Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Macrophage function and COPD
Macrophage diversity in COPD
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(2):298-308. doi:10.7150/ijms.100160 This issue Cite
Review
Advances on the Role of Lung Macrophages in the Pathogenesis of Chronic Obstructive Pulmonary Disease in the Era of Single-Cell Genomics
Xiaohua Li1†, Hui Zhang2†, Xianhong Chi1, Weibin Ruan1, Xia Meng1, Jiehua Deng1, Mianluan Pan1, Tingting Ma3, Jianquan Zhang1 ![]()
1. Department of Respiratory and Critical Medicine, the Eighth Affiliated Hospital, Sun Yat-Sen University, Shenzhen 518000, Guangdong Province, China.
2. Department of Respiratory and Critical Medicine, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, China.
3. Department of Respiratory and Critical Medicine, Zhuhai People's Hospital (Zhuhai Hospital affiliated with Jinan University), Zhuhai, Guangdong 519000, China.
†Equal contributors.
Received 2024-6-26; Accepted 2024-11-7; Published 2025-1-1
Abstract

Chronic Obstructive Pulmonary Disease (COPD) is a heterogeneous respiratory disorder characterized by persistent airflow limitation. The diverse pathogenic mechanisms underlying COPD progression remain incompletely understood. Macrophages, serving as the most representative immune cells in the respiratory tract, constitute the first line of innate immune defense and maintain pulmonary immunological homeostasis. Recent advances have provided deeper insights into the phenotypic and functional alterations of pulmonary macrophages and their role in COPD pathogenesis. Notably, the advent of single-cell RNA sequencing has revolutionized our understanding of macrophage molecular heterogeneity in COPD. Herein, we review principal investigations concerning the sophisticated mechanisms through which pulmonary macrophages influence COPD, encompassing inflammatory mediator production, protease/antiprotease release, and phagocytic activity. Additionally, we synthesize findings from available literature regarding all identified pulmonary macrophage sub-populations in COPD, thereby advancing our comprehension of macrophage heterogeneity's significance in the complex pathophysiological mechanisms of COPD.
Keywords: macrophages, chronic obstructive pulmonary disease, immune function, scRNAseq, cigarette smoke
Introduction
Chronic Obstructive Pulmonary Disease (COPD) represents a complex respiratory disorder characterized by persistent airflow limitation and respiratory manifestations. These clinical features stem from two primary pathological changes: airway structural abnormalities (bronchitis and bronchiolitis) and parenchymal destruction (emphysema) [1]. The global burden of COPD continues to escalate, with particularly concerning trends in low- and middle-income countries. This increasing prevalence stems from demographic shifts toward an aging population, combined with inadequate regulation of tobacco use and environmental pollutants, resulting in substantial socioeconomic impact [2,3]. COPD has become the third leading cause of death worldwide [4], emphasizing the critical need to elucidate its pathogenic mechanisms and identify novel therapeutic targets.
The pathological processes underlying COPD development encompass several interconnected mechanisms, including inflammatory response [5,6], dysregulation of protease-antiprotease balance [7], and tissue destruction [8]. Current evidence demonstrates remarkable tissue-specific heterogeneity among macrophages, as exemplified by microglia, osteoclasts, and Kupffer cells. Nevertheless, the extensive overlap in surface marker expression has impeded the precise identification and classification of different macrophage populations [9].
The emergence of single-cell RNA sequencing (scRNAseq) has transformed our understanding of macrophage diversity. This advanced methodology employs next-generation sequencing platforms to conduct comprehensive transcriptome analysis at single-cell resolution, enabling detailed examination of cellular gene expression patterns. In comparison to conventional bulk RNA sequencing approaches, scRNAseq offers superior resolution and accuracy in characterizing individual cell transcriptional profiles, facilitating the identification of both prevalent and rare cellular populations [10]. The application of scRNAseq has expanded across multiple disease contexts, providing valuable insights into macrophage diversity and immunological changes in endocrine disorders [11], hematological malignancies [12], neurological conditions [13], gastrointestinal diseases [14,15,16], and cardiovascular pathologies [17]. However, a comprehensive review of scRNAseq applications in COPD pathogenesis is lacking.
This review presents a three-dimensional analysis of macrophage involvement in COPD pathogenesis and summarizes the underlying regulatory mechanisms to identify potential therapeutic interventions. Furthermore, we provide a detailed characterization of macrophage sub-populations identified through scRNAseq, illustrating their roles in various mechanisms of COPD and underscoring the importance of macrophage heterogeneity in COPD initiation and progression.
Macrophage function and COPD
This review endeavors to elucidate the complex pathogenic mechanisms of COPD from three distinct perspectives: chronic inflammation, protease/antiprotease balance, and phagocytic activity, with particular emphasis on macrophage involvement in these processes. As depicted in Figure 1, macrophages assume diverse roles across different mechanisms, and macrophages exhibiting similar functions may represent distinct cellular sub-populations.
The role of lung macrophage in the progression of COPD (Created in BioRender: https://BioRender.com/b40o000).
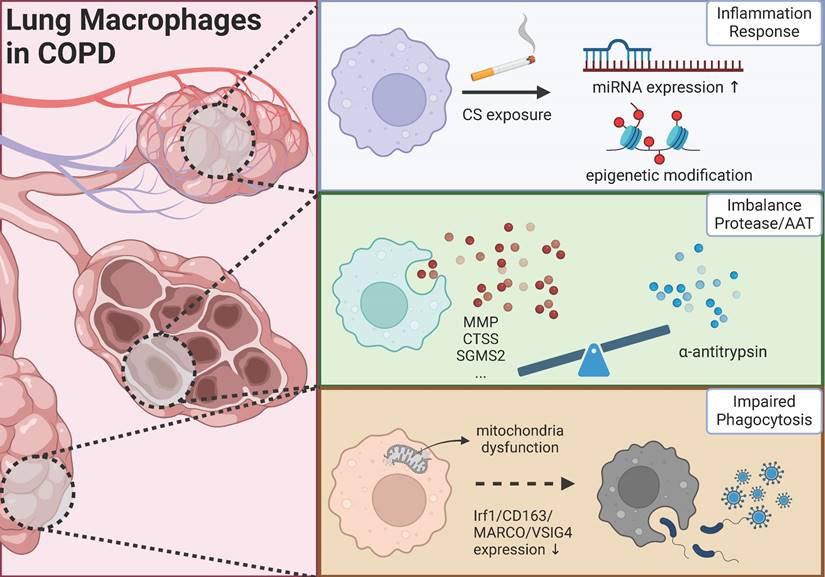
Gene expression regulation in macrophages: a critical component of COPD inflammatory mechanisms
For one thing, as a key immune cell type, macrophages play a broad role in the pathophysiology of chronic inflammatory diseases. Following chemokine signals, macrophages migrate to inflammatory sites and undergo polarization through complex signaling networks, thus coordinating inflammatory responses. For another, chronic inflammation remains a major focus in understanding COPD mechanisms. Current treatments for COPD are mainly limited to symptom management, such as airway dilation, highlighting the pressing need for new therapeutic approaches. With recent advances in biological therapies, many studies have focused on inflammatory cells and factors as potential therapeutic targets for COPD, and several clinical trials of COPD-specific biological agents are ongoing. However, the complete picture of how macrophages regulate inflammation in COPD remains unclear. We review recent studies from a new perspective of gene regulation, examining how macrophages control COPD inflammation through miRNA and epigenetic modifications.
MicroRNAs (miRNAs) are small, non-coding RNA molecules, typically comprising approximately 23 nucleotides in length, that lack the capacity for translation into proteins [18]. Extensive research has demonstrated that miRNA are widely involved in cellular biological processes, thereby maintaining tissue and organ homeostasis. Conversely, dysregulation of miRNA expression can lead to disease onset and progression. Recent studies have begun to uncover the specific mechanisms by which miRNA in pulmonary macrophages regulate chronic lung inflammation, particularly in COPD. Specifically, miR-21-3P exhibits heightened expression levels in alveolar macrophages (AMs) of mice exposed to cigarette smoke (CS) [19]. Moreover, elevated levels of miR-21 and miR-155 are observed in lung samples from both smokers with and without COPD, as well as in macrophages exposed to cigarette smoke extract (CSE) [20,21]. Significantly, epithelial cell exocytosis is found to decrease the secretion of miR-21 in response to higher concentrations of CSE [22], which suggests a potential role for miR-21 in regulating apoptosis and inflammation, thereby implicating its involvement in the pathogenesis of CS-induced COPD. Notably, variations in miRNA expression profiles among lung macrophages across distinct lung regions, attributed to variances in oxygen levels, indicate the necessity of integrating spatial miRNA levels for a comprehensive evaluation of macrophage functionality [23]. Investigating the mechanistic involvement of miRNA in macrophages in COPD may serve as a valuable approach for targeted diagnostic and therapeutic strategies, leveraging the inherent stability of miRNAs and the advanced technology of miRNA high-throughput sequencing to enhance the precision and sensitivity of miRNA detection. Furthermore, the analysis of miRNA-mRNA interactions may offer insights into cellular interactions within the context of COPD. For instance, a study employs next-generation sequencing and bioinformatics analyses to identify the miR6511a-5p-NT5E interaction [24], highlighting the intimate relationship between bronchial epithelial cells and macrophages in the airway micro-environment of COPD. Based on these collective findings, we propose that investigating the mechanistic involvement of miRNAs in COPD macrophages can be enhanced by leveraging both the inherent stability of miRNA and advanced high-throughput sequencing technologies, which together improve the precision and sensitivity of miRNA detection. Furthermore, miRNA-containing extracellular vesicles are emerging as promising novel biomarkers for early COPD diagnosis and severity assessment [25]. However, current studies provide limited evidence regarding the regulatory role of macrophage-derived, miRNA-carrying extracellular vesicles in COPD inflammatory mechanisms.
Furthermore, epigenetic modifications also play a significant role in the pathogenesis of COPD. Specifically, N6-methyladenosine (m6A) is an epigenetic modification that regulates gene expression and function by impacting post-transcriptional RNA modifications [26]. The increased gene of m6A modifications in lung tissues of both stable COPD and acute exacerbation of COPD (AECOPD) mice indicates that differentially methylated genes are implicated in signaling pathways related to immune function [26]. Methylation in macrophage genes in individuals with COPD plays a significant role in the development of the disease [1,27]. Analysis of genome-wide methylation patterns in sputum cells indicates that methylation of genes in macrophages in COPD primarily affects those associated with Major Histocompatibility Complex (MHC) class I and class II molecules linked to human leukocyte antigen (HLA) genes [28]. These findings underscore the potential impact of methylation on the inflammatory response within the lungs. Methylation is intricately linked to monocyte recruitment in COPD, as indicated by epigenetic networks that propose the involvement of the macrophage epigenetic factor protein arginine methyltransferase 7 (PRMT7) in the transcription and monomethylation of the RAP1A-regulated histone protein, which is crucial for monocyte recruitment [29,30]. Furthermore, in conjunction with methylation alterations, acetylation modifications play a significant role in the development of COPD. Through the examination of transcriptomic, proteomic, and acetylomic data obtained from mice model of COPD induced by CS, it is observed that aldolase A (ALDOA) is notably downregulated and hyperacetylated in the lung tissues of both COPD patients and mice [31], which suggests that ALDOA may serve as a promising diagnostic tool and/or biomarker for COPD. The decreased expression of key enzymes involved in the novo NAD+ synthesis pathway in macrophages, including HAAO, KMO, KYNU, and QPRT, in the chronic particulate matter (PM)-induced mouse model of COPD results in reduced expression of the kynurenine pathway and subsequent histone acetylation, and it implied that histone acetylation is responsible for the increased expression of proinflammatory genes in macrophages exposed to PM [32]. Targeting metabolic pathways in macrophages and manipulating epigenetic reprogramming represent potential novel strategies for treating COPD [32]. While epigenetic editing is anticipated to induce enduring changes in gene expression, further investigation is required to comprehensively elucidate the impact on the intrinsic chromatin landscape.
Macrophage-associated proteases/protease inhibitors and COPD
In 1997, Hautamaki et al. [33] first demonstrated in animal models that pulmonary macrophages release elastase, a key factor in CS-induced emphysema. This discovery prompted an investigation into the types and production mechanisms of these elastases. Subsequently, Finlay et al. [34] found that AMs in bronchoalveolar lavage fluid (BALF) samples from emphysema patients secreted high levels of proteases, including matrix metalloproteinases (MMPs), leading to elastolysis and damage of alveolar tissue.
With technological advances, the diversity of these proteases and their associated mechanisms have been progressively revealed. Currently, MMPs, primarily synthesized by macrophages, are widely recognized as the principal proteases in emphysema, particularly MMP-12 [35]. Patients with COPD show elevated MMP-12 protein levels in sputum compared to non-COPD individuals [36], and their lungs contain a higher number of MMP-12-positive macrophages [37]. Mechanistically, MMP-12 enhances placental growth factor (PGF) expression and upregulates its downstream signaling molecules, resulting in bronchial epithelial cell apoptosis and emphysema development [38]. Targeting the activity of transient receptor potential mucolipin 3 (TRPML3) affects endolysosomal trafficking and phagocytosis of MMP-12 in pulmonary macrophages [39], ultimately reducing COPD-related emphysema incidence. Beyond MMP-12, other MMP family members including MMP-9 and MMP-1 have been documented in the literature [40,41]. Notably, some researchers have proposed that macrophages with high MMP-12 expression represent distinct cellular sub-populations in atherosclerosis [42]. We suggest that AMs might similarly be classified into sub-populations, although COPD research has yet to establish clear definitions for such classifications.
Other proteases merit significant attention. Cathepsin S (CTSS), an elastolytic protease, shows increased expression in macrophages during COPD progression, leading to exacerbated lung tissue damage [43]. Additionally, proteinases with a disintegrin and a metalloproteinase domain (ADAMs) are intricately linked with macrophages. Specifically, in smokers and even more so in COPD patients, lung macrophages exhibit heightened and detrimental expression of ADAM9 [44], often resulting in tissue damage and promoting emphysema development. Notably, ADAM8 [45], an enzyme known for its protective properties, is found to be reduced in individuals with COPD, Additionally, ADAM8 deficiency has been shown to exacerbate lung inflammation by inhibiting CS-induced activation of the intrinsic apoptosis pathway in macrophages, although its expression in macrophages is still undetermined.
Conversely, protease inhibitors play a vital role in maintaining protease balance. Alpha-1 antitrypsin (AAT), encoded by the SERPINA1 gene, functions as an elastase inhibitor that modulates pulmonary inflammatory responses. A large-scale, multicenter retrospective study demonstrated significant survival benefits of plasma-purified AAT intravenous therapy in patients with severe AAT deficiency (AATD) [46]. However, research indicates that individuals with AATD, particularly those with genetic mutations resulting in insufficient AAT levels, exhibit elevated expression of matriptase (a membrane-bound serine protease) in their AMs [47]. This elevation leads to extracellular matrix degradation and pulmonary tissue destruction. Additionally, matriptase may regulate MMP-14 activity, indirectly contributing to lung extracellular matrix degradation. These findings not only partially explain the limitations of AAT supplementation therapy but also identify potential therapeutic targets.
Impaired phagocytosis of macrophages in COPD
Lung macrophages play a dual role in maintaining pulmonary homeostasis by facilitating the clearance of apoptotic cells to prevent the release of intracellular toxic substances and promoting tissue repair, while also participating in the phagocytosis of particles and microorganisms to orchestrate the immune response during infection.
In individuals with COPD, the phagocytosis of lung macrophage is often compromised [48]. Proteomic analysis of lung tissues has revealed a downregulation in the expression of CD163, MARCO, and VSIG4 in macrophages, indicating a decrease in their phagocytic activity [49]. Recent scRNAseq studies have identified various causes of macrophage phagocytic impairment, including smoking, microenvironment, methylation etc. [50]. Specifically, smoking has been shown to disrupt the bronchial-mucus barrier, leading to changes in cellular composition and transcriptome as well as increased mucus production [51]. The alteration in the microenvironment results in epigenetic reprogramming of airway macrophages, ultimately affecting the expression of Irf1 and subsequently reducing macrophage autophagy and phagocytosis [52]. Reduced methylation has been found to potentially play a role in the defective efferocytosis of AMs in patients with COPD [53]. Numerous studies have indicated that mitochondrial dysfunction is a significant factor in the impaired phagocytosis of macrophages in COPD, characterized by proton leakage and reductions in maximal and spare respiratory capacity, coupling efficiency, and the bioenergetic health index, reflecting the overall status of mitochondria [54]. The upregulation of Iron-responsive element-binding protein 2 (IRP2) results in increased mitochondrial iron accumulation and cytochrome c oxidase (COX) levels, ultimately leading to mitochondrial dysfunction and the development of experimental COPD [55]. The NADPH-generating enzyme malic enzyme 1 (ME1) has been shown to restore mitochondrial function and energy metabolism, effectively reversing phagocytic dysfunction in lung macrophages affected by COPD. ME1 is identified as a target of Nuclear factor erythroid 2-related factor 2 (NRF2), with selective activation of NRF2 resulting in the reduction of reactive oxygen species (ROS), enhancement of energy status, maintenance of redox homeostasis, and restoration of mitochondrial biological characteristics and energy metabolism [56,57]. The findings that mice treated with mitochondrial complexing agents or placed on a low-iron diet were shielded from CS-induced COPD indicate a significant involvement of the mitochondrial iron axis in COPD pathogenesis, highlighting its potential as a therapeutic target [55]. Furthermore, the expedited degradation and reduced half-life of Rubicon, a crucial protein in phagosomal vesicle formation via the lysosomal pathway, impaired phagocytosis in macrophages [58]. The improvement of the Wnt5a-rac1-disheveled autophagy pathway linked to phagocytosis in macrophages leads to the restoration and enhancement of macrophage phagocytosis of bacteria [59]. However, the use of corticosteroids commonly utilized in the treatment of COPD does not ameliorate lung macrophage phagocytosis dysfunction [60], and the underlying cause of this phenomenon remains uncertain.
The compromised phagocytic function of macrophages in individuals with COPD contributes to the colonization of microbial pathogens in the lower respiratory tract. For instance, patients with COPD exhibit impaired clearance of Haemophilus influenzae by AMs [61], and one factor is the delayed secretion of the antimicrobial peptide S100A8/A9 by epithelial cells, which possesses phagocytosis-enhancing properties [62]. Notably, AECOPD is commonly precipitated by respiratory infections, most of which are viral [63]. In a retrospective study in Korea, approximately 41.2% of severe acute exacerbations are attributed to viral infections, with human rhinovirus (HRV) being one of the most commonly identified viruses [64]. HRV impairs the ability of lung macrophages in patients with COPD to phagocytose bacteria while having no impact on phagocytosis in macrophages of healthy individuals [65]. Moreover, the impact of the human immunodeficiency virus (HIV) on phagocytosis in lung macrophages in COPD is evident through the increased presence of poorly phagocytosed double-negative (CD40- CD163-) lung macrophages [66]. It implies that viral infections exacerbate phagocytosis deficiencies of macrophages in patients with COPD. Notably, derivatives of Tanshinones (TS) have been shown to mitigate the exacerbation of COPD caused by viral infection, with TS demonstrating beneficial effects dependent on hemagglutinin in macrophages [67]. However, further research is needed to fully understand the causal relationship between viruses and phagocytosis dysfunction in macrophages in COPD. All in all, the colonization of microbiota in individuals with COPD varies depending on the underlying causes, with an observed correlation between the degree of impairment in phagocytosis and the increased load of respiratory pathogens [68]. It highlights the importance of investigating microbial colonization patterns based on the clinical phenotypes of COPD. The heightened cytophagocytosis of black carbon (BC) or carbon content by airway macrophages as potential predictors of respiratory symptoms and impaired lung function following exposure to environmental pollutants like indoor PM2.5 or CS exposure presents a novel avenue for diagnosis of COPD [69,70]. Further research is warranted to investigate the intricate relationship and underlying mechanisms between pathogenic microorganisms and the phagocytosis of lung macrophages in COPD. Addressing the dysfunction of phagocytosis in lung macrophages may potentially mitigate the advancement of COPD. Restoring macrophage phagocytosis, in conjunction with innovative strategies such as novel antimicrobials, vaccines, or phage therapeutics to rebalance the airway microbiota, could present a promising avenue for future therapeutic interventions.
Macrophage diversity in COPD
Although the inflammatory mechanisms of COPD have been increasingly detailed, current therapeutic approaches remain largely limited to symptomatic treatments such as bronchodilators and ventilators, rather than targeting inflammation directly. This limitation partly stems from the marked heterogeneity of pulmonary inflammation in COPD, characterized by variations in inflammatory chemokines, predominant inflammatory cells, and inflammation intensity. These factors create complex inflammatory networks, potentially explaining the differential treatment responses among patients.
Classical classification of macrophages in COPD
Previous studies have identified four distinct macrophage phenotypes: M0 (non-polarized), M1, M2, and M1-M2 (double-positive) [71-74]. M1 macrophages exhibit pro-inflammatory characteristics and are typically associated with inflammatory micro-environments [74], while M2 macrophages are characterized by their anti-inflammatory properties and are predominantly found in normal lung tissue and during inflammation resolution [73,74]. For instance, Mycobacterium tuberculosis infection intensifies macrophage polarization responses to CS toward both M1 and M2 phenotypes [75]. In COPD, significant increases in the proportion of both M1 and M2 macrophages may indicate disease progression [76,77].
Numerous studies have not only confirmed this finding but also revealed potential targets within M2 macrophages. COPD severity can be indirectly assessed by monitoring levels of TREM-2, an immunoregulatory receptor involved in M2 macrophage polarization and phenotype switching, and the TREM-2/TREM-1 ratio [78,79]. Peroxisome proliferator-activated receptor γ (PPARγ) agonists enhance M2 macrophage function by inhibiting JAK-STAT, MAPK, and NF-κB pathways [76,80]. However, conflicting studies suggest that PPARγ transcription may be impaired in macrophages [80]. Increased M2 macrophage proportions lead to enhanced epithelial-mesenchymal transition (EMT), a process closely associated with CS-activated TGF-β/Smad signaling in M2 macrophages [79]. Notably, bronchial epithelial cells counteract reduced EMT by decreasing miR-21 content in extracellular vesicles (EVs), thereby reducing M2 macrophage polarization [22].
These findings highlight the limitations of the classical M1/M2 polarization balance theory in explaining macrophage alterations in COPD patients' lungs. Some studies directly suggest that a substantial proportion of macrophages cannot be classified within this dichotomous framework, particularly in healthy individuals and COPD patients [48, 81-82]. Significantly, Yu et al. [42] synthesized recent single-cell RNA sequencing data from atherosclerosis studies, offering new insights into the traditional M1/M2 classification while proposing additional macrophage sub-populations. This suggests the need to seek new perspectives in understanding the complex macrophage polarization regulatory mechanisms in COPD, with scRNAseq potentially offering a promising approach.
Macrophage diversity in COPD revisited in the era of single-cell omics
In 2009, Tang et al. [83] achieved the first parallel detection of single-cell mRNA through scRNAseq, marking the beginning of the single-cell sequencing era. Subsequently, scRNAseq technology has rapidly evolved and been increasingly applied to cell atlas construction, exploration of cellular heterogeneity and sub-populations, and biomarker discovery. This advancement has expanded our understanding of macrophage heterogeneity: Mulder et al. [84] utilized scRNAseq to reveal macrophage diversity in health and disease, suggesting this technology's potential in designing personalized macrophage immunotherapy strategies. Furthermore, scRNAseq has elucidated how various pulmonary cells participate in COPD pathogenesis at single-cell resolution, revealing additional potential therapeutic targets. Lee et al. [85] conducted a meta-analysis of scRNAseq datasets, mapping the associations of diverse immune cells in COPD etiology, thereby enhancing our understanding of COPD pathological pathways. Xu et al. [86] employed scRNAseq to identify a cathepsin L (CTSL)-secreting eosinophil population crucial in mouse emphysema development, proposing CTSL as a potential therapeutic target. Consequently, we believe that the emergence of scRNAseq technology can address some limitations of classical macrophage classification by identifying additional macrophage sub-populations, ultimately revealing more comprehensive immunoregulatory mechanisms. Here, we present a multi-dimensional analysis of how scRNAseq demonstrates pulmonary macrophage diversity in COPD patients, while also highlighting current limitations in this research direction. Additionally, we have compiled these studies in Table 1 to provide readers with a more intuitive understanding of the rich diversity of macrophage sub-populations.
Overview of macrophage subsets and correspondent marked genes by scRNAseq.
| Reference | Sample Source | Macrophage Subsets: Marker Genes (Function) |
|---|---|---|
| Liégeois et al. 2022 [81] | BALF in healthy nonsmokers, smokers without COPD, and smokers with COPD | Cluster 1: FAPB4, AKR1C1, LIMA1 (lipid metabolism) Cluster 2: NCF1, ALOX5AP, CES1 (response to toxic substances) Cluster 3:SPP1, SELENOP, MMP9 (matrix components): Cluster 4: MKI67, PCLAF, CDK1 (cell cyclic) |
| Hu et al. 2023 [82] | Explanted lung tissue of COPD | COPD-predominant: AMs: Cluster 0: FABP4 (lipid metabolism) Control-predominant: non-AMs: Cluster 1: FABP4, CCL2, IFI6 Cluster 3: MT1G Cluster 4: SFTPC Cluster 5: VSIG4 Cluster 7: SPP1, S100A8/9 Cluster 9: BAG3 Cluster 13: SERPINB2 AMs: Cluster 12: FABP4 Cluster 14: MALAT1 Mixed: non-AMs: Cluster 6: RGS1 |
| Baßler et al. 2022 [48] | BALF and peripheral blood from early-stage COPD | MФ3: C1QA-C, SERPINA1 (protease inhibition) MФ5 : VCAN, S100A8, CCL2, CHIT1 (monocyte attractant) MФ6 : HLA-DQ (antigen presentation) MФ7 : IFIT1, IFIT2 (viral defense) MФ8 : MKI67, TOP2A, NUSAP1, HIST1H4C, HIST1H1D (proliferation) MФ9 : HLA-DR (antigen presentation) MФ12 : HBA2, HBA1, HBB (erythrocytes or oxygen transport) |
| Wan et al. 2024 [88] | Lung tissues with ACOS and without ACOS | AMs: C1QA, C1QB, CD68, APOC1 IMs: LGMN, RNASE1, CCL2 |
| Murano et al. 2024 [102] | Lung in patient with COPD and mice exposed to CS | MoAMs : LPLAT9 (pro-inflammation factor PAF) |
| Wohnhaas et al. 2024 [103] | Lung of eight-week-old female C57BL/6J mice with CS | MoAMs (pro-inflammation/tissue remolding) ResAMs (lipid metabolism) |
| Li et al. 2023 [50] | Lung tissues with non-COPD and COPD (mild/moderate) | M1 : SOD2, GPX4 (resistance to ferroptotic death) M2 : HO-1 (suspectial to ferroptotic death) |
Abbreviations: AMs, Alveolar Macrophages; ACOS, Asthma-COPD Overlap Syndrome; BALF, Bronchoalveolar Lavage Fluid; COPD, Chronic Obstructive Pulmonary Disease; CS, Cigarette Smoking; IMs, Interstitial Macrophages; MoAMs, Monocyte-derived Alveolar Macrophages; ResAMs, Resident Alveolar Macrophages.
A study conducted scRNAseq on BALF samples from healthy non-smokers and COPD patients, reveals the presence of four distinct macrophage sub-populations in COPD [81], and the classification of CD206+ macrophages based on their autofluorescence levels was achieved by integrating flow cytometry and bulk RNA sequencing (bulk-seq) methodologies, resulting in the identification of CD206+ autofluorescenthigh AMs (tissue resident-derived) and CD206+ autofluorescentlow AMs (monocyte origin) [81]. In a research investigation involving 18 patients diagnosed with COPD and 28 control subjects, the monocytes/macrophages present in the pulmonary tissues of the participants were observed to be classified into 16 distinct sub-populations, among these sub-populations, three primary classes of monocyte/macrophage populations were found to be prevalent in individuals with COPD, including elevated expression levels of FABP4, CD52, and IL-1β respectively, implying a severe inflammatory reaction [82]. These findings indicate that scRNAseq offers a superior level of resolution in comparison to conventional methodologies. Furthermore, the diversity of lung macrophages is observed to vary across different stages of COPD. Analysis of scRNAseq from BALF in early COPD revealed the presence of distinct macrophage sub-populations exhibiting elevated expression of genes related to proliferation, histocompatibility II, and hemoglobin following the phagocytosis of erythrocytes [48]. This phenomenon has been corroborated in other studies [87].
ScRNAseq is utilized for pseudo-temporal analysis to infer cell trajectories and RNA speeds, providing a deeper examination of the dynamic changes in macrophages during various disease stages of COPD or its different sub-populations, which is advantageous for identifying potential therapeutic targets and compounds. For instance, through a comparison of cell dynamics in healthy and COPD samples, novel therapeutic targets for COPD were discovered in this study, for example, inhaled corticosteroids and other compounds can modulate the transcriptomic profile of distinct lung macrophages and monocytes in patients with COPD [82]. But the research contributes valuable insight, because in comparison to research in other disciplines, there is a scarcity of studies on the various stages of COPD development, warranting further investigation. In addition, COPD frequently presents comorbidities with other diseases, typically exhibiting heterogeneity of macrophages. One such comorbidity is Asthma and COPD Overlap Syndrome (ACOS). ScRNAseq reveals that monocytes/macrophages are the predominant cell type in lung tissues of patients with ACOS, constituting over 50% of the cells examined [88].
Numerous academic studies have demonstrated that the integration of various omics techniques has enhanced the efficacy of diagnosing and treating diseases. In the context of COPD, utilizing induced sputum metabolomics/lipidomics can yield a diagnostic framework that effectively discriminates between asthma and COPD [89]. By refining and amalgamating protein-protein interaction (PPI), proteomics, and transcriptomics data pertinent to COPD, predictive models for disease classification can be further developed [90], thereby enhancing the precision of analyses conducted on limited sample sizes utilizing multi omics data, and previous studies have also reported such findings [91]. Omics approaches—including transcriptomics, epigenetics, whole-exome and wholegenome sequencing—have identified rare genetic determinants of COPD. These findings enhance our understanding of disease susceptibility and the heterogeneity observed among patients [92]. The integration of scRNAseq is anticipated to enhance the accuracy of disease diagnosis and treatment [93]. In the future, the integration of scRNAseq technology with other omics techniques will be instrumental in elucidating the pathogenesis of macrophages in COPD.
The utilization of scRNAseq in the medical field has experienced notable growth over the last decade. Emerging technologies derived from scRNAseq, such as spatial transcriptomics (ST) platforms like 10x Visium and NanoString GeoMx, offer intricate insights into the spatial heterogeneity of lung macrophage sub-populations [94,95]. However, the restricted resolution of these platforms hinders the comprehensive characterization of lung macrophages. It is noteworthy that Stereoseq and DBiT Seq exhibit enhanced resolution and diversity [96-98], thereby offering more comprehensive spatial information regarding mRNA and protein molecules at the individual cell level. Furthermore, ATAC-seq represents a novel methodology utilizing transposase to pinpoint chromosomal regions suitable for transcription [99,100]. A study utilizes ATAC-seq to assess the enrichment of COPD risk variants and investigate the regulatory impact of chromatin-level information on lung cells [101]. These methodologies offer insights into the spatial and functional heterogeneity of various cell sub-populations.
Henceforth, it is imperative to integrate additional novel technologies in order to investigate the spatial diversity, and molecular and functional diversity of lung macrophages, and gain a more comprehensive understanding of the intricate regulation of lung macrophages sub-populations in COPD.
To sum up, there is a limited number of current studies utilizing scRNAseq to analyze macrophages in COPD, with small sample sizes being a common limitation. A consensus on the classification of macrophages in COPD has yet to be established. Future research should focus on expanding the use of scRNAseq in conjunction with other methodologies to compare macrophage populations from various origins, as well as refining and standardizing criteria for monocyte/macrophage classification. Simultaneously, it is imperative to thoroughly consider the diverse nature of COPD and its associated comorbidities in order to investigate the role of macrophages in the pathogenesis of the disease. This exploration may lead to the identification of potential therapeutic targets for precise intervention and modification of the disease's trajectory, thereby enhancing the progress of personalized therapy.
Conclusion
This review comprehensively examines the role of pulmonary macrophages in COPD pathogenesis, with particular emphasis on the impact of altered macrophage immune functions, including inflammatory mediator secretion, protease/antiprotease release, and phagocytic activity on COPD progression. Furthermore, we highlight the diversity of pulmonary macrophages in the COPD microenvironment revealed through scRNAseq. The discovery of these novel sub-populations has broadened our understanding of immune regulation in COPD. We propose that leveraging innovative technologies and methodologies to enhance our comprehension of pulmonary macrophage mechanisms in COPD is crucial for developing precision medicine strategies for COPD treatment.
Acknowledgements
Funding
This work was supported by grants from the Natural Science Foundation of Guangdong Province (No: 2024A1515011073), the Shenzhen Science Technology Program (JCYJ 20230807110914029), Futian Healthcare Research Project (FTWS2022019), the Natural Science Foundation of Guangdong Province (No: 2023A1515012987), Shenzhen Science Technology Program (JCYJ20210324115000002), Zhuhai People's Hospital Cultivation Project (NO:2019PY-18),Beijing Norman Bethune Public Welfare Foundation (No: BJ-RW2020022J) and Clinical Key Specialty Project in Shenzhen's Futian District (No: QZDZK-202406).
Author contributions
Author Contributions XL and HZ contributed equally to the writing of the manuscript. XC, WR, XM, JD, MP and MT participated in literature research and editing. JZ conceived and designed the work. All authors contributed to the article and approved the submitted version.
Consent for publication
All co-authors have read and agreed with the content of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Agustí A, Celli BR, Criner GJ. et al. Global Initiative for Chronic Obstructive Lung Disease 2023 Report: GOLD Executive Summary. Arch Bronconeumol. 2023;61:2300239
2. Rossaki FM, Hurst JR, van Gemert F. et al. Strategies for the prevention, diagnosis and treatment of COPD in low- and middle- income countries: the importance of primary care. Expert Rev Respir Med. 2021;15(12):1563-77
3. Vos Theo. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1204-22
4. Safiri S, Carson-Chahhoud K, Noori M. et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: Results from the global burden of disease study 2019. BMJ. 2022;378:e069679
5. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. 2021;76(2):188-95
6. Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74(7):1249-56
7. Wang C, Zhou J, Wang J. et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. 2020;5(1):248
8. Liu G, Philp AM, Corte T. et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. 2021;225:107839
9. Geissmann F, Gordon S, Hume DA. et al. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10(6):453-460
10. Sin DD. What Single Cell RNA Sequencing Has Taught Us about Chronic Obstructive Pulmonary Disease. Tuberc Respir Dis (Seoul). 2024;87(3):252-260
11. Sharifiaghdam M, Shaabani E, Faridi-Majidi R. et al. Macrophages as a therapeutic target to promote diabetic wound healing. Mol Ther. 2022;30(9):2891-908
12. Li W, Wang F, Guo R. et al. Targeting macrophages in hematological malignancies: recent advances and future directions. J Hematol Oncol. 2022;15(1):110
13. Zhou K, Han J, Wang Y. et al. The therapeutic potential of bone marrow-derived macrophages in neurological diseases. CNS Neurosci Ther. 2022;28(12):1942-1952
14. Yunna C, Mengru H, Lei W. et al. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090
15. Vonderlin J, Chavakis T, Sieweke M, Tacke F. The multifaceted roles of macrophages in NAFLD pathogenesis. Cell Mol Gastroenterol Hepatol. 2023;15(6):1311-1324
16. Hegarty LM, Jones GR, Bain CC. Macrophages in intestinal homeostasis and inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2023;20(8):538-553
17. Xue S, Su Z, Liu D. Immunometabolism and immune response regulate macrophage function in atherosclerosis. Ageing Res Rev. 2023;90:101993
18. Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell. 2009;136(2):215-233
19. Chen Z, Wu H, Fan W. et al. Naringenin suppresses BEAS-2B-derived extracellular vesicular cargoes disorder caused by cigarette smoke extract thereby inhibiting M1 macrophage polarization. Front Immunol. 2022;13:930476
20. Kim RY, Sunkara KP, Bracke KR. et al. A microRNA-21-mediated SATB1/S100A9/NF-κB axis promotes chronic obstructive pulmonary disease pathogenesis. Sci Transl Med. 2021;13(621):eaav7223
21. De Smet EG, Van Eeckhoutte HP, Avila Cobos F. et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. 2020;13(3):423-36
22. He S, Chen D, Hu M. et al. Bronchial epithelial cell extracellular vesicles ameliorate epithelial-mesenchymal transition in COPD pathogenesis by alleviating M2 macrophage polarization. Nanomedicine. 2019;18:259-71
23. Armstrong DA, Nymon AB, Ringelberg CS. et al. Pulmonary microRNA profiling: implications in upper lobe predominant lung disease. Clin Epigenetics. 2017;9:56
24. Chang WA, Tsai MJ, Jian SF. et al. Systematic analysis of transcriptomic profiles of COPD airway epithelium using next-generation sequencing and bioinformatics. Int J Chron Obstruct Pulmon Dis. 2018;13:2387-98
25. Lv J, Xiong X. Extracellular vesicle microRNA: A promising biomarker and therapeutic target for respiratory diseases. Int J Mol Sci. 2024;25(17):9147
26. Hu T, Xu L, Jiang M. et al. N6-methyladenosine-methylomic landscape of lung tissues of mice with chronic obstructive pulmonary disease. Front Immunol. 2023;14:1137195
27. Konigsberg IR, Yang IV. Differential Methylation of Chronic Obstructive Pulmonary Disease Lung Macrophage Genes Sheds Light on Disease Pathogenesis. Am J Respir Cell Mol Biol. 2022;66(6):589-90
28. Groth EE, Weber M, Bahmer T. et al. Exploration of the sputum methylome and omics deconvolution by quadratic programming in molecular profiling of asthma and COPD: the road to sputum omics 2.0. Respir Res. 2020;21(1):274
29. Halabelian L, Barsyte-Lovejoy D. Structure and Function of Protein Arginine Methyltransferase PRMT7. Life (Basel). 2021;11(8):768
30. Günes Günsel G, Conlon TM, Jeridi A. et al. The arginine methyltransferase PRMT7 promotes extravasation of monocytes resulting in tissue injury in COPD. Nat Commun. 2022;13(1):1303
31. Gao J, Liu H, Wang X. et al. Associative analysis of multi-omics data indicates that acetylation modification is widely involved in cigarette smoke-induced chronic obstructive pulmonary disease. Front Med (Lausanne). 2022;9:1030644
32. Noh M, Sim JY, Kim J. et al. Particulate matter-induced metabolic recoding of epigenetics in macrophages drives pathogenesis of chronic obstructive pulmonary disease. J Hazard Mater. 2023;464:132932
33. Hautamaki RD, Kobayashi DK, Senior RM. et al. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002-2004
34. Finlay GA, O'Driscoll LR, Russell KJ. et al. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med. 1997;156(1):240-247
35. Bracke K, Cataldo D, Maes T. et al. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int Arch Allergy Immunol. 2005;138(2):169-79
36. Demedts IK, Morel-Montero A, Lebecque S. et al. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax. 2006;61(3):196-201
37. Molet S, Belleguic C, Lena H. et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res. 2005;54(1):31-6
38. Hou HH, Wang HC, Cheng SL. et al. MMP-12 activates protease-activated receptor-1, upregulates placenta growth factor, and leads to pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol. 2018;315(3):L432-l42
39. Spix B, Butz ES, Chen CC. et al. Lung emphysema and impaired macrophage elastase clearance in mucolipin 3 deficient mice. Nat Commun. 2022;13(1):318
40. Joos L, He JQ, Shepherdson MB. et al. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum Mol Genet. 2002;11(5):569-76
41. Skjøt-Arkil H, Clausen RE, Nguyen QH. et al. Measurement of MMP-9 and -12 degraded elastin (ELM) provides unique information on lung tissue degradation. BMC Pulm Med. 2012;12:34
42. Yu L, Zhang Y, Liu C. et al. Heterogeneity of macrophages in atherosclerosis revealed by single-cell RNA sequencing. FASEB J. 2023;37(3):e22810
43. Doherty DF, Nath S, Poon J. et al. Protein Phosphatase 2A Reduces Cigarette Smoke-induced Cathepsin S and Loss of Lung Function. Am J Respir Crit Care Med. 2019;200(1):51-62
44. Wang X, Polverino F, Rojas-Quintero J. et al. A Disintegrin and A Metalloproteinase-9 (ADAM9): A Novel Proteinase Culprit with Multifarious Contributions to COPD. Am J Respir Crit Care Med. 2018;198(12):1500-18
45. Polverino F, Rojas-Quintero J, Wang X. et al. A Disintegrin and Metalloproteinase Domain-8: A Novel Protective Proteinase in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2018;198(10):1254-67
46. Fraughen DD, Ghosh AJ, Hobbs BD. et al. Augmentation therapy for severe alpha-1 antitrypsin deficiency improves survival and is decoupled from spirometric decline—A multinational registry analysis. Am J Respir Crit Care Med. 2023;208(9):964-974
47. Krotova K, Marek GW, Wang RL. et al. Alpha-1 Antitrypsin-Deficient Macrophages Have Increased Matriptase-Mediated Proteolytic Activity. Am J Respir Cell Mol Biol. 2017;57(2):238-47
48. Baßler K, Fujii W, Kapellos TS. et al. Alveolar macrophages in early stage COPD show functional deviations with properties of impaired immune activation. Front Immunol. 2022;13:917232
49. Liu Y, Liu H, Li C. et al. Proteome Profiling of Lung Tissues in Chronic Obstructive Pulmonary Disease (COPD): Platelet and Macrophage Dysfunction Contribute to the Pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2020;15:973-80
50. Li Y, Yang Y, Guo T. et al. Heme oxygenase-1 determines the cell fate of ferroptotic death of alveolar macrophages in COPD. Front Immunol. 2023;14:1162087
51. Rathnayake SNH, Ditz B, van Nijnatten J. et al. Smoking induces shifts in cellular composition and transcriptome within the bronchial mucus barrier. Respirology. 2022;28(2):132-142
52. Hey J, Paulsen M, Toth R. et al. Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat Commun. 2021;12(1):6520
53. Barnawi J, Jersmann H, Haberberger R. et al. Reduced DNA methylation of sphingosine-1 phosphate receptor 5 in alveolar macrophages in COPD: A potential link to failed efferocytosis. Respirology. 2017;22(2):315-21
54. O'Beirne SL, Kikkers SA, Oromendia C. et al. Alveolar Macrophage Immunometabolism and Lung Function Impairment in Smoking and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2020;201(6):735-39
55. Cloonan SM, Glass K, Laucho-Contreras ME. et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22(2):163-74
56. Ryan EM, Sadiku P, Coelho P. et al. NRF2 Activation Reprogrammes Defects in Oxidative Metabolism to Restore Macrophage Function in COPD. Am J Respir Crit Care Med. 2023;207(8):998-1011
57. Bewley MA, Budd RC, Ryan E. et al. Opsonic Phagocytosis in Chronic Obstructive Pulmonary Disease Is Enhanced by Nrf2 Agonists. Am J Respir Crit Care Med. 2018;198(6):739-50
58. Asare PF, Hurtado PR, Tran HB. et al. Reduction in Rubicon by cigarette smoke is associated with impaired phagocytosis and occurs through lysosomal degradation pathway. Clin Exp Med. 2023;23(7):4041-4055
59. Jati S, Kundu S, Chakraborty A. et al. Wnt5A Signaling Promotes Defense Against Bacterial Pathogens by Activating a Host Autophagy Circuit. Front Immunol. 2018;9:679
60. Higham A, Scott T, Li J. et al. Effects of corticosteroids on COPD lung macrophage phenotype and function. Clin Sci (Lond). 2020;134(7):751-63
61. Berenson CS, Garlipp MA, Grove LJ. et al. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. 2006;194(10):1375-84
62. Skronska-Wasek W, Durlanik S, Le H. et al. The antimicrobial peptide S100A8/A9 produced by airway epithelium functions as a potent and direct regulator of macrophage phenotype and function. Eur Respir J. 2022;59(4):2002732
63. Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet. 2007;370(9589):786-96
64. Jang JG, Ahn JH, Jin HJ. Incidence and Prognostic Factors of Respiratory Viral Infections in Severe Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Int J Chron Obstruct Pulmon Dis. 2021;16:1265-73
65. Finney LJ, Belchamber KBR, Fenwick PS. et al. Human Rhinovirus Impairs the Innate Immune Response to Bacteria in Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2019;199(12):1496-507
66. Akata K, Leung JM, Yamasaki K. et al. Altered Polarization and Impaired Phagocytic Activity of Lung Macrophages in People With Human Immunodeficiency Virus and Chronic Obstructive Pulmonary Disease. J Infect Dis. 2022;225(5):862-67
67. Yang N, Zhang L, Tian D. et al. Tanshinone increases Hemopexin expression in lung cells and macrophages to protect against cigarette smoke-induced COPD and enhance antiviral responses. Cell Cycle. 2023;22:645-665
68. Ghosh B, Gaike A, Pyasi K. et al. Bacterial load and defective monocyte-derived macrophage bacterial phagocytosis in biomass smoke-related COPD. Eur Respir J. 2019;53(2):1702273
69. Tejwani V, Woo H, Liu C. et al. Black carbon content in airway macrophages is associated with increased severe exacerbations and worse COPD morbidity in SPIROMICS. Respir Res. 2022;23(1):310
70. Yin G, Wu X, Wu Y. et al. Evaluating carbon content in airway macrophages as a biomarker of personal exposure to fine particulate matter and its acute respiratory effects. Chemosphere. 2021;283:131179
71. Yamasaki K, Eeden SFV. Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD. Int J Mol Sci. 2018;19(2):582
72. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13
73. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090
74. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787-95
75. Le Y, Cao W, Zhou L. et al. Infection of Mycobacterium tuberculosis Promotes Both M1/M2 Polarization and MMP Production in Cigarette Smoke-Exposed Macrophages. Front Immunol. 2020;11:1902
76. He S, Tian R, Zhang X. et al. PPARγ inhibits small airway remodeling through mediating the polarization homeostasis of alveolar macrophages in COPD. Clin Immunol. 2023;250:109293
77. Bazzan E, Turato G, Tinè M. et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir Res. 2017;18(1):40
78. Byers DE, Wu K, Dang-Vu G. et al. Triggering Receptor Expressed on Myeloid Cells-2 Expression Tracks With M2-Like Macrophage Activity and Disease Severity in COPD. Chest. 2018;153(1):77-86
79. He S, Xie L, Lu J, Sun S. Characteristics and potential role of M2 macrophages in COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:3029-39
80. Feller D, Kun J, Ruzsics I. et al. Cigarette Smoke-Induced Pulmonary Inflammation Becomes Systemic by Circulating Extracellular Vesicles Containing Wnt5a and Inflammatory Cytokines. Front Immunol. 2018;9:1724
81. Liégeois M, Bai Q, Fievez L. et al. Airway Macrophages Encompass Transcriptionally and Functionally Distinct Subsets Altered by Smoking. Am J Respir Cell Mol Biol. 2022;67(2):241-52
82. Hu Y, Shao X, Xing L. et al. Single-Cell Sequencing of Lung Macrophages and Monocytes Reveals Novel Therapeutic Targets in COPD. Cells. 2023;12(24):2771
83. Tang F, Barbacioru C, Wang Y. et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377-82
84. Mulder K, Patel AA, Kong WT. et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54(8):1883-1900.e5
85. Lee Y. Meta-analysis of single-cell RNA-sequencing data for depicting the transcriptomic landscape of chronic obstructive pulmonary disease. Comput Biol Med. 2023;167:107685
86. Xu X, Yu T, Dong L. Eosinophils promote pulmonary matrix destruction and emphysema via cathepsin L. Signal Transduct Tar. 2023;8(1):390
87. Huang Q, Wang Y, Zhang L. et al. Single-cell transcriptomics highlights immunological dysregulations of monocytes in the pathobiology of COPD. Respir Res. 2022;23(1):367
88. Wan R, Srika P, Xie S. et al. PPARγ Attenuates Cellular Senescence of Alveolar Macrophages in Asthma- COPD Overlap. Respir Res. 2024;25(1):174
89. Correnti S, Preianò M, Gamboni F. et al. An integrated metabo-lipidomics profile of induced sputum for the identification of novel biomarkers in the differential diagnosis of asthma and COPD. J Transl Med. 2024;22(1):301
90. Zhuang Y, Xing F, Ghosh D. et al. Deep learning on graphs for multi-omics classification of COPD. PLoS One. 2023;18(4):e0284563
91. Li CX, Wheelock CE, Sköld CM, Wheelock Å M. Integration of multi-omics datasets enables molecular classification of COPD. Eur Respir J. 2018;51(5):1701930
92. Silverman EK. Applying Functional Genomics to Chronic Obstructive Pulmonary Disease. Ann Am Thorac Soc. 2018;15(Suppl 4):S239-s42
93. Pei Y, Wei Y, Peng B. et al. Combining single-cell RNA sequencing of peripheral blood mononuclear cells and exosomal transcriptome to reveal the cellular and genetic profiles in COPD. Respir Res. 2022;23(1):260
94. Shen X, Zhao Y, Wang Z, Shi Q. Recent advances in high-throughput single-cell transcriptomics and spatial transcriptomics. Lab Chip. 2022;22(24):4774-4791
95. Chen TY, You L, Hardillo JAU, Chien MP. Spatial Transcriptomic Technologies. Cells. 2023;12(16):2042
96. Xu J, Guo P, Hao S. et al. A spatiotemporal atlas of mouse liver homeostasis and regeneration. Nature genetics. 2024;56(5):953-969
97. Chen C, Guo Q, Liu Y. et al. Single-cell and spatial transcriptomics reveal POSTN(+) cancer-associated fibroblasts correlated with immune suppression and tumour progression in non-small cell lung cancer. Clin Transl Med. 2023;13(12):e1515
98. Su G, Qin X, Enninful A. et al. Spatial multi-omics sequencing for fixed tissue via DBiT-seq. STAR Protoc. 2021;2(2):100532
99. Luo L, Gribskov M, Wang S. Bibliometric review of ATAC-Seq and its application in gene expression. Brief Bioinform. 2022;23(3):bbac061
100. Xiao C, Chen Y, Meng Q. et al. Benchmarking multi-omics integration algorithms across single-cell RNA and ATAC data. Brief Bioinform. 2024;25(2):bbae095
101. Benway CJ, Liu J, Guo F. et al. Chromatin Landscapes of Human Lung Cells Predict Potentially Functional Chronic Obstructive Pulmonary Disease Genome-Wide Association Study Variants. Am J Respir Cell Mol Biol. 2021;65(1):92-102
102. Murano H, Inoue S, Hashidate YT. et al. Lysophospholipid acyltransferase 9 promotes emphysema formation via platelet-activating factor. Am J Respir Cell Mol Biol. 2024;70(6):482-492
103. Wohnhaas CT, Baßler K, Watson CK. et al. Monocyte-derived alveolar macrophages are key drivers of smoke-induced lung inflammation and tissue remodeling. Front Immunol. 2024;15:1325090
Author contact
![]() Corresponding author: Jianquan Zhang; Email: jqzhang2002com.
Corresponding author: Jianquan Zhang; Email: jqzhang2002com.