Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(1):71-86. doi:10.7150/ijms.104932 This issue Cite
Research Paper
Receptor-Interacting Protein Kinase 3-Mediated Modulation of Endothelial Cell Necroptosis and Mitochondrial Dysfunction through AMPK/Drp1 Signaling Pathway: Insights into the Pathophysiological Mechanisms of Lipopolysaccharide-Induced Acute Lung Injury
Zhaoning Zhao1,2,*, Pingjun Zhu1,2,3,*, Yue Lou4, Ruoyu Hou1,5, Heqiang Sun6, Yingzhen Du2,7, ![]() , Guogang Xu1,
, Guogang Xu1, ![]()
1. Health Management Institute, The Second Medical Center & National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing 100853, China.
2. Chinese PLA General Hospital, Medical School of Chinese PLA, Beijing 100853, China.
3. Department of Respiratory and Critical Care Medicine, The Second Medical Center & National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing 100853, China.
4. The Second Medical Center, Chinese PLA General Hospital, Beijing 100853, China.
5. School of Biology, University of St Andrews, St Andrews, KY16 9ST, UK.
6. Department of Laboratory Medicine, The Second Medical Center & National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing 100853, China.
7. Department of Disease Control and Prevention, The Second Medical Center & National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing 100853, China.
*Zhaoning Zhao and Pingjun Zhu contributed equally to this work.
Received 2024-10-9; Accepted 2024-11-8; Published 2025-1-1
Abstract

Receptor-interacting protein 3 (Ripk3) plays a crucial part in acute lung injury (ALI) by regulating inflammation-induced endothelial damage in the lung tissue. The precise mechanisms through which Ripk3 contributes to the endothelial injury in ALI still remain uncertain. In the current research, we employed Ripk3-deficient (Ripk3-/-) mice to examine the role of Ripk3 in ALI progression, focusing on its effects on endothelial cells (ECs), mitochondrial damage and necroptosis. Our study observed significant Ripk3 upregulation in lipopolysaccharide- (LPS-) treated lung tissues, as well as in murine pulmonary microvascular endothelial cells (PMVECs). Ripk3 deletion improved lung tissue morphology, reduced inflammation, oxidative stress and endothelial dysfunction under LPS challenge. It also mitigated LPS-induced necroptosis and mitochondrial damage in PMVECs. Ripk3 upregulation suppressed the AMP-activated protein kinase (AMPK) pathway and activated Drp1-mediated mitochondrial fission, increasing mitochondrial permeability transition pore (mPTP) opening and PMVEC necroptosis. Conversely, Ripk3 deletion activated the AMPK/Drp1-mitochondrial fission pathway, preventing mPTP opening and PMVEC necroptosis in ALI. These findings demonstrated that Ripk3 promotes necroptosis through the AMPK/Drp1/mPTP opening pathway, identifying a potential therapeutic target for ALI treatment.
Keywords: Ripk3, Acute lung injury, Cell necroptosis, Mitochondrial damage
Introduction
Acute respiratory distress syndrome (ARDS) is a widespread lung injury that arises from several pathological stresses such as ischemia, anoxia, severe infection, and trauma [1, 2]. ARDS is defined by an exaggerated inflammatory response that leads to the breakdown of the alveolar-capillary barrier (ACB) and fluid accumulation in the lungs, causing pulmonary edema [3, 4]. It is further characterized by persistent hypoxemia, reduced pulmonary compliance, and the presence of abnormal shadows on chest imaging [5], considered to be a prevalent refractory complication with a substantial mortality rate (30-40%) [6] among severely ill patients [7, 8]. Currently, there is a dearth of effective therapy for ARDS. Based on the pathogenic mechanism of ARDS, disseminating stimulation of inflammation influences pulmonary microvascular endothelium first, which leads to an increase in its permeability to an abnormal state [9, 10]. As the major component of ACB, pulmonary microvascular endothelium typically maintains its permeability at an appropriate level to serve its function [11]. This hyperpermeability of pulmonary microvascular endothelial cells (PMVECs) harms the integrity of ACB and allows the fluid to undergo osmosis from capillary to alveoli. This leakage brings inflammatory cells to alveolar space, and leads to edema in lung tissues eventually [12]. Thus, it is crucial to discover practical and effective strategies to enhance the impairment of barrier function in PMVECs for treating ARDS.
Mitochondria, acting as the cellular powerhouse, regulate various physiological processes, including signaling transmission, oxidative stress, cell growth and death [13], particularly in some severe conditions, for instance ARDS. The involvement of mitochondrial-derived reactive oxygen species (ROS) in ARDS has garnered significant interest due to its regulatory function [14]. Excessive ROS have a permanent detrimental impact on endothelial cells [15]. Moreover, oxidative stress in endothelial cells intensifies apoptosis, promotes disruption of the protective barrier, thus causes damage to the lungs [16]. In cases of lipopolysaccharide- (LPS-) induced acute lung injury (ALI), there is a reduction in the production of mitochondrial adenosine triphosphate (ATP), while ROS levels increase due to the concentration of neutrophils in the lung tissues [17]. These data suggest that mitochondrial dysfunction plays a part in ALI, while the specific mechanisms remain incompletely understood. The morphology of mitochondria is sustained by the dynamic and contrasting actions of mitochondrial fusion and fission [18]. Mitochondrial fusion facilitates the dispersion of harmful substances, while mitochondrial fission permits the separation and eventual breakdown of damaged and depolarized mitochondria. An imbalance in the process of mitochondrial fission and fusion has been associated with a diverse range of disorders [19]. Recent data have indicated that changes in mitochondrial morphology are preceded by mitochondrial damage [20, 21]. Augmented mitochondrial fragmentation serves as an initial indication of mitochondrial membrane potential (MMP) depolarization and overproduction of mitochondrial ROS. Although the significant contribution of mitochondrial fusion to ARDS is widely acknowledged, there is still limited knowledge on alteration of mitochondrial homeostasis in ARDS progression.
ARDS may involve several forms of cellular demise, such as autophagy, apoptosis, and necroptosis [22]. Necroptosis, in particular, enhances inflammation and has a significant impact on ARDS. Receptor-interacting protein 3 (Ripk3) takes control of necroptosis, a programmed cell death that has been recently identified [23]. Necroptosis depends on the phosphorylation of Ripk3, which acts on mixed lineage kinase domain-like pseudokinase (MLKL) as its substrate [24]. Necroptosis is thought to play a part in endothelial dysfunction [25], involving the progressive stimulation of the inherent immune response and blood clotting, which enhances endothelial cell permeability [26]. Nevertheless, the precise mechanism by which necroptosis occurs in ARDS remains unidentified. Several studies demonstrate that lung injury in severe ARDS triggered by large doses of LPS is mainly caused by Ripk3-MLKL-mediated necroptosis and endothelial dysfunction, suggesting its potential role in the development of ARDS [26, 27]. In addition, some studies have observed that Ripk3 has a crucial impact on the occurrence of mitochondrial dysfunction, including mitochondrial fission and mitochondrial oxidative stress in pathological conditions, for instance ALI [28]. In light of the correlation between necroptosis and mitochondrial injury, our objective is to investigate whether the initiation of necroptosis results from changes in mitochondrial morphology and consequently causes lung injury associated with ARDS. We hypothesize that Ripk3 influences mitochondrial fission, leading to the initiation of necroptosis in the development of ARDS.
Materials and Methods
Animal model
The laboratory animals at this facility were handled in accordance with the Guidelines for the Care and Use of Laboratory Animals. Ethics approval was granted by the Chinese PLA General Hospital Ethics Committee in Beijing, China. The C57BL/6 mice used in this study were genetically modified to be Ripk3-deficient (Ripk3-/-). The ages and sexes of all experimental mice were matched. LPS (5 mg/kg) was injected intratracheally for 24 hours to induce ALI in the animal model [29]. The control group (Sham) received injections of phosphate-buffered saline (PBS) and was observed for 24 hours. The bronchoalveolar lavage fluid (BALF) and lung tissue were harvested for subsequent assessment.
Analysis of histology
Preserved lung tissues were embedded in paraffin blocks after being treated with a 4% fixative solution (Sigma-Aldrich). Each specimen was sliced into 5μm thick, then stained with hematoxylin and eosin (H&E, Sigma-Aldrich). The areas of interest were analyzed with Image J software.
Lung wet to dry ratio (W/D)
Edema severity was determined by calculating the W/D ratio. The lung tissues were measured in terms of weight following the removal of the left lung and the extraction of blood. After 4 days of drying at 80°C, the specimen reached a constant weight.
Evens Blue assay
Evans Blue dye (EB, 35 mg/kg, Sigma-Aldrich) was administered to the mice intraperitoneally two hours before euthanasia. Following this, the pulmonary circulation was flushed with normal saline for 2 minutes to remove any remaining EB in the blood vessels. Then, the lungs were removed, homogenized in 1 mL PBS, and incubated in 2 mL formalin at 60°C for 24 hours. The supernatants containing the extracted EB dye were collected, and the absorbance of the resulting supernatant was measured at 620 nm to quantify the concentration of EB dye.
Analysis of protein content and cell count in BALF
Murine BALF was obtained by injecting PBS intratracheally after mice euthanization. The Bradford Protein Quantification Kit (Beyotime, China) was applied to quantifying the protein concentration in BALF following centrifugation at 1500 rpm at 4°C for 10 minutes. A hemocytometer and Wright-Giemsa staining (Beyotime, China) were conducted to quantify the total cell count and several types of hemocytes, such as macrophages, neutrophils, and leukocytes [30]. The levels of proinflammatory cytokines (tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1)) were estimated with ELISA kits (R&D, USA).
Cell culture and treatment
The mouse pulmonary microvascular endothelial cell line was acquired from Shanghai Univ, China. DMEM (Gibco, USA) was used to culture PMVECs added with 10% fetal bovine serum (FBS, Gibco, USA). The cells were cultivated in a humidified incubator maintained at 37°C with 5% carbon dioxide. The PMVECs were incubated under LPS (10 μg/ml, Sigma-Aldrich, USA) for 12 h to cause ALI [31].
Gene-specific knockdown
A lentivirus system (OBiO, Shanghai, China) was employed to suppress the expression level of Ripk3, following the manufacturer's protocol. A total of 105 cells each well were cultured in 12-well plates and infected with lentiviral shRNA at 50 multiplicity of infection (MOI) for 24 h. Subsequently, cells were grown in DMEM containing 10% FBS for three days. Nonsilencing scrambled short hairpin RNA (shRNA) free of EGFP served as the negative control. The transfected cells were subjected to LPS treatment as previously mentioned, and then collected for further analysis.
Cell necroptosis
Necroptosis was identified by flow cytometry with a kit (Vazyme Biotech). Following treatment, the cells were gathered, rinsed with PBS, then resuspended in 100μL binding buffer containing 5μL Annexin V-FITC and PI, and incubated at ambient temperature for 10 minutes. After incubation, flow cytometry (Thermo Fisher Scientific, USA) was employed to identify necroptosis.
Western blotting
4-12% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) were applied for protein samples (25-35μg). Then polyvinylidene fluoride (PVDF) membrane (EMD Millipore) covered the SDS-PAGE. Reference was taken from the pertained protein ladder (Thermo Fisher Scientific, 26616). 5% skim milk submerged the membranes later. After the 1 hour coverage, primary antibodies and secondary antibodies were added, incubated overnight under 4°C and for 1 hour at RT respectively. TBST was required to wash membranes after two incubations. The signals were detected using ECL Detection Reagent (GE Healthcare). The luminosity of the bands was analyzed with LAS 400 system (GE Healthcare), and normalized to GAPDH.
Transmission electron microscopy (TEM)
Following LPS treatment, a 2-hour fixation process on the samples was conducted initially with 2.5% glutaraldehyde at 4°C, followed by post-fixation in 1 % osmium tetroxide at RT for an additional 2 hours. Subsequently, the samples underwent dehydration in a serial diluted ethanol group (65 %, 70 %, 75 %, 80 %, and 95 % ethanol for 10 minutes each) before being placed in epoxy resin. Afterward, uranyl acetate and lead citrate (Ted Pella, USA) were used to stain the ultrathin section (70nm). Finally, images were visualized utilizing a TEM microscope (JEM1400, Japan).
Isolation of mitochondrial and cytosolic fractions
Fractions of mitochondria and cytosol were isolated using a kit (Beyotime) as described by the protocol of manufacturer. Cells were collected and resuspended in an isolation buffer with 1mM PMSF, then left to incubate for 10 minutes on ice. This suspension was homogenized and centrifuged at 1000 × g at 4°C for 10 minutes. Next, the supernatant was gathered and subjected to centrifugation at 11000 × g at 4°C for 10 minutes. The mitochondrial and cytosolic fractions were distinguished by resuspending the deposit in the mitochondrial lysis buffer after separating the supernatant.
Analyses of mitochondrial membrane potential and mitochondrial permeability transition pore (mPTP) opening
JC-1 probes (Beyotime) were used to measure mitochondrial membrane potential. The fluorescence intensity of tetramethylrhodamine ethyl ester (TMRE) reflects the haphazard opening rate of mPTP [32]. As a negative control, Cyclosporin A (CsA, 10μM, Sigma Aldrich) was used to inhibit the opening of mPTP.
ROS, glutathione (GSH), superoxide dismutase (SOD), and malondialdehyde (MDA) assays
GSH level, SOD activity, and MDA level were assessed following the manufacturer's guidelines. Previously reported technique such as 2,7′-dichlorofluorescein-diacetate (DCFHDA, Beyotime, China) staining [33] was used to detect ROS.
Single-cell RNA sequencing (scRNA-seq) analysis
The samples were integrated by anchors method from the R package "Seurat" [34], and core cells were acquired by filtering scRNA-seq. The first step involved the removal of low-quality data through three methods: filtering out single cells with less than 5 genes expressed, eliminating cells expressing less than 100 genes, and excluding cells with more than 5% mitochondrial genes. Subsequently, gene expression of core cells was normalized using a linear regression model, and then the top 2000 genes with highly variable characteristics were screened by analysis of variance (ANOVA). Principal component analysis (PCA) was performed on single-cell samples, and the top 20 principal components (PC) were selected for subsequent analysis. Cell clustering analysis was performed using the "FindNeighbors" and "FindClusters" functions, and T-distributed random neighborhood embedding (t-SNE) was carried out through the "RunTSNE" function for visualization. Gene expression differences in different samples were assessed utilizing the Wilcoxon-Mann-Whitney test. Using the "singleR" package from R [35], three databases including HumanPrimaryCellAtlasData, BlueprintEncodeData, and ImmuneCellExpressionData were utilized as the reference for auxiliary annotation, followed by the CellMarker database [36] and previous studies to find marker genes for manual annotation of different clusters.
Analyses of statistics
The involvement of unpaired t-test aimed to construct a comparison between two clusters. The one-way ANOVA was applied to compare discrepancies between at least three independent experiments. In all cases, results were demonstrated in the form of mean ± standard deviation (SD), and the significance was set at P < 0.05.
Results
Ripk3 is increased in lung ECs following LPS challenge
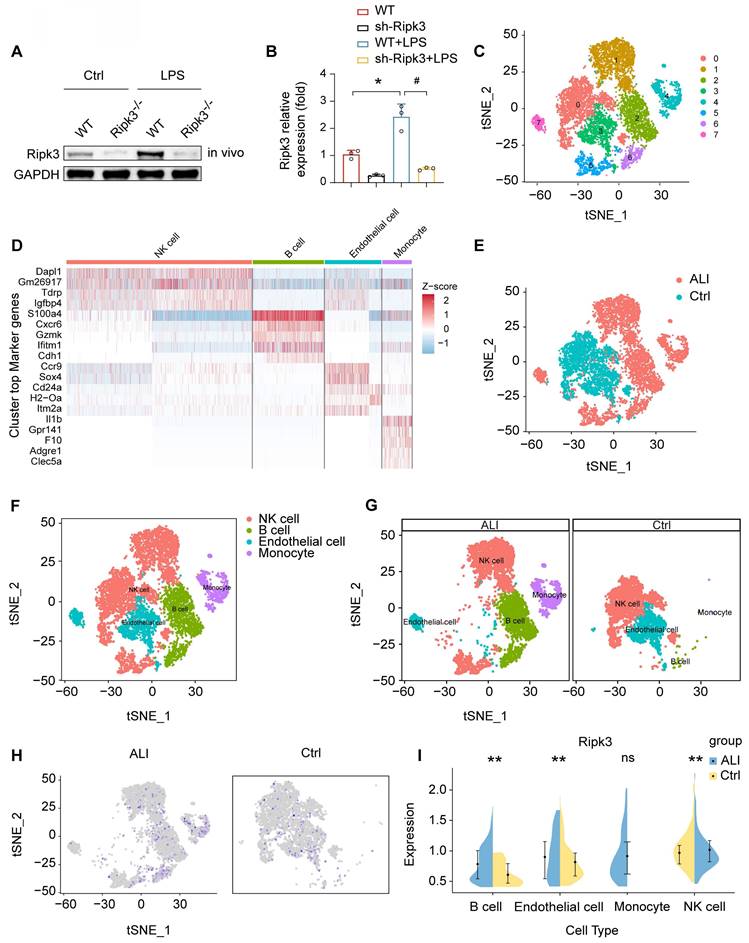
Analysis of Ripk3 expression in lung tissues was conducted using Western blotting. The findings revealed an upregulation of Ripk3 in lung tissues after LPS treatment compared to baseline levels (Fig. 1A-B). To determine the main cellular source of Ripk3 in lung tissue, we performed single-cell analyses using the Gene Expression Omnibus (GEO) database, which contains single-cell profiles from mouse lung samples, with and without septic stress. The results indicated a high abundance of Ripk3 in ECs (Fig. 1C-I). Suggestion could be drawn that Ripk3 is highly involved during acute lung injury pathogenesis, meriting further exploration as a potential therapeutic target.
LPS upregulates Ripk3 in lung ECs. (A-B) The change of Ripk3 expression in vivo was quantified using western blotting. (C-I) Ripk3 expression in different cell types from the lung was determined using single-cell sequencing analysis. Mean ± SEM, ∗p < 0.05 vs. the wild-type (WT) group; #p < 0.05 vs. the LPS group.
Ripk3 disrupts the pulmonary vascular endothelial barrier in ALI mice
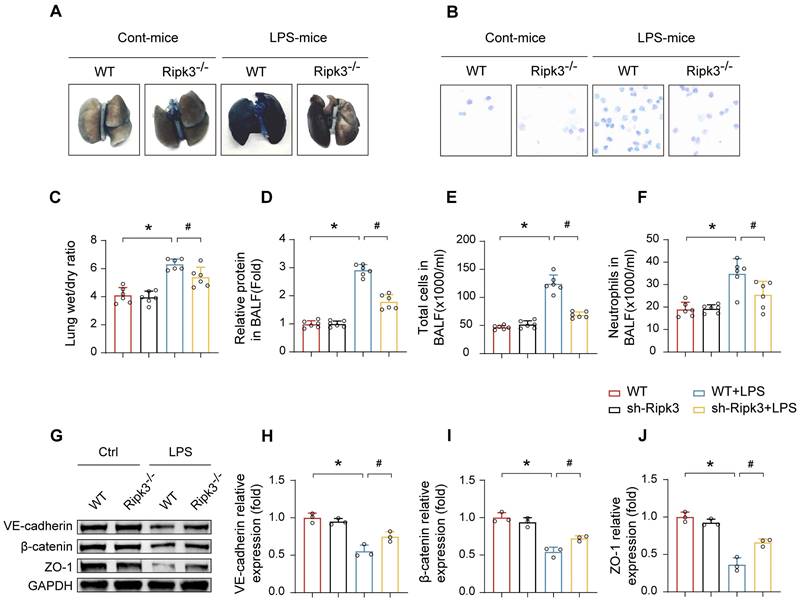
To investigate the relationship between elevated Ripk3 expression and LPS-induced lung injury, Ripk3-/- mice were employed. The results demonstrated that LPS exposure led to augmented pulmonary vascular permeability, as evidenced by significant increases in Evans Blue extravasation, lung W/D weight ratio, BALF protein concentration, total cell count, and neutrophils in the BALF (Fig. 2A-F). To assess microvascular barrier function, the expressions of key intercellular junction proteins in lung tissues including VE-cadherin, β-catenin, and ZO-1, were examined by Western blot analysis, which revealed a marked downregulation following LPS administration. However, these alternations were apparently reversed in Ripk3 knockout mice (Fig. 2G-J). These results suggest that Ripk3 participates intensively in compromising pulmonary vascular endothelial barrier integrity during ALI.
Effect of Ripk3 deletion on the alveolar-capillary barrier in LPS-induced ALI mice. (A) Representative images of Evans Blue (EB) extravasation in the lungs. (B-F) The ratio of wet weight to dry weight of the lungs, the protein content, the number of total cells and neutrophils in BALF were analyzed to assess lung permeability. (G-J) The protein levels of VE-cadherin, β-catenin, and ZO-1 in the lung tissues were measured using western blot and quantitative analysis. Mean ± SEM, ∗p < 0.05 vs. the WT group; #p < 0.05 vs. the LPS group.
Ripk3 deletion reduces LPS-induced pulmonary pathological damage, inflammation, and oxidative injury
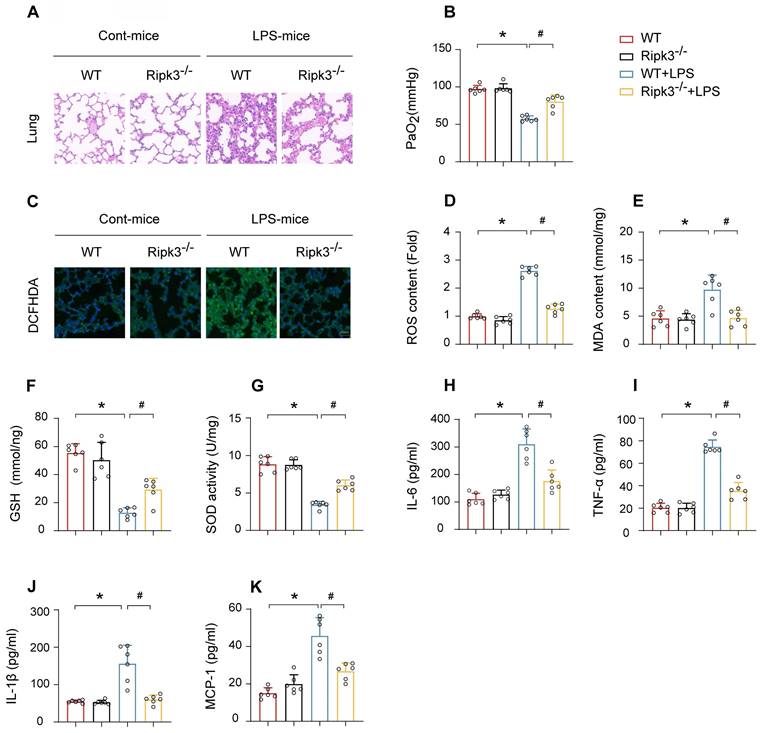
HE staining of lung tissues from mice treated with LPS revealed significant pathological alterations, including inflammatory cell infiltration, interstitial and alveolar edema, hemorrhage, and diffuse alveolar damage. The alterations were notably attenuated in Ripk3-deficient mice (Fig. 3A). Furthermore, Ripk3 ablation improved the partial pressure of arterial oxygen (PaO2) in response to LPS challenge (Fig. 3B). Improvement in PaO2 accelerated the repair process of pulmonary vascular endothelial cells and improved respiratory function while reducing the risk of multiple organ dysfunction syndrome (MODS). Given the important role of oxidative stress in ALI, we investigated whether Ripk3 deletion protects against oxidative injury. DCFHDA staining demonstrated elevated ROS levels in the lung tissue of mice after LPS challenge, which reduced to baseline levels in Ripk3-deficient mice (Fig. 3C-D). Additionally, LPS stimulation increased MDA production and decreased the levels of antioxidant factors such as GSH and SOD. Ripk3 deletion normalized these changes (Fig. 3E-G). LPS induced elevated expression of inflammatory cytokines (IL-6, TNF-α, IL-1β, and MCP-1) in lung tissues. Ripk3 deletion demonstrated anti-inflammatory effects by lowering these cytokine levels following LPS exposure (Fig. 3H-K). In conclusion, Ripk3 deletion protects against LPS-induced ALI by alleviating inflammation, oxidative stress, and tissue damage.
Ripk3 deletion attenuates LPS-mediated oxidative stress and inflammation response in acute lung injury. (A) Pathological alterations of lung parenchyma observed by HE staining after acute lung injury. (B) Measurement of the partial pressure of arterial oxygen (PaO2). (C-D) DCFHDA staining was used to detect ROS content. (E-G) MDA level, SOD activity, GSH level were also measured in lung tissue. (H-K) Protein levels of IL-6, TNF-α, IL-1β, and MCP-1 were quantified by ELISA. Mean ± SEM, ∗p < 0.05 vs. the WT group; #p < 0.05 vs. the LPS group.
Ripk3 evokes lung ECs necroptosis via promoting mPTP opening
ALI is characterized by the loss of functional cells due to cell death. To explore the potential protective effect of Ripk3 deletion against LPS-triggered vascular endothelial cell necroptosis, a series of experiments were conducted. Deficiency of Ripk3 was found to attenuate cellular necroptosis in lung tissue caused by LPS, as evidenced by reduced levels of phosphoglycerate mutase 5 (PGAM5) and phosphorylated mixed lineage kinase domain-like protein (p-MLKL). Cellular experiments further supported the protective role of Ripk3 deficiency in acute lung injury. In PMVECs, LPS exposure increased the necroptosis index, as indicated by raised PGAM5 and p-MLKL levels. Nevertheless, this effect was nullified in Ripk3 knockdown cells (Fig. 4A-C). Annexin V/PI staining showed that LPS increased necroptosis in PMVECs, which was significantly reduced by Ripk3 inhibition (Fig. 4D). These findings collectively suggest that LPS stimulates the activation of necroptosis in PMVECs by increasing Ripk3 expression.
Ripk3 knockdown reduces PMVEC necroptosis by inhibiting mPTP opening. (A-C) Western blots were used to analyze the expression of PGAM5, and p-MLKL. (D) Necroptosis of PMVECs was quantified by flow cytometry with Annexin V/PI staining. The necroptosis group: the percentage of PI+ cells. Cyclosporin A (CsA) was used as the inhibitor to prevent mitochondrial permeability transition pore (mPTP) opening. (E) Evaluation of the rate of mPTP opening. (F-G) Changes of mitochondrial membrane potential were identified using JC-1 staining. Mean ± SEM, ∗p < 0.05 vs. the Ctrl group; #p < 0.05 vs. the LPS group; &p < 0.05 vs. the LPS+CsA group.
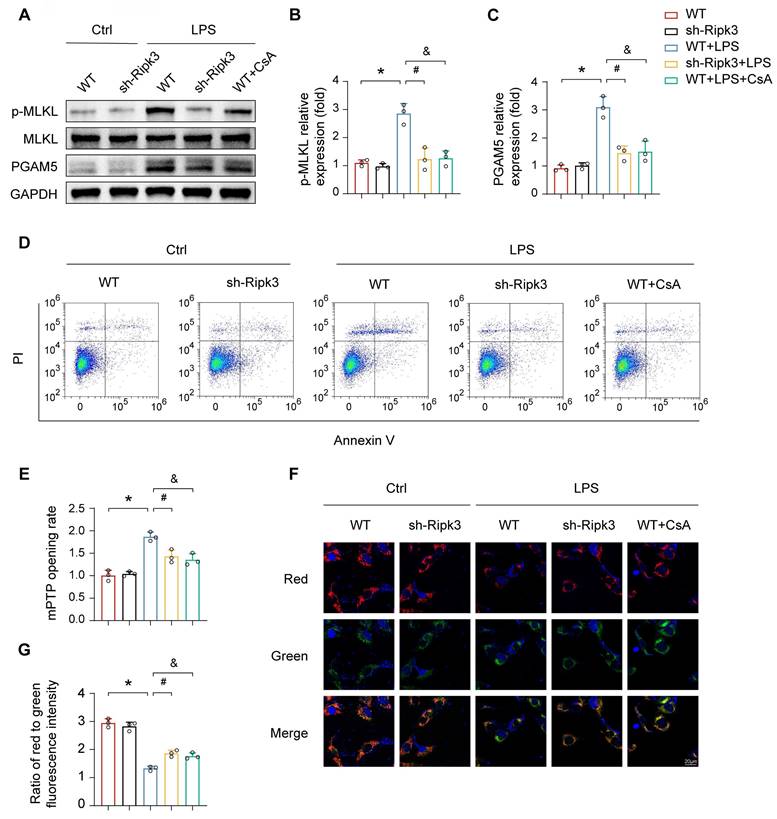
To clarify the mechanism of Ripk3-mediated necroptosis, the study paid attention to mPTP opening, which has been recognized as an upstream trigger of cellular necroptosis. As shown in Fig. 4E, LPS significantly prolonged mPTP opening time, an effect that was attenuated in Ripk3-deficient cells. Mitochondrial membrane potential, which typically dissipates upon mPTP opening, showed similar trends. CsA, an mPTP opening inhibitor, was used as a negative control. CsA increased cellular viability under LPS stress, similar to the protective effect of Ripk3 inhibition (Fig. 4F-G). These findings suggest that Ripk3-initiated necroptosis occurs through mPTP opening in ECs.
Ripk3 facilitates mPTP opening via Drp1-related mitochondrial fission
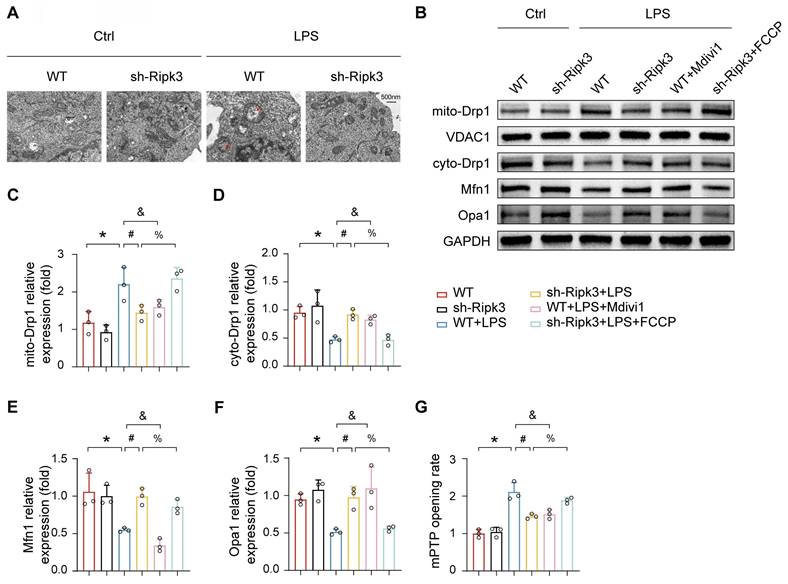
Previous research has demonstrated that Ripk3 deficiency helps maintain mitochondrial balance by normalizing mitochondrial dynamics, including fission and mitophagy. Additionally, enhanced mitochondrial fission has been linked to mPTP opening. Based on these observations, we investigated whether Ripk3 induces mPTP opening by promoting mitochondrial fission. TEM imaging revealed that LPS treatment caused significant alterations in mitochondrial morphology, which were characterized by partial mitochondrial fragmentation, loss of cristae, vacuole formation, and irregular arrangement (Fig. 5A). Ripk3 knockdown cells showed reversal of LPS-induced mitochondrial abnormalities. Western blot analysis revealed that LPS increased mitochondrial Drp1 expression, while decreasing cytoplasmic Drp1 expression. (Fig. 5B-D). Interestingly, Ripk3 inhibition reversed this phenomenon. Furthermore, Ripk3 deficiency resulted in elevated expression levels of mitofusin-1 (Mfn1) and optic atrophy 1 (Opa1), both of which are involved in mitochondrial fusion (Fig. 5B, E-F). These findings suggest that LPS excessively stimulates mitochondrial fission, possibly due to increased Ripk3 expression. To assess whether Ripk3 knockdown protects against mPTP opening in LPS-treated PMVECs by inhibiting mitochondrial fission, we used the fission inhibitor Mdivi1 (20μM). Conversely, we treated Ripk3-deficient cells with the fission activator FCCP (5μM) under LPS conditions (Fig. 5G). The results confirm that LPS and FCCP triggered mPTP opening, while Ripk3 knockdown and Mdivi1 inhibited it. These results indicate that Ripk3 induces mPTP opening through Drp1-mediated mitochondrial fission.
Ripk3 knockdown inhibits mPTP opening via Drp1-related mitochondrial fission. (A) Representative transmission electron microscopy (TEM) images depicting changes in mitochondrial morphology. Red arrow: damaged and swollen mitochondria. (B-F) The expression levels of mito-Drp1, cyto-Drp1, Mfn1, and Opa1 were quantified by Western blotting. The mitochondrial fission inhibitor, Mdivi1, was utilized to impede mitochondrial fission in WT cells under LPS administration. The mitochondrial fission activator, FCCP, was employed to promote mitochondrial fission in Ripk3-deficient cells under LPS treatment. (G) The change of mPTP opening time. Mean ± SEM, ∗p < 0.05 vs. the Ctrl group; #p < 0.05 vs. the LPS group; &p < 0.05 vs. the LPS+Mdivi1 group; %p < 0.05 vs. the LPS+ FCCP group.
Ripk3 regulates Drp1 activation via the AMPK pathway
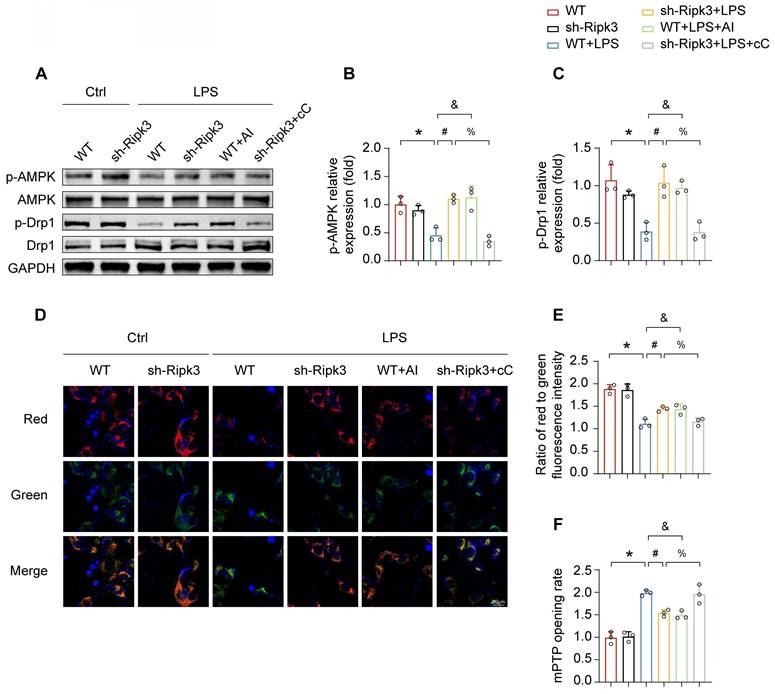
The mechanism by which Ripk3 triggers Drp1-mediated mitochondrial fission is still not fully understood. Multiple studies have asserted that the AMPK pathway is crucial in activating Drp1 phosphorylation at Ser637 and inhibits mitochondrial fission [37, 38]. Therefore, our study examined whether the AMPK pathway was involved in Ripk3-induced mitochondrial fission. In order to address this question, AICAR (AI, 1mM), the AMPK pathway activator, was employed as the positive control, whereas compound C (cC, 10μM), the AMPK pathway inhibitor, was employed as the negative control, both in the presence of LPS. LPS inhibited AMPK pathway activation, evidenced by reduced phospho-AMPK expression, which was reversed by Ripk3 knockdown. (Fig. 6A-B). In order to investigate the involvement of AMPK in the activation of mitochondrial fission, the level of phospho-Drp1 expression was measured. LPS inhibited the expression of phospho-Drp1 at Ser637. Nevertheless, the alterations were reversed as a result of Ripk3-knockdown, which had a resemblance to the administration of AICAR. In addition, the upregulation of phospho-Drp1 at Ser637 in Ripk3-knockdown cells was diminished by compound C (Fig. 6A, C). The findings supported our hypothesis that Ripk3 controls mitochondrial fission related to Drp1 through the AMPK pathway. Moreover, the presence of LPS stress and compound C induced cellular oxidative stress, which was reduced by the knockdown of Ripk3 and the administration of AICAR. Meanwhile, we assessed the mitochondrial membrane potential. Compound C caused a reduction in mitochondrial membrane potential, which was subsequently restored by AICAR (Fig. 6D-E). We additionally evaluated the rate of mPTP opening. The administration of LPS and compound C resulted in an elongated time of mPTP opening in WT cells, which was counteracted in Ripk3-deficient cells and treated with AICAR (Fig. 6F). Overall, these findings indicate that Ripk3 knockdown lessens damage to the mitochondria via activating the phosphorylation of Drp1 at Ser637 through AMPK.
Ripk3 knockdown alleviates mitochondrial injury through promoting AMPK-mediated Drp1 phosphorylation. (A-C) The phosphorylation of Drp1 and AMPK was measured using western blots. The AMPK pathway activator, AICAR (AI), was employed to activate AMPK pathway in WT cells under LPS treatment. The AMPK pathway inhibitor, compound C (cC), was utilized to block the AMPK pathway in Ripk3-deficient cells under LPS injury. (D-E) Impact of AMPK on mitochondrial membrane potential. (F) The change of mPTP opening time. Mean ± SEM, ∗p < 0.05 vs. the Ctrl group; #p < 0.05 vs. the LPS group; &p < 0.05 vs. the LPS+AI group; %p< 0.05 vs. the LPS+cC group.
Discussion
ALI, or its clinical manifestation, ARDS, is a significant health concern in developing countries, accounting for 30% of all deaths. ALI is characterized by noncardiogenic pulmonary edema with oxidative stress and inflammation affecting alveolar epithelial cells and pulmonary mesenchyme [39, 40]. The aetiology of ARDS involves multiple factors such as ischemia, anoxia, severe infection, and trauma. These etiologies lead to lung injury through different mechanisms. In ARDS caused by viral pneumonia, viral invasion of lung cells activates the host's immune response. Viral nucleic acids are recognized as pathogen-associated molecular patterns (PAMPs) by intracellular pattern recognition receptors (PRRs), for example Toll-like receptors (TLRs) [41]. This recognition process activates the Ripk3 signaling pathway and induces necroptosis in alveolar epithelial cells and pulmonary vascular endothelial cells. In ARDS caused by bacterial pneumonia, components such as LPS can stimulate inflammatory cells (such as macrophages) by releasing cytokines for example TNF-α [42], which can activate Ripk3 via the death receptor pathway when bound to cell surface receptors. In ARDS related to aspiration of gastric contents, acidic inhalant from the stomach will cause chemical damage to lung tissue after inhalation [43]. This acidic substance directly harms alveolar epithelial cells and vascular endothelial cells, leading to cell damage and death. Meanwhile, damage-associated molecular patterns (DAMPs) are released, stimulating inflammatory cells in lung tissue, which in turn activate the Ripk3 signaling pathway, further damaging the structure and function of lung tissue and promoting the formation of ARDS. For sepsis-associated ARDS, toxins released by pathogens such as bacteria enter the blood circulation and cause systemic inflammatory responses [44]. A large number of inflammatory factors such as TNF-α and IL-1β can activate Ripk3 through multiple signaling pathways, leading to necroptosis of lung cells, affecting the function of vascular endothelial cells, and promoting the formation of microthrombi, thus exacerbating pulmonary circulation disorders. After severe trauma, tissue damage releases large numbers of DAMPs, which trigger the body's immune response [45]. At the same time, massive blood transfusion may cause transfusion-related acute lung injury (TRALI) [46]. Inflammatory cell activation and increased oxidative stress due to trauma and blood transfusion may contribute to necroptosis of lung cells through activation of Ripk3, disrupting the alveolar-capillary barrier and ultimately leading to ARDS. The primary therapeutic approaches for ALI encompass antioxidant [47], anti-inflammatory [48], ventilation, and mesenchymal stem cell therapies [49]; ARDS is a multifaceted condition that lacks targeted treatment options, although recent advancements in clinical care have led to better outcomes [50]. Exploring new therapeutic methods and mechanisms is essential for developing more effective treatments for the devastating syndrome. Our findings contribute to this effort by identifying potential targets for novel ARDS therapies.
ARDS is characterized by dysfunction of the pulmonary microvascular endothelial barrier, a pathophysiological process significantly impacted by oxidative stress, which contributes to the breakdown of the barrier in PMVECs during the development of ARDS [51, 52]. While a proper quantity of ROS is essential for facilitating signal transmission under physiological conditions, exposure to inflammatory substances can cause PMVECs to produce an excessive amount of ROS, leading to cytoskeleton contraction via myosin light chain (MLC) and the reduction of intercellular junction proteins. This results in the formation of gaps between cells, which brings increased permeability, fluid leakage, and the development of edema eventually [51]. LPS is a primary causative factor of ALI, initially inducing mitochondrial injury, stimulating ROS overload, inhibiting oxidative phosphorylation, and causing elevated oxidative stress and damage to lung tissue [53, 54]. The present research has determined Ripk3 to be a key regulator of LPS-induced ALI, providing compelling proof for the role of Drp1-induced mitochondrial fission and PMVEC necroptosis in the progression of ALI induced by LPS.
Mitochondria are recognized as organelles that function as a powerhouse, producing ATP to support various cellular processes, and also serve as a central hub for signaling and regulating various biological processes, such as metabolism, cell proliferation, and immune response. The respiratory chain of mitochondria is crucial for the transportation of electrons and the conversion of energy, and any disruption to this process can have an impact on energy metabolism and result in the disruption of tissue balance. Research has demonstrated that damaged mitochondria can disrupt the metabolic health of lung epithelial cells, resulting in various lung disorders including asthma, ARDS, lung cancer, and COPD [55, 56]. The data provided strongly demonstrate the significance of preserving mitochondrial integrity to ensure tissue homeostasis and hinder the development of severe lung diseases. Mitochondria are targeted to trigger ALI under LPS challenge, since mitochondrial dysfunction has been shown to activate the NF-κB signaling pathway in lung tissue [57]. Impaired regulation of mitochondrial quality is believed to be the primary cause of excessive ROS generation and oxidative stress in tubular cells during LPS-induced acute kidney injury [58]. Septic cardiomyopathy is characterized by a decrease in mitophagy activity and a reduction in mitochondrial biogenesis. These factors contribute to a disturbance in cardiomyocyte energy metabolism and a failure in cardiac contractility [59]. Consistent with these results, our investigation also observed impaired mitochondrial function and structural abnormalities in the mitochondria of lung tissue treated with LPS. Actually, extensive researches have been carried out to explore the role of mitochondrial dysfunction in ARDS. For example, ALI is characterized by metabolic changes regulated by mitochondria [60]. Lung tissues treated with LPS have shown a decrease in ATP synthesis of mitochondria and an augment in ROS production of mitochondria [61]. Interestingly, our current research has revealed significant changes in mitochondria morphology in lung tissue under LPS stress. In terms of mitochondrial injury, damage to mitochondria morphology is regarded as the initial sign, compared with mitochondrial function disorder. However, the hypothesis has not been verified in cases of ALI. Our research revealed that changes in mitochondrial morphology directly impact mitochondrial function in PMVECs treated with LPS. The discovery offers a fresh perspective on the molecular mechanisms involved in ALI induced by LPS.
Necroptosis is a newly discovered form of regulated cell death which is distinct from apoptosis, causing inflammation and damage to tissues. Ripk3 is a widely recognized protein kinase that promotes necroptosis, regardless of whether cell death is induced by interferons, TLR3, TLR4, or TNF superfamily death receptors. The level of Ripk3 expression plays a crucial role in deciding the specific model of cell death [62]. Cells lacking Ripk3 are resistant to necroptosis. In acute lung injury, impaired alveolar epithelial and endothelial permeability involves multiple essential mechanisms, such as the activation of myosin light chain kinase (MLCK), RhoA and tyrosine kinases; increased calcium influx; and endothelial cell apoptosis [63, 64]. It has been shown that the Nrf2-MafF/ARE signaling pathway inhibits apoptosis of endothelial cells under extensive inflammation and reduces oxidative damage in ALI/ARDS [65]. In addition, mitoQ greatly reduces the high permeability of human pulmonary microvascular endothelial cells monolayer through regulating the expression of anti-apoptosis protein Bcl-2 and pro-apoptosis BAK, and decreases the activity of caspase 3 in endothelial cells, which are consistent with previous report that NFκB/NLRP3 pathway inhibits ROS and excessive autophagy in CSE-induced HUVEC injury [66]. Recently, a correlated study has demonstrated that mitoQ inhibits apoptosis in lung tissues by activating the PI3K/Akt/GSK-3 β/mTOR pathway, protecting lung function [67]. Unlike apoptosis, the understanding of the specific necroptosis mechanism is still limited in ARDS. The current study reveals that both apoptosis and necroptosis are stimulated by LPS, suggesting that necroptosis may have been overlooked as a causative element in ARDS. In addition, our findings further demonstrate that the Ripk3/Drp1 signaling pathway regulates LPS-induced necroptosis, and suppression of Ripk3 can delay the activation of necroptosis. However, there are still some unresolved matters that need to be addressed. Firstly, it remains uncertain whether necroptosis interacts with apoptosis in ARDS. Furthermore, it remains ambiguous whether they are controlled by a shared signaling pathway or upstream protein.
An association between Ripk3 upregulation and mPTP opening has been illustrated in several studies, which corroborates the necessity of mPTP for Ripk3-mediated necroptosis [68, 69]. The necroptosis process following Ripk3 activation leads to an imbalance in intracellular calcium homeostasis. When the concentration of calcium ions in the mitochondrial matrix reaches a certain threshold, mPTP opening is triggered. At the same time, the necroptosis process is accompanied by an increase in ROS. In mitochondria, ROS can oxidatively modify key components of mPTP, such as adenine nucleotide translocator (ANT) and voltage-dependent anion channel (VDAC) proteins. This oxidative modification lowers the threshold for mPTP opening and drives the opening of the pore. In addition, it has been shown that MLKL may interact with proteins such as mitochondrial VDAC, which in turn alters the permeability of the mitochondrial membrane and affects the opening state of mPTP. During necroptosis, mitochondrial oxidative phosphorylation is impaired and ATP production is reduced, resulting in a decrease in mitochondrial membrane potential, which affects the stability of mPTP, making it easier to open. Our findings suggest that necroptosis is provoked by Ripk3 and executed by mPTP opening, answering the underlying mechanism of Ripk3 regulating mPTP opening, especially in the ALI setting.
In addition to mitochondrial fission, mitochondrial fusion also plays a crucial part in controlling the morphology and quantity of mitochondria, and these two activities usually maintain a constant balance. Drp1 is typically located in the cytoplasm and undergoes bidirectional movement between the cytoplasm and the outer surface of mitochondria [70]. During periods of stress, including starvation and hypoxia, Drp1 can move from the cytoplasm to the mitochondria through phosphorylation [71]. The molecules then congregate toward the outer membrane of the mitochondria, where they form oligomers and facilitate mitochondria fission [72]. Our study confirms that Ripk3 enhances Drp1-mediated mitochondrial fission in acute lung injury generated by LPS. In addition, it has been verified that Ripk3 suppresses the process of mitochondrial fusion in septic cardiomyopathy [73]. Further research is necessary to ascertain the impact of Ripk3 on mitochondrial fusion in lung injury linked to inflammation.
Drp1-mediated mitochondrial fission is controlled through various post-translational modifications, including SUMOylation, ubiquitination, and phosphorylation [74]. Phosphorylation is known to regulate the activity of Drp1. Phosphorylation at Ser637 inhibits mitochondrial fission, which ultimately reduces necroptosis [75]. AMPK, a key detector of energy strain, can be activated (predominantly phosphorylation at Thr172) by various mitochondrial stimuli and promptly affects mitochondrial fission [76, 77]. Thus, AMPK is an essential focus for the management of ALI. There is ongoing debate regarding the impact of AMPK on the process of mitochondrial fission. A recent research has found that AMPK stimulates mitochondrial fission through upregulating Mff [78], while some research has indicated that AMPK hinders mitochondrial fission by regulating Drp1 expression in aortic endothelial cells [79]. Our data showed that the inhibition of Ripk3 via a lentivirus system activated the AMPK pathway by causing phosphorylation at Thr172 in PMVECs. AMPK activation promotes the phosphorylation of Drp1 (Ser637) and also prevents Drp1-mediated mitochondrial fission. Our research uncovered the role of the AMPK/Drp1/mitochondrial fission axis in controlling LPS-triggered necroptosis in ALI.
It's worth noting that the environment of cell culture in vitro is not exactly the same as the environment of the complex cellular population in vivo, and in vitro culture lacks regulation by the nervous and endocrine systems. Lacking these regulatory controls, the metabolism of in vitro cells is more constant than that of in vivo cells, but this is not a true representation of the tissue from which the cells originate. In addition, compared with in vivo cells, cells cultured in vitro, especially those that have been repeatedly passed on and cultured for a long time, have undergone certain changes in morphology and function, and may undergo the phenomenon of dedifferentiation and screening, resulting in the loss of some biological properties of the original cells. Moreover, cells cultured in vitro lack interaction with other different types of cells, which makes the physiological response ability of cells become single and has certain limitations.
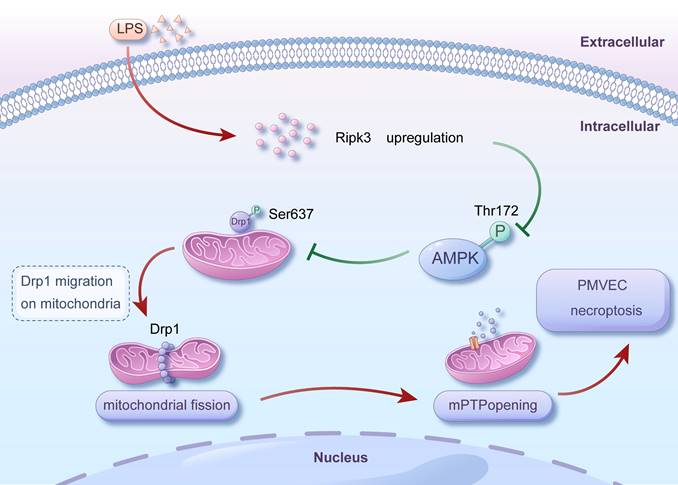
Given the above, our studies have revealed a new role of Ripk3 in ALI induced by LPS. Specifically, Ripk3 controls mitochondrial fission mediated by Drp1 through the AMPK signaling pathway. The buildup of excessive mitochondrial fragments results in function disorder of mitochondria and the opening of mPTP, ultimately triggering necroptosis in PMVECs and impairing migration (Fig. 7). These discoveries provide insight into the therapeutic potential of targeting Ripk3 as a remedy for ALI. By inhibiting the activity of Ripk3, necroptosis can be blocked, thereby attenuating lung injury. At present, some inhibitors targeting Ripk3 have been developed and have shown certain efficacy in animal experiments. For example, a newly developed Ripk3 inhibitor, UH15-38, can effectively and selectively block necroptosis of alveolar epithelial cells triggered by influenza A virus in vivo, improve lung inflammation, and prevent death after infection, which provides a certain reference for drug development of ALI [80]. Meanwhile, Ripk3 inhibitors have the potential to be used in combination with other therapeutic agents for ALI, such as anti-inflammatory drugs and antioxidant drugs, with a view to playing a better role in the treatment of ALI.
Mechanism diagram for the role of Ripk3-AMPK-Drp1-mPTP in LPS-mediated acute lung injury via initiating necroptosis. LPS injury caused upregulation of Ripk3, which resulted in a reduction in the phosphorylation of AMPK. Inactivating AMPK hindered the phosphorylation of Drp1 at Ser637, which triggered fatal mitochondrial fission. Excessive fission caused the opening of mPTP, ultimately leading to the necroptosis of PMVECs. Nevertheless, the recovery of AMPK activity could halt the excessive fission, offering prosurvival effects for the lung in the presence of LPS injury. Our findings revealed a novel role for Ripk3 in LPS-induced acute lung injury through regulating Drp1-mediated mitochondrial fission via the AMPK signaling pathway. These results suggest that targeting Ripk3 could be a promising therapeutic approach for treating acute lung injury in clinical practice.
Initiation of the Ripk3 signaling pathway promotes fibroblast activation and proliferation in lung tissue, increases extracellular matrix deposition and influences extracellular matrix remodeling, providing a cellular basis for the development of pulmonary fibrosis [81]. Appropriate regulation of Ripk3 during ALI regression helps to reduce the infiltration of inflammatory cells into lung tissue. By inhibiting Ripk3, its downstream necroptosis signaling pathway is blocked, which decreases inflammatory mediators released by inflammatory cells (such as neutrophils and macrophages) due to necroptosis, and at the same time alleviates the occurrence of necroptosis in alveolar epithelial cells, which contributes to maintaining the integrity of the alveolar epithelial cell-to-cell junctions, and promotes the recovery of alveolar function.
Given that the function of mitochondria is controlled by various activities such as mitophagy, mitochondrial fusion and fission, the precise molecular mechanism of acute lung injury requires more clarification.
Conclusion
In summary, acute lung injury is correlated with elevated Ripk3 expression, resulting in inhibition of AMPK. The reduction in AMPK subsequently hinders the phosphorylation of Drp1 (Ser637), leading to excessive stimulation of mitochondrial fission, increasing the opening of mPTP and resulting in cellular necroptosis in acute lung injury. Therefore, by genetically eliminating Ripk3, the AMPK/Drp1-mitochondrial fission pathway can be activated to prevent mPTP opening and PMVEC necroptosis. As a result, acute lung injury can be finally avoided. The outcomes of our study offer a hopeful target for the management of ALI.
Acknowledgements
This study was financially supported by grants from the Beijing Natural Science Foundation (No. 7242028) and the National Natural Science Foundation of China (No. 82341089 and 32470800).
Data availability
The original data presented in the study are included in the article; further inquiries can be directed to the corresponding authors. The scRNA-seq data are available on the National Center for Biotechnology Information Gene Expression Omnibus database under accession no. GSE186839.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chambers ED, White A, Vang A, Wang Z, Ayala A, Weng T. et al. Blockade of equilibrative nucleoside transporter 1/2 protects against Pseudomonas aeruginosa-induced acute lung injury and NLRP3 inflammasome activation. Faseb j. 2020;34:1516-31
2. Liu M, Chen Y, Wang S, Zhou H, Feng D, Wei J. et al. α-Ketoglutarate Modulates Macrophage Polarization Through Regulation of PPARγ Transcription and mTORC1/p70S6K Pathway to Ameliorate ALI/ARDS. Shock. 2020;53:103-13
3. Hu Q, Zhang S, Yang Y, Yao JQ, Tang WF, Lyon CJ. et al. Extracellular vesicles in the pathogenesis and treatment of acute lung injury. Mil Med Res. 2022;9:61
4. Siegel ER, Croze RH, Fang X, Matthay MA, Gotts JE. Inhibition of the lipoxin A4 and resolvin D1 receptor impairs host response to acute lung injury caused by pneumococcal pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. 2021;320:L1085-l92
5. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. 2021;398:622-37
6. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A. et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18
7. Abedi F, Hayes AW, Reiter R, Karimi G. Acute lung injury: The therapeutic role of Rho kinase inhibitors. Pharmacol Res. 2020;155:104736
8. Yan J, Wang A, Cao J, Chen L. Apelin/APJ system: an emerging therapeutic target for respiratory diseases. Cell Mol Life Sci. 2020;77:2919-30
9. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW. et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21:319-29
10. Millar FR, Summers C, Griffiths MJ, Toshner MR, Proudfoot AG. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax. 2016;71:462-73
11. Pugliese NR, Masi S. The emerging role of endothelial function in cardiovascular oncology. Eur J Prev Cardiol. 2020;27:604-7
12. He YQ, Zhou CC, Yu LY, Wang L, Deng JL, Tao YL. et al. Natural product derived phytochemicals in managing acute lung injury by multiple mechanisms. Pharmacol Res. 2021;163:105224
13. Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415
14. Forrester SJ, Xu Q, Kikuchi DS, Okwan-Duodu D, Campos AC, Faidley EA. et al. Poldip2 deficiency protects against lung edema and vascular inflammation in a model of acute respiratory distress syndrome. Clin Sci (Lond). 2019;133:321-34
15. Huang Y, Song C, He J, Li M. Research progress in endothelial cell injury and repair. Front Pharmacol. 2022;13:997272
16. Ta HQ, Teman NR, Kron IL, Roeser ME, Laubach VE. Steen solution protects pulmonary microvascular endothelial cells and preserves endothelial barrier after lipopolysaccharide-induced injury. J Thorac Cardiovasc Surg. 2023;165:e5-e20
17. Grazioli S, Dunn-Siegrist I, Pauchard LA, Blot M, Charles PE, Pugin J. Mitochondrial alarmins are tissue mediators of ventilator-induced lung injury and ARDS. PLoS One. 2019;14:e0225468
18. Gu H, Yang K, Wu Q, Shen Z, Li X, Sun C. A link between protein acetylation and mitochondrial dynamics under energy metabolism: A comprehensive overview. J Cell Physiol. 2021;236:7926-37
19. Di Nottia M, Verrigni D, Torraco A, Rizza T, Bertini E, Carrozzo R. Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes (Basel). 2021;12:247
20. Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P. et al. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening. J Am Heart Assoc. 2017;6:e005328
21. Wang J, Toan S, Zhou H. Mitochondrial quality control in cardiac microvascular ischemia-reperfusion injury: New insights into the mechanisms and therapeutic potentials. Pharmacol Res. 2020;156:104771
22. Dong J, Liu W, Liu W, Wen Y, Liu Q, Wang H. et al. Acute lung injury: a view from the perspective of necroptosis. Inflamm Res. 2024;73:997-1018
23. Yi Y, Gao K, Zhang L, Lin P, Wang A, Jin Y. Zearalenone Induces MLKL-Dependent Necroptosis in Goat Endometrial Stromal Cells via the Calcium Overload/ROS Pathway. Int J Mol Sci. 2022;23:10170
24. Petrie EJ, Birkinshaw RW, Koide A, Denbaum E, Hildebrand JM, Garnish SE. et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc Natl Acad Sci U S A. 2020;117:8468-75
25. Singla S, Sysol JR, Dille B, Jones N, Chen J, Machado RF. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am J Respir Cell Mol Biol. 2017;57:307-14
26. Chen H, Li Y, Wu J, Li G, Tao X, Lai K. et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 2020;27:2568-85
27. Zhang J, Pan Z, Zhou J, Zhang L, Tang J, Gong S. et al. The myosin II inhibitor, blebbistatin, ameliorates pulmonary endothelial barrier dysfunction in acute lung injury induced by LPS via NMMHC IIA/Wnt5a/β-catenin pathway. Toxicol Appl Pharmacol. 2022;450:116132
28. Boytard L, Hadi T, Silvestro M, Qu H, Kumpfbeck A, Sleiman R. et al. Lung-derived HMGB1 is detrimental for vascular remodeling of metabolically imbalanced arterial macrophages. Nat Commun. 2020;11:4311
29. Du Y, Zhu P, Wang X, Mu M, Li H, Gao Y. et al. Pirfenidone alleviates lipopolysaccharide-induced lung injury by accentuating BAP31 regulation of ER stress and mitochondrial injury. J Autoimmun. 2020;112:102464
30. Qiao X, Ding Y, Altawil A, Yin Y, Wang Q, Wang W. et al. Roles of noncoding RNAs in chronic obstructive pulmonary disease. J Transl Int Med. 2023;11:106-10
31. Zhang N, Tang S, Zhang J, Pei B, Pang T, Sun G. The dipeptidyl peptidase-4 inhibitor linagliptin ameliorates LPS-induced acute lung injury by maintenance of pulmonary microvascular barrier via activating the Epac1/AKT pathway. Biomed Pharmacother. 2022;155:113704
32. Zhu P, Wan K, Yin M, Hu P, Que Y, Zhou X. et al. RIPK3 Induces Cardiomyocyte Necroptosis via Inhibition of AMPK-Parkin-Mitophagy in Cardiac Remodelling after Myocardial Infarction. Oxid Med Cell Longev. 2021;2021:6635955
33. Luan Y, Huang E, Huang J, Yang Z, Zhou Z, Liu Y. et al. Serum myoglobin modulates kidney injury via inducing ferroptosis after exertional heatstroke. J Transl Int Med. 2023;11:178-88
34. Gribov A, Sill M, Lück S, Rücker F, Döhner K, Bullinger L. et al. SEURAT: visual analytics for the integrated analysis of microarray data. BMC Med Genomics. 2010;3:21
35. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163-72
36. Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C. et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721-d8
37. Liu J, Yan W, Zhao X, Jia Q, Wang J, Zhang H. et al. Sirt3 attenuates post-infarction cardiac injury via inhibiting mitochondrial fission and normalization of AMPK-Drp1 pathways. Cell Signal. 2019;53:1-13
38. Jeong HY, Kang JM, Jun HH, Kim DJ, Park SH, Sung MJ. et al. Chloroquine and amodiaquine enhance AMPK phosphorylation and improve mitochondrial fragmentation in diabetic tubulopathy. Sci Rep. 2018;8:8774
39. Du HL, Zhai AD, Yu H. Synergistic effect of halofuginone and dexamethasone on LPS-induced acute lung injury in type II alveolar epithelial cells and a rat model. Mol Med Rep. 2020;21:927-35
40. Nadeem A, Al-Harbi NO, Ahmad SF, Al-Harbi MM, Alhamed AS, Alfardan AS. et al. Blockade of interleukin-2-inducible T-cell kinase signaling attenuates acute lung injury in mice through adjustment of pulmonary Th17/Treg immune responses and reduction of oxidative stress. Int Immunopharmacol. 2020;83:106369
41. Shirey KA, Blanco JCG, Vogel SN. Targeting TLR4 Signaling to Blunt Viral-Mediated Acute Lung Injury. Front Immunol. 2021;12:705080
42. Zhu P, Wang J, Du W, Ren J, Zhang Y, Xie F. et al. NR4A1 Promotes LPS-Induced Acute Lung Injury through Inhibition of Opa1-Mediated Mitochondrial Fusion and Activation of PGAM5-Related Necroptosis. Oxid Med Cell Longev. 2022;2022:6638244
43. Li T, Zou C. The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. Int J Mol Sci. 2020 21
44. Ding WC, Chen J, Liao HY, Feng J, Wang J, Zhang YH. et al. [Mechanism of Xuebijing Injection in treatment of sepsis-associated ARDS based on network pharmacology and in vitro experiment]. Zhongguo Zhong Yao Za Zhi. 2023;48:3345-59
45. Xiao Y, Fang H, Zhu Y, Zhou J, Dai Z, Wang H. et al. Multifunctional Cationic Hyperbranched Polyaminoglycosides that Target Multiple Mediators for Severe Abdominal Trauma Management. Adv Sci (Weinh). 2024;11:e2305273
46. Tung JP, Chiaretti S, Dean MM, Sultana AJ, Reade MC, Fung YL. Transfusion-related acute lung injury (TRALI): Potential pathways of development, strategies for prevention and treatment, and future research directions. Blood Rev. 2022;53:100926
47. Huang Q, Ren Y, Yuan P, Huang M, Liu G, Shi Y. et al. Targeting the AMPK/Nrf2 Pathway: A Novel Therapeutic Approach for Acute Lung Injury. J Inflamm Res. 2024;17:4683-700
48. Jiang L, Guo P, Ju J, Zhu X, Wu S, Dai J. Inhalation of L-arginine-modified liposomes targeting M1 macrophages to enhance curcumin therapeutic efficacy in ALI. Eur J Pharm Biopharm. 2023;182:21-31
49. Liang D, Liu C, Yang M. Mesenchymal stem cells and their derived exosomes for ALI/ARDS: A promising therapy. Heliyon. 2023;9:e20387
50. Battaglini D, Robba C, Pelosi P, Rocco PRM. Treatment for acute respiratory distress syndrome in adults: a narrative review of phase 2 and 3 trials. Expert Opin Emerg Drugs. 2022;27:187-209
51. Karki P, Birukov KG. Rho and Reactive Oxygen Species at Crossroads of Endothelial Permeability and Inflammation. Antioxid Redox Signal. 2019;31:1009-22
52. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383:2255-73
53. Di A, Mehta D, Malik AB. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium. 2016;60:163-71
54. Liu Z, Ying Y. The Inhibitory Effect of Curcumin on Virus-Induced Cytokine Storm and Its Potential Use in the Associated Severe Pneumonia. Front Cell Dev Biol. 2020;8:479
55. Riou M, Alfatni A, Charles AL, Andrès E, Pistea C, Charloux A. et al. New Insights into the Implication of Mitochondrial Dysfunction in Tissue, Peripheral Blood Mononuclear Cells, and Platelets during Lung Diseases. J Clin Med. 2020;9:1253
56. Cho HY, Kleeberger SR. Mitochondrial biology in airway pathogenesis and the role of NRF2. Arch Pharm Res. 2020;43:297-320
57. Xu P, Zhang WQ, Xie J, Wen YS, Zhang GX, Lu SQ. Shenfu injection prevents sepsis-induced myocardial injury by inhibiting mitochondrial apoptosis. J Ethnopharmacol. 2020;261:113068
58. Sabry MM, Ahmed MM, Maksoud OMA, Rashed L, Morcos MA, El-Maaty AA. et al. Carnitine, apelin and resveratrol regulate mitochondrial quality control (QC) related proteins and ameliorate acute kidney injury: role of hydrogen peroxide. Arch Physiol Biochem. 2022;128:1391-400
59. Yang Y, Zhu Y, Xiao J, Tian Y, Ma M, Li X. et al. Maresin conjugates in tissue regeneration 1 prevents lipopolysaccharide-induced cardiac dysfunction through improvement of mitochondrial biogenesis and function. Biochem Pharmacol. 2020;177:114005
60. Robinson MJ, Krasnodembskaya AD. Therapeutic targeting of metabolic alterations in acute respiratory distress syndrome. Eur Respir Rev. 2020;29:200114
61. Ten VS, Ratner V. Mitochondrial bioenergetics and pulmonary dysfunction: Current progress and future directions. Paediatr Respir Rev. 2020;34:37-45
62. Tummers B, Green DR. Mechanisms of TNF-independent RIPK3-mediated cell death. Biochem J. 2022;479:2049-62
63. Lu H, Poirier C, Cook T, Traktuev DO, Merfeld-Clauss S, Lease B. et al. Conditioned media from adipose stromal cells limit lipopolysaccharide-induced lung injury, endothelial hyperpermeability and apoptosis. J Transl Med. 2015;13:67
64. Yu Y, Lv N, Lu Z, Zheng YY, Zhang WC, Chen C. et al. Deletion of myosin light chain kinase in endothelial cells has a minor effect on the lipopolysaccharide-induced increase in microvascular endothelium permeability in mice. Febs j. 2012;279:1485-94
65. Cen M, Ouyang W, Zhang W, Yang L, Lin X, Dai M. et al. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol. 2021;41:101936
66. Chen S, Wang Y, Zhang H, Chen R, Lv F, Li Z. et al. The Antioxidant MitoQ Protects Against CSE-Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells. Int J Biol Sci. 2019;15:1440-51
67. Li R, Ren T, Zeng J. Mitochondrial Coenzyme Q Protects Sepsis-Induced Acute Lung Injury by Activating PI3K/Akt/GSK-3β/mTOR Pathway in Rats. Biomed Res Int. 2019;2019:5240898
68. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA. et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658-62
69. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H. et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652-8
70. Jhun BS, J OU, Adaniya SM, Cypress MW, Yoon Y. Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology. Antioxidants (Basel). 2018;7:195
71. Pegadraju H, Abby Thomas J, Kumar R. Mechanistic and therapeutic role of Drp1 in the pathogenesis of stroke. Gene. 2023;855:147130
72. Yang X, Wang H, Ni HM, Xiong A, Wang Z, Sesaki H. et al. Inhibition of Drp1 protects against senecionine-induced mitochondria-mediated apoptosis in primary hepatocytes and in mice. Redox Biol. 2017;12:264-73
73. Zhong J, Tan Y, Lu J, Liu J, Xiao X, Zhu P. et al. Therapeutic contribution of melatonin to the treatment of septic cardiomyopathy: A novel mechanism linking Ripk3-modified mitochondrial performance and endoplasmic reticulum function. Redox Biol. 2019;26:101287
74. Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. 2021;42:655-64
75. Sun J, Liu X, Shen C, Zhang W, Niu Y. Adiponectin receptor agonist AdipoRon blocks skin inflamm-ageing by regulating mitochondrial dynamics. Cell Prolif. 2021;54:e13155
76. Xiao YY, Xiao JX, Wang XY, Wang T, Qu XH, Jiang LP. et al. Metformin-induced AMPK activation promotes cisplatin resistance through PINK1/Parkin dependent mitophagy in gastric cancer. Front Oncol. 2022;12:956190
77. Chan CM, Sekar P, Huang DY, Hsu SH, Lin WW. Different Effects of Metformin and A769662 on Sodium Iodate-Induced Cytotoxicity in Retinal Pigment Epithelial Cells: Distinct Actions on Mitochondrial Fission and Respiration. Antioxidants (Basel). 2020;9:1057
78. Peng F, Jiang D, Xu W, Sun Y, Zha Z, Tan X. et al. AMPK/MFF Activation: Role in Mitochondrial Fission and Mitophagy in Dry Eye. Invest Ophthalmol Vis Sci. 2022;63:18
79. Tong W, Leng L, Wang Y, Guo J, Owusu FB, Zhang Y. et al. Buyang huanwu decoction inhibits diabetes-accelerated atherosclerosis via reduction of AMPK-Drp1-mitochondrial fission axis. J Ethnopharmacol. 2023;312:116432
80. Gautam A, Boyd DF, Nikhar S, Zhang T, Siokas I, Van de Velde LA. et al. Necroptosis blockade prevents lung injury in severe influenza. Nature. 2024;628:835-43
81. Quan MY, Yan X, Miao W, Li X, Li J, Yang L. et al. Metformin alleviates benzo[a]pyrene-induced alveolar injury by inhibiting necroptosis and protecting AT2 cells. Ecotoxicol Environ Saf. 2024;272:116094
Author contact
![]() Corresponding authors: Dr Yingzhen Du; zhenzhen52com, and Prof. Guogang Xu; gxuorg.
Corresponding authors: Dr Yingzhen Du; zhenzhen52com, and Prof. Guogang Xu; gxuorg.