. Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487-94
52. Carter JL, Hege K, Yang J, Kalpage HA, Su Y, Edwards H. et al. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther. 2020;5:288
53. Molica M, Breccia M, Foa R, Jabbour E, Kadia TM. Maintenance therapy in AML: The past, the present and the future. Am J Hematol. 2019;94:1254-65
54. Xie Q, Tong C, Xiong X. An overview of the co-transcription factor NACC1: Beyond its pro-tumor effects. Life Sci. 2024;336:122314
55. Gao M, Wu RC, Herlinger AL, Yap K, Kim JW, Wang TL. et al. Identification of the NAC1-regulated genes in ovarian cancer. Am J Pathol. 2014;184:133-40
56. Yeasmin S, Nakayama K, Ishibashi M, Katagiri A, Iida K, Purwana IN. et al. Expression of the bric-a-brac tramtrack broad complex protein NAC-1 in cervical carcinomas seems to correlate with poorer prognosis. Clin Cancer Res. 2008;14:1686-91
57. Li L, Yu H, Ren Q. MiR-218-5p Suppresses the Progression of Retinoblastoma Through Targeting NACC1 and Inhibiting the AKT/mTOR Signaling Pathway. Cancer Manag Res. 2020;12:6959-67
58. Darici S, Alkhaldi H, Horne G, Jørgensen HG, Marmiroli S, Huang X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. J Clin Med. 2020;9:2934
59. Nepstad I, Hatfield KJ, Grønningsæter IS, Reikvam H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int J Mol Sci. 2020;21:2907
60. Gruszka AM, Valli D, Alcalay M. Wnt Signalling in Acute Myeloid Leukaemia. Cells. 2019;8:1403
61. Lee JT Jr, McCubrey JA. The Raf/MEK/ERK signal transduction cascade as a target for chemotherapeutic intervention in leukemia. Leukemia. 2002;16:486-507
62. Duffy MJ, McKiernan E, O'Donovan N, McGowan PM. Role of ADAMs in cancer formation and progression. Clin Cancer Res. 2009;15:1140-4
63. Wu E, Croucher PI, McKie N. Expression of members of the novel membrane linked metalloproteinase family ADAM in cells derived from a range of haematological malignancies. Biochem Biophys Res Commun. 1997;235:437-42
64. Oria VO, Lopatta P, Schilling O. The pleiotropic roles of ADAM9 in the biology of solid tumors. Cell Mol Life Sci. 2018;75:2291-301
65. Moriwaki M, Le TT, Sung SY, Jotatsu Y, Yang Y, Hirata Y. et al. Relevance of A Disintegrin and Metalloproteinase Domain-Containing (ADAM)9 Protein Expression to Bladder Cancer Malignancy. Biomolecules. 2022;12:791
66. Shintani Y, Higashiyama S, Ohta M, Hirabayashi H, Yamamoto S, Yoshimasu T. et al. Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res. 2004;64:4190-6
67. Lin YS, Kuo TT, Lo CC, Cheng WC, Chang WC, Tseng GC. et al. ADAM9 functions as a transcriptional regulator to drive angiogenesis in esophageal squamous cell carcinoma. Int J Biol Sci. 2021;17:3898-910
Author contact
![]() Corresponding author: Dr. Beizhong Liu; Email: liubeizhongedu.cn.
Corresponding author: Dr. Beizhong Liu; Email: liubeizhongedu.cn.