Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Milestones of the discovery of...
Cell death mechanisms in PAH
Summary and Future Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(10):1840-1851. doi:10.7150/ijms.93902 This issue Cite
Review
Cell Death in Pulmonary Arterial Hypertension
Xia Li1,2,3, JunLan Tan2,3, JiaJing Wan2,3, BeiBei Cheng2,3, Yu-Hong Wang4, Aiguo Dai1,2,3 ![]()
1. Hunan Academy of Chinese Medicine, Changsha 410208, Hunan, People's Republic of China.
2. Department of Respiratory Diseases, Medical School, Hunan University of Chinese Medicine, Changsha 410208, Hunan, People's Republic of China.
3. Hunan Provincial Key Laboratory of Vascular Biology and Translational Medicine, Changsha 410208, Hunan, People's Republic of China.
4. Science and Technology Innovation Center, Hunan University of Chinese Medicine, Changsha 410208, China.
Received 2024-1-4; Accepted 2024-5-22; Published 2024-7-14
Abstract

Pulmonary arterial hypertension (PAH) is a severe pulmonary vascular disease characterized by increased pulmonary vascular resistance because of vascular remodeling and vasoconstriction. Subsequently, PAH leads to right ventricular hypertrophy and heart failure. Cell death mechanisms play a significant role in development and tissue homeostasis, and regulate the balance between cell proliferation and differentiation. Several basic and clinical studies have demonstrated that multiple mechanisms of cell death, including pyroptosis, apoptosis, autophagy, ferroptosis, anoikis, parthanatos, and senescence, are closely linked with the pathogenesis of PAH. This review summarizes different cell death mechanisms involved in the death of pulmonary artery smooth muscle cells (PASMCs) and pulmonary artery endothelial cells (PAECs), the primary target cells in PAH. This review summarizes the role of these cell death mechanisms, associated signaling pathways, unique effector molecules, and various pro-survival or reprogramming mechanisms. The aim of this review is to summarize the currently known molecular mechanisms underlying PAH. Further investigations of the cell death mechanisms may unravel new avenues for the prevention and treatment of PAH.
Keywords: pulmonary arterial hypertension, pulmonary artery smooth muscle cells, pulmonary artery endothelial cells, cell death
Introduction
Pulmonary hypertension (PAH) is a complex disease characterized by a progressive increase in the pulmonary vascular resistance (PVR) and the pulmonary artery pressure (PAP). Vascular remodeling is a central feature of the pathophysiology of PAH and is characterized by structural and functional changes in the pulmonary artery wall. This leads to increased muscularization of the pulmonary artery wall and the peripheral non-muscular vessels of the respiratory acinar artery, and the formation of neointima and plexiform lesions. The global prevalence of PAH is 93 cases per 1 million subjects with a male-to-female prevalence ratio of 1:2.3 and a 3-year survival rate of only 55-65% [1,2]. The interplay between various cell death mechanisms and other cellular processes drives the complex etiology, pathogenesis, and progression of PAH. This review describes various cell death mechanisms, specific cell death effector molecules, and associated signaling pathways in PAH. It also highlights the pro-survival or reprogramming mechanisms for circumventing cell death. The aim of this review is also to highlight future directions in this field and identify novel therapeutic targets for the prevention and treatment of PAH (Table 1).
Multiple types of cell death mechanisms in PAH
| Cell death mechanisms | Stimulation | Features | Morphology | Molecular mechanisms |
|---|---|---|---|---|
| Apoptosis | Physiology or pathology | Absence of inflammatory response; formation of apoptotic bodies | Chromatin aggregates, divides, localizes to the nuclear membrane; cytoplasm condenses; finally the nuclear membrane ruptures; cells form many apoptotic bodies by budding. | AKT, ERK1/2, NF-κB and STAT3 signaling pathways; imbalance of mitochondrial dynamics imbalance; Warburg effect. |
| Autophagy | Physiology or pathology | Autophagosome formation | The autophagosome is a vacuole-like structure with double or multilayer membrane and contains cytoplasmic components, including mitochondria, endoplasmic reticulum, and ribosomes. | AMPK-mediated mTOR autophagy signaling pathway; BNIP3-dependent Beclin-1 autophagy signaling pathway; NF- κ B signaling pathway |
| Pyroptosis | Pathological stimulation | inflammatory reaction; pyroptotic body formation | Under electron microscope, rupture of the plasma membrane is clearly seen; large number of vesicles or pyroptotic bodies seen; pores are formed in the cell membrane; the cell membrane bursts and releases the intracellular contents. | Non-Classic caspase--4/-5/-11 signaling pathway;Classic caspase-1signaling pathwayand GSDMD/GSDME signaling pathway |
| Ferroptosis | Pathological stimulation | Intracellular accumulation of iron ions; lipid peroxidation; elevated ROS. | Cell membrane rupture and blistering; mitochondrial atrophy; decrease or even disappearance of the mitochondrial ridge; increased membrane density; normal nuclear morphology; lack of chromatin condensation | HMGB1-mediatedsignaling pathway; TLR4/NLRP3signaling pathway |
| Parthanatos | Pathological stimulation | Activated PARP1 binds to AIF/M1 and mediates the translocation of AIF/M1 complex from the mitochondria to the nucleus; MI stimulates DNA fragmentation. | Chromatin condensation; DNA fragmentation (large) | AIF/M1 dependent and AIF/M1 independent signaling pathway |
| Anoikis | Pathological stimulation | It only occurs when adherent cells lose their adhesion | Cell volume is reduced; organelles shrink; formation of apoptotic bodies. | Intrinsic pathway (mitochondrial events induced by cell stress) and extrinsic pathway (mediated by TNF and first apoptosis signal (Fas-ligand) |
| Cell senescence | Physiology or pathology | Cells shrink and become smaller in size; reduced metabolic rate | Cell water content decreases; volume is reduced; cell membrane fragility increases; and permeability decreases | Mechanisms driving cellular senescence include DNA damage, telomere shortening, FXN loss, and elevated SASP |
Milestones of the discovery of the role of cell death mechanisms in PAH
In 1996, Jones and Rabinovitch reported the occurrence of endothelial cell apoptosis during the early stages of monocrotaline (MCT)-induced pulmonary arterial hypertension (PAH) in the adult Sprague-Dawley rats [3]. In 2008, Marsboom et al. reported that chronic hypoxia exacerbated PAH by inducing senescence of the circulating endothelial progenitor cells (EPCs) in the C57Bl6/N mice [4]. In 2011, Lee et al. reported that autophagy was upregulated in the lung tissues of patients with idiopathic pulmonary hypertension (IPAH), in vitro hypoxia-treated human primary endothelial cells (PAECs) and smooth muscle cells (PASMCs), and the chronic hypoxia-induced PAH model mice [5]. In 2018, Chen et al. reported that PASMCs underwent anoikis, a novel type of apoptosis, because of detachment of PASMCs from the extracellular matrix due to reduced levels of integrin α5 and β1 during PAH [6]. In 2020, Zhang et al. reported that the expression levels of interleukin-18 (IL-18), cysteine protease-1 (caspase-1), and high mobility group protein B1 (HMGB1) transcripts and proteins were elevated in the lung tissues of the MCT-induced PAH model rats; this linked pyroptosis to the upregulation of the programmed death receptor 1 (PD-1) in the human PASMCs [7]. In 2021, Zhang and Liu performed transcriptome analysis of the lung tissue samples from 15 PAH patients and 11 normal controls, and identified 514 differentially expressed genes (DEG) based on the Wilcoxon rank sum test and weighted gene correlation network analysis. Subsequently, they used the FerrDb database and identified eight ferroptosis-related DEGs; this demonstrated that ferroptosis was upregulated in the lung tissues during PAH and was a potential therapeutic target [8]. In 2021, Lv et al. analyzed blood samples from 50 healthy controls and 88 patients with PAH, including those with idiopathic pulmonary hypertension (IPAH), chronic obstructive pulmonary disease-associated pulmonary hypertension (COPD-PH) and chronic thromboembolic pulmonary hypertension (CTEPH), and reported the association of PAH pathogenesis with the upregulation of parthanatos, a poly-ADP ribose polymerase 1 (PARP1)-dependent and apoptosis inducing factor (AIF)-mediated caspase-independent cell death mechanism; higher PARP1 and AIF expression levels were associated with increased morbidity and mortality of PAH patients [9] (Figure 1).
Milestones of the discovery of the roles of multiple cell death mechanisms in PAH.
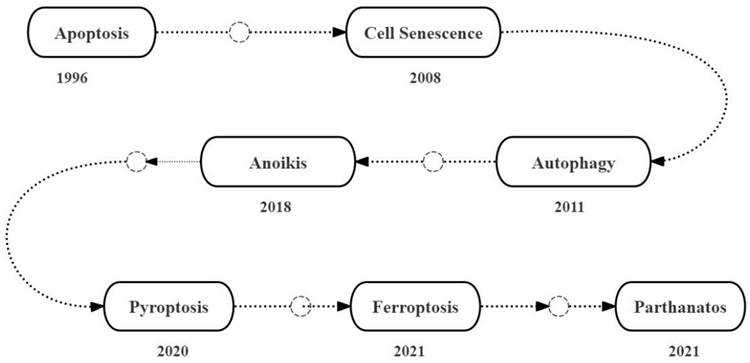
Cell death mechanisms in PAH
Apoptosis and PAH
Apoptosis is a programmed cell death mechanism that plays a crucial role in development and tissue homeostasis by removing abnormal cells in the multicellular organisms [10]. Apoptosis is a complex process regulated by multiple genes, including Bcl-2 and caspase family of proteins, oncogenes, and tumor suppressor genes [11,12]. The dysregulation of apoptosis is linked to various human diseases, including PAH.
Tissue injury and hypoxia promotes pulmonary vascular remodeling and PAH development by disrupting the balance between cell survival and apoptosis [13].
Several studies have demonstrated that apoptotic PAECs play a critical role in the pathogenesis of PAH. Many diseases and environmental factors induce pulmonary vascular endothelial injury by enhancing endothelial cell apoptosis and dysfunction. Subsequently, increased proliferation of the smooth muscle cells and fibroblasts promote pulmonary arterial remodeling and thrombosis. Furthermore, upregulated apoptosis of PAECs causes severe PAH and triggers excessive proliferation of apoptosis-resistant endothelial cells and subsequent formation of plexiform lesions and pulmonary vascular occlusions [13]. Therefore, inhibition of PAEC apoptosis at various stages of PAH development represents a novel therapeutic strategy. In general, the rates of endothelial cell proliferation and apoptosis rates are very low. The dynamic balance between proliferation and apoptosis is required for maintaining a constant number of endothelial cells and normal vascular function. Endothelial injury in the pulmonary precapillary arterioles serves as the initial trigger for PAH pathogenesis, whereas excessive apoptosis of the endothelial cells is a key mechanism of vascular endothelial injury and dysfunction [14]. Pulmonary vascular remodeling, plexiform lesions, and in-situ thrombosis represent the primary pathological changes in response to aberrant endothelial cell apoptosis in the pulmonary vascular system [15]. PAECs are localized in a stable internal environment. However, genetic mutations in the endothelial cells and other extrinsic factors induce endothelial cell apoptosis and pulmonary vascular endothelial damage in the genetically susceptible individuals that are exposed to environmental factors such as viruses, inflammation, hypoxia, blood flow shear force, or certain drugs [16,17]. Taraseviciene-Stewart et al. reported severe PAH in rats treated with chronic hypoxia and vascular endothelial growth factor (VEGF) receptor blocker SU-5416; the PAH model rats showed significant PAEC apoptosis in the early stages followed by precapillary arterial occlusion because of endothelial cell hyper proliferation; monocrotaline-induced PAH model also confirmed that endothelial cell apoptosis played a significant role in the pathogenesis of PAH [18,19]. These results demonstrated that apoptosis of PAECs was an important early event in PAH pathogenesis.
PAH is characterized by elevated peripheral pulmonary vascular resistance because of vascular tension, smooth muscle cell contraction, and vascular remodeling. The hypertrophy, proliferation, migration, and anti-apoptotic properties of the PASMCs are key factors that contribute to these vascular changes [20]. Increased activation of the anti-apoptotic mechanisms in the PASMCs promote abnormal smooth muscle cell proliferation and pulmonary vascular remodeling, which is histologically manifested by increased number of PASMCs, disordered arrangement, thickening of the media, significant narrowing of the peripheral pulmonary artery lumen, and complete occlusion of pulmonary arterioles in the advanced stages of PAH. Functionally, impaired regulation of vascular tension, decreased diastolic ability, increased systolic sensitivity, and dysfunction of the systolic and diastolic phases lead to a persistent increase in the pulmonary vascular resistance and mean pulmonary artery pressure [21,22]. Therefore, inhibition of anti-apoptotic mechanisms, including suppression of Bcl2 expression delays disease progression [23]. Furthermore, inhibition of myelin and lymphocyte protein (MAL) suppressed hypoxia-induced PASMCs proliferation and promoted apoptosis [24]. Moreover, pro-apoptotic proteins such as Bcl-xL induced PASMCs apoptosis and reversed pulmonary vascular remodeling [25,26]. The delicate balance between proliferation and apoptosis of the PASMCs plays a crucial role in the pathogenic pulmonary vascular remodeling underlying PAH. Therefore, targeted inhibition of PASMCs and/or induction of PASMCs apoptosis represents a novel therapeutic approach to reverse pulmonary vascular remodeling and manage PAH.
Several genetic factors have been associated with an increased risk of pulmonary arterial hypertension (PAH). Functional mutations in the bone morphogenetic protein receptor 2 (BMPR2) gene are commonly associated with an increased risk of PAH. Germline mutations in BMPR2 are found in 70% of cases with familial PAH (FPAH) and 10% to 30% of cases with idiopathic PAH (IPAH) [27]. Research showed that puerarin administration exerted significant protective effects in both hypoxia and MCT-induced experimental PAH rodent models, evidenced by significantly reduced right ventricular systolic pressure (RVSP) and lung injury, improved pulmonary artery blood flow as well as pulmonary vasodilation and contraction function, inhibited inflammatory responses in lung tissues, improved resistance to apoptosis and abnormal proliferation in lung tissues, attenuated right ventricular injury and remodeling, and maintained normal function of the right ventricle. Revealed that MCT and hypoxia treatment significantly downregulated BMPR2/Smad signaling in the lung tissues and PPARγ/PI3K/Akt signaling in the lung tissues and right ventricles, which were restored by puerarin administration [28]. BMPR2 mediates anti-apoptotic effects in the endothelial cells. Genetic deletion of endothelial BMPR2 promotes PAH by enhancing endothelial cell apoptosis [29]. BMP2 also mediates anti-proliferative and pro-apoptotic effects in the pulmonary arterial smooth muscle cells (PASMCs) by modulating the expression and function of the voltage-gated K+ channels [30-31].
In the pulmonary vascular cells, mitochondria play a crucial role in maintaining energy balance and regulation of cell death [32]. The dynamic balance between fusion and fission of mitochondria plays a key role in cellular apoptosis [33]. Increased mitochondrial fission is linked with elevated apoptosis in pathological conditions such as pulmonary arterial hypertension (PAH). In PAH, upregulation of mitochondrial fission proteins such as MiD and Drp1 accelerate mitosis in the PASMCs and reduce apoptosis. Conversely, silencing of MiD49 or MiD51 promotes apoptosis by restoring mitochondrial fusion, increasing Bak expression, and reducing Akt activation [34]. The epigenetic regulation of mitochondrial dynamics contributes to apoptosis resistance in both the human and experimental animal and cellular models of PAH [35,36].
The Warburg effect plays a significant role in the development of pulmonary arterial hypertension (PAH) by promoting excessive cellular proliferation and apoptosis resistance [37]. Mitochondria in the PASMCs act as oxygen sensors and trigger hypoxic pulmonary vasoconstriction under low oxygen concentrations. Acquired mitochondrial abnormalities, including elevated expression and activities of pyruvate dehydrogenase kinase (PDK) and pyruvate kinase muscle subtype 2 (PKM2) promote uncoupled glycolysis (Warburg effect), which is linked with PAH development [35]. The glycolysis inhibitor 3-bromopyruvate (3-BrPA) inhibits the mitochondria-localized hexokinase-2 (HK-2) and promotes activation of the apoptotic pathway through the release of cytochrome C into the cytosol and activation of caspase-3 (Casp-3) [38]. Furthermore, 3-BrPA reversed hypoxia-induced PAH in the monocrotaline (MCT)-induced PAH model rats by inhibiting glycolysis and significantly reducing various pathological processes associated with PAH development [39]. This suggested that 3-BrPA exerted its beneficial effects on PAH by specifically targeting HK-2 to induce apoptosis and inhibit inflammation. Therefore, 3-BrPA is a promising therapeutic option for PAH. Furthermore, reduced oxidative phosphorylation and increased glycolysis in the endothelial cells and smooth muscle cells decreases the threshold for apoptosis activation by lowering the levels of mitochondrial reactive oxygen species (ROS) and increasing the mitochondrial membrane potential [40]. Therefore, a deeper understanding of the molecular mechanisms governing mitochondrial metabolism and dynamics could reveal new biomarkers and therapeutic targets for PAH.
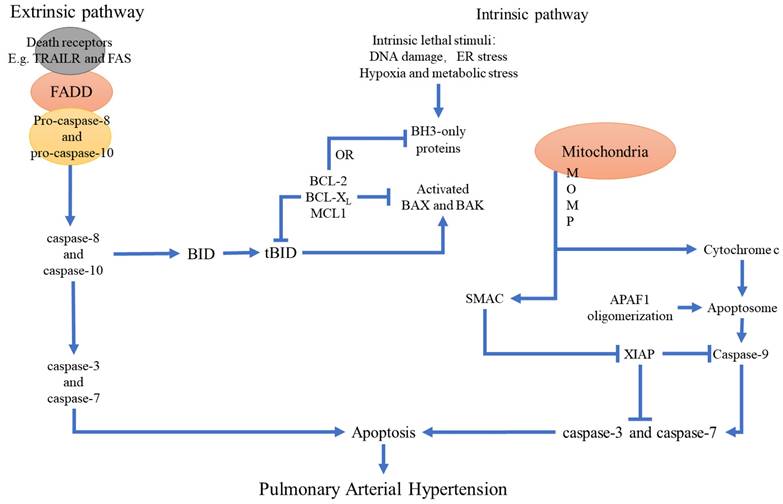
The result of abnormal extracellular signaling is the activation of anti-apoptotic cell signaling pathways, such as AKT, ERK1/2, NF-κB and STAT3 [41-44], and the inactivation of the pro-apoptotic FOXO signaling pathway [45]. In the hypoxia and MCT-induced experimental models of PAH, ERK1/2 and AKT pathways play a key role in the initiation of apoptosis by regulating the expression levels of Bcl2 and the proteins of the caspase family [46,47]. Furthermore, in the hypoxia-induced PAH models, cIAP and BCL2 levels are increased and the levels of BAX and BIM are decreased in the endothelial cells and the smooth muscle cells. Moreover, inhibition of the PI3K/AKT pathway, anti-apoptotic BCL2 protein, and cIAP, as well as activation of FOXO signaling alleviates PAH in the animal models [48]. Loss-of-function mutations in BMPR2 promote apoptosis resistance in the smooth muscle cells through the STAT3 signaling pathways [30]. These findings demonstrate that targeting the apoptosis and anti-apoptotic pathways in the pulmonary vascular cells may potentially abrogate PAH progression and offer novel avenues for preventing PAH (Figure 2).
The relationship between apoptosis and the main pathological changes in PAH. Apoptosis plays a key role in pulmonary hypertension. Stimulation of various apoptotic or pro-apoptotic factors because of injury and/or hypoxia promotes changes in the structure and function of the pulmonary artery wall and generates an imbalance between cell survival and apoptosis. This leads to pulmonary vascular remodeling and eventually PAH.
Autophagy and PAH
Autophagy is a type of programmed cell death that is required for maintaining tissue homeostasis and involves a process of “self-digestion”, which is characterized by the engulfment and degradation of damaged cells, organelles, proteins, and pathogens through the lysosomal system [49]. Autophagy is categorized into the following three types based on the mechanisms by which the intracellular materials are transported to the lysosomes: chaperone-mediated autophagy, microautophagy, and macroautophagy [50]. Aberrant up- or down-regulation of autophagy is implicated in several human diseases such as PAH [51].
Autophagy dysregulation in the PASMCs and PAECs is implicated in pulmonary vascular remodeling and the pathogenesis of PAH [52]. For example, estradiol directly inhibits vascular endothelial cell proliferation, improves hemodynamics, and mitigates PAH development by enhancing autophagy, especially mitophagy [53]. Beclin-1 knockdown showed defective autophagy and increased endothelial angiogenesis in the pulmonary artery [54]. Moreover, in HIV-related PAH, autophagy promotes transition of PAECs from an apoptotic phenotype to a hyperproliferative phenotype [55]. This suggested a close relationship between the onset of PAH and increased autophagy in the pulmonary endothelium. Therefore, autophagy inhibition is a promising therapeutic option for the PAH patients, but further validation is required through basic and clinical research.
In the monocrotaline (MCT)-induced PAH model, inhibition of autophagy in the PASMCs suppressed lysosomal degradation of bone morphogenetic protein receptor type 2, downregulated the DNA-binding inhibitory factor 1 (Id1), and suppressed the progression of PAH [51].
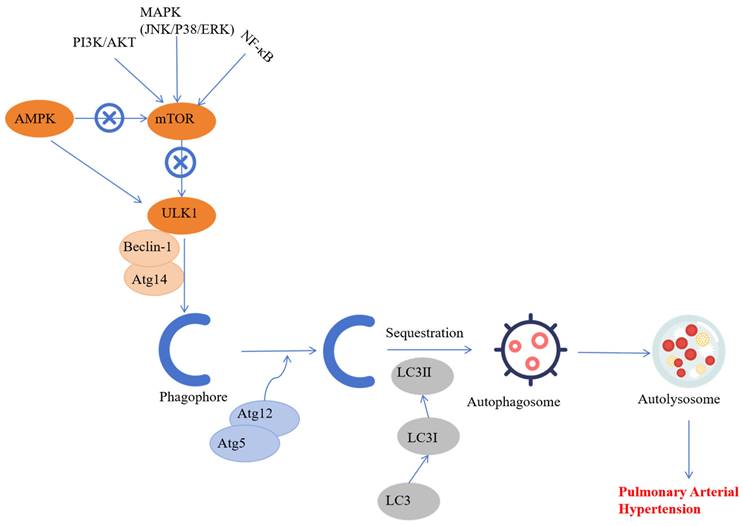
Conversely, MCT treatment increased autophagy in the PASMCs and was associated with significantly higher PASMCs proliferation and migration, pulmonary artery remodeling, and development of PAH [51]. Moreover, increased expression of LC3B and Beclin1 in the hypoxia-induced PAH models suggested that autophagy played a significant role in PAH pathogenesis [56]. The induction of autophagy is a multifaceted process and is influenced by various extrinsic and intrinsic factors such as drugs, cigarette smoke extracts, and mitochondrial dysfunction [57-58]. The regulatory mechanisms of autophagy vary in the PAH model rats and include the AMPK-mediated mTOR signaling pathway, BNIP3-dependent Beclin-1 signaling pathway, and the NF-κB signaling pathway [59]. Therefore, targeted drugs to inhibit these autophagy-related signaling pathways are promising avenues for treating human diseases with enhanced autophagy, including PAH.
These findings indicate that autophagy is activated in various cell types during PAH through multiple signaling pathways. Although several signaling pathways related to PAH-induced autophagy have been identified, the precise molecular mechanism remains unclear. Therefore, further research is required to determine the molecular mechanisms that regulate PAH-associated autophagy (Figure 3).
The role of autophagy in the pathology underlying pulmonary artery hypertension. Autophagy is activated in several cell types of the pulmonary artery during PAH through multiple signaling pathways. Although several autophagy pathways related to pulmonary hypertension have been reported, the precise molecular mechanism that regulates dysfunctional autophagy during PAH development and progression remains to be determined.
Pyroptosis and PAH
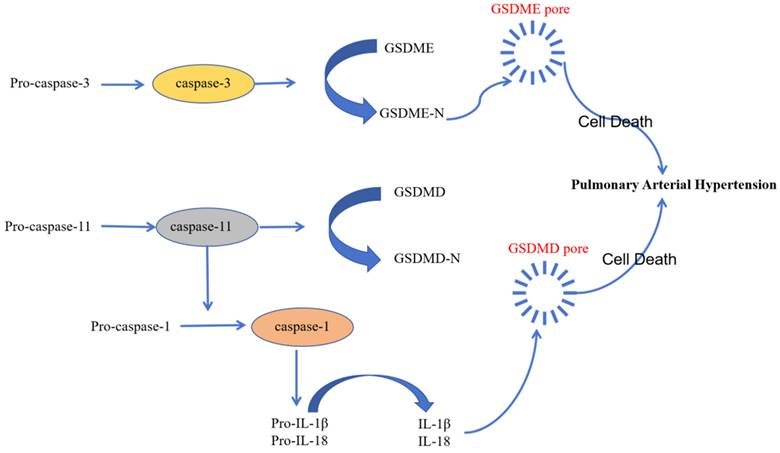
Pyroptosis is a novel type of inflammation-activated programmed cell death during which inflammasome sensors in the human body recognize pathogenic signals and damage-related molecules related and activate caspase-1/4/5/11. Subsequently, gasdermin-D and other family members of the gasdermin family of proteins are cleaved by the activated caspase-1/4/5/11into a N-terminal pore-forming domain (PFD) and a C-terminal repressor domain (RD). PFD induces cell swelling and rupture by generating macropores in the cell membrane [60]. Several studies have demonstrated that pyroptosis plays a key role in the development of PAH and inhibition of pyroptosis reduces the severity of PAH [61-63].
Activation of inflammatory caspase is a critical event in the initiation of pyroptosis. Caspase-1/4/5/11 plays a significant role in the development of PAH [61]. NLRP3, a prominent inflammasome, triggers pyroptosis in the pulmonary arterial endothelial cells by generating activated pro-inflammatory cytokines, which subsequently promote PAH progression [62]. The classic caspase-1-related pyroptosis pathway involves cytosolic pattern-recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) and assemble ASC and pro-caspase-1 to form functional inflammasome complex; pro-caspase-1 is then cleaved to generate the activated cleaved caspase-1, which cleaves gasdermin-D to generate PFD; pores generated by PFD in the cell membrane stimulate the release of activated pro-inflammatory factors [63]. The inflammation promotes vascular remodeling and disease progression in PAH. Inhibition of caspase-1-mediated canonical inflammasomes mitigates PAH [64]. Circ-Calm4 is a specific circular RNA that modulates pyroptosis-related signaling pathways and targets in the PASMCs, thereby regulating PAH; moreover, inhibition of circ-Calm4 significantly reduced the levels of pyroptosis-related proteins such as NLRP3, caspase-1, IL-1β, and IL-18 in the PASMCs [65].
The non-classical pyroptosis pathway is mediated by direct binding of caspase-4/5/11 to the lipopolysaccharides and subsequent cleavage of gasdermin-D to form the pore-forming domain (PFD) and release of the activated pro-inflammatory factors. This process mediates endothelial dysfunction and leads to enhanced vascular inflammation, vascular remodeling, and right ventricular failure, thereby contributing to the pathogenesis of PAH [63]. Endothelial dysfunction in PAH is triggered by the tumor necrosis factor-α (TNF-α), which is elevated in patients with PAH and the animal models of PAH. TNF-α stimulation mitigated PAH development by inhibiting caspase-4/5/11 [66]. Moreover, pyroptosis involved direct binding of the caspase-4/5/11 to capsase-3 and activation of the caspase-3-GSDME axis; cleavage of GSDM-E by caspase-3 induced pyroptosis [66]. Therefore, inhibition of caspase-3 expression is another potential therapeutic strategy to prevent PAH development.
Caspase-8 promotes synthesis and processing of pro-IL-1β through the non-classical and classical pathways by regulating the formation of the functional NLRP3 inflammasome and cleaved caspase-1. Experiments in the caspase-8 gene knockout SD rats and mice demonstrated that caspase-8 was required for the secretion of IL-1β via the NLRP3 inflammasome and caspase-1 pathways in the macrophages. Subsequent proliferation of the PASMCs and infiltration of the inflammatory cells promoted development of PAH. However, in the Caspase-8 knockout rats and mice, the progression of PAH was inhibited [62].
The diverse recognition of various pathogens and injury-related molecular patterns by the inflammatory sensors demonstrates the pivotal role of pyroptosis in the body's defense against various pathogens. Pyroptosis initiates release of cellular contents from infected cells. This acts as a signal for initiating the inflammatory cascade. The localized inflammation promotes recruitment and activation of immune cells, which assist in the elimination of pathogens from the human body. Therefore, investigating the relationship between pyroptosis and PAH may unravel novel signaling components and pathway mechanisms, thereby revealing new avenues for the prevention and treatment of PAH (Figure 4).
The role of pyroptosis in the pathology of PAH. Pyroptosis plays an significant role in the pathogenesis of PAH. The occurrence and development of pulmonary hypertension is mediated by the classical caspase-1 pathway, non-classical caspase-4/5/11 pathway, and the caspase-3-GSDME pathway of pyroptosis.
Ferroptosis and PAH
Ferroptosis is a novel form of non-apoptotic cell death that is dependent on iron and reactive oxygen species (ROS) and is characterized by a reduction in cell size and thickening of the mitochondrial membrane [67]. During ferroptosis, excessive accumulation of iron in the cells generates excessive reactive oxygen species (ROS), which leads to the oxidation of polyunsaturated fatty acids, damage to the cell membrane structure, and cell death [68]. Aberrant changes in ferroptosis are associated with PAH. Ferroptosis is upregulated in the pulmonary arterial endothelial cells (PAECs) of the MCT-induced PAH rat models, and is characterized by lipid peroxidation, increased cellular iron levels, mitochondrial damage, abnormal expression of GPX4, ferritin heavy chain 1 (FTH1), and NADPAH oxidase 4 (NOX4), activation of inflammatory factors, and severe pulmonary artery remodeling. Furthermore, inhibition of ferrostatin-1 (an iron death inhibitor) delayed pulmonary vascular remodeling and protected right ventricular function in the PAH patients [67]. Moreover, PRDX6 is an important driver of PAH progression by mediating ferroptosis in the PAECs through the release of HMGB1 and activation of the TLR4/NLRP3 inflammatory signaling pathway [69]. PASMCs are another key target cell for ferroptosis in PAH. Solute carrier family 7 member 11 (SLC7A11) is up-regulated in the Sugen5416/hypoxia-induced PAH rats and the PAH patients. Overexpression of SLC7A11 inhibits ferroptosis and promotes PASMC proliferation [70]. Proteomic analysis showed that the ferroptosis pathway was enriched in patients with severe PAH. Furthermore, the presence of the SNP rs1444732 in GPX4 was associated with severe PAH [71]. These findings demonstrate that ferroptosis played a key role in the pathogenesis of PAH. Therefore, ferroptosis inhibitors or targeted drugs against ferroptosis-regulatory genes are potential therapeutic targets in PAH. However, further basic and clinical studies are necessary.
Parthanatos and PAH
Parthanatos is a form of programmed cell death that is initiated by the hyperactivation of poly(ADP-ribose) polymerase 1 (PARP-1) and downstream events involving activation of the apoptosis-inducing factor (AIF), DNA fragmentation, and cell death [72]. This process is independent of the caspases and Bcl-2, and is characterized by mitochondrial dysfunction, excessive generation of reactive oxygen species (ROS), and alterations in the calcium channel activity., Parthanatos is characterized rupture of the plasma membrane but does not involve formation of apoptotic bodies and cell swelling as observed during apoptosis [73]. The Parthanatos cascade involves four main steps: (1) poly-ADP ribose polymer (PAR) accumulation because of hyperactivation of PARP-1; (2) subsequent release of AIF from mitochondria due to excess PAR; (3) binding of AIF with MIF and translocation of the AIF/MIF complex to the nucleus; and (4) MIF-induced DNA fragmentation in the nucleus [74,75]. Parthanatos is triggered not only by severe DNA damage but also by oxidative stress, hypoxia, hypoglycemia, and inflammation [76]. A previous study demonstrated that treatment of rats with MCT and Su5416/hypoxia (SuHx) enhanced right ventricular myocardial apoptosis through various pathways and subsequently lead to PAH [77]. A recent clinical study reported that circulating levels of PARP1, PAR, AIF, and MIF were higher in the non-surviving patients with PAH compared with the surviving patients with PAH. The plasma levels of PARP1, PAR, AIF, and MIF correlated significantly with the mean pulmonary arterial pressure (mPAP), mean right atrial pressure (mRAP), pulmonary vascular resistance (PVR), and the cardiac index (CI) of the PAH subjects. Elevated levels of PARP-1 and AIF were associated with morbidity and mortality of PAH patients and are considered as strong predictors of increased PAH risk [9]. These data demonstrated that the key molecules involved in the Parthanatos pathway, including PARP-1, AIF, and MIF are potential therapeutic targets for PAH.
Anoikis and PAH
Anoikis is a form of apoptotic cell death caused by detachment of cells from the extracellular matrix because of disruptions in the integrin connections [78]. Integrins are cell adhesion receptor proteins that mediate intercellular interactions as well as interactions between the extracellular matrix and proteins of cell cytoskeleton such as connexins. Integrins regulate cell-cell adhesion and transmit important intracellular signals for cell survival, proliferation, gap junction and motility, and are critical determinants of donor cell survival in case of stem cell transplantation [79,80].
Stem cells have emerged as promising candidates for the treatment of patients with PAH because of their ability to self-renew and differentiate into multiple cell types. However, the low survival rate of the transplanted stem cells (typically less than 1%) has significantly hindered the success of stem cell therapy [81]. The low survival and engraftment rates of stem cells post-transplantation are primarily attributed to the activation of anoikis, a novel mechanism of cell death [82-85]. Therefore, enhancing the adhesion properties of the stem cells at the injury site is critical for promoting their survival and engraftment, as well as, suppressing the activation of anoikis. Previous studies have shown that the levels of integrin α5 and integrin β1 are significantly reduced in the pulmonary arterial smooth muscle cells (PASMC) during PAH [86]. Moreover, integrins facilitate endothelium-dependent vasodilation through the nitric oxide (NO) pathway [87]. Therefore, NO is a promising treatment for PAH because it can increase the resistance against anoikis and improve cell migration [88-90]. Chen et al. transfected recombinant bone marrow mesenchymal stem cells (rBMSCs) with a lentiviral vector encoding ITGA5B1 and reported that the recombinant BMSCs with ITGA5B1 showed enhanced cell adhesion, viability, and NO production and reduced anoikis [6,86]. Since the levels of ITGA5B are reduced in PAH, the transgenic expression of ITGA5B in the MSCs may offer a promising therapeutic strategy to alleviate PAH in the future.
Cell senescence and PAH
Cell senescence is an irreversible cell cycle arrest caused by a variety of physiological and pathological stressors, including reactive oxygen species, DNA damage, active oncogenes, and metabolic stress [91]. The senescent cells do not proliferate but have metabolic activity and showing characteristic morphological and physiological features, including enlarged flat shape, accumulation of β-galactosidase, and a secretory phenotype. The senescent cells regulate the tissue micro-environment in a paracrine manner. In general, tissue remodeling after transient induction of senescence is beneficial for eliminating damaged cells [92]. However, prolonged senescence or inability to eliminate the senescent cells is harmful. The concept of cell senescence was first reported by Hayflick and Moorhead in 1961 based on a permanent loss of cell division in the normal human embryonic lung fibroblasts in vitro; similar effects were later confirmed in various other cell types [93-95]. The involvement of senescence in lung diseases was first reported in patients with chronic obstructive pulmonary disease (COPD), especially those with emphysema [96]. Subsequently, senescent pulmonary artery smooth muscle cells were reported in patients with COPD-PH [97]. More recent evidence has suggested that the presence of senescent pulmonary artery smooth muscle cells and endothelial cells in patients with pulmonary arterial hypertension (PAH) is associated with pulmonary artery remodeling and PAH development [98].
The senescence of PASMCs plays a crucial role in the pathogenesis of PAH. PASMCs are key components of the pulmonary vasculature and are responsible for regulating vascular tone and maintaining a normal contractile function of the blood vessels. Extrinsic factors such as smoke, dust, hypoxia, and bacteria, as well as intrinsic factors such as inflammation and cellular stress accelerate the senescence of PASMCs. The senescent PASMCs exhibit enlarged and flattened cell morphology, expression of senescence-associated β-galactosidase, osteopontin, and cyclin-dependent kinase inhibitors, and telomere shortening [97,99]. In the hypoxia-treated mice and monocrotaline-induced PAH rats, p53 expression is reduced in the aging PASMCs and may contribute to pulmonary vascular remodeling by promoting PASMC proliferation and calcium influx [100]. Furthermore, PASMCs from idiopathic PAH patients show reduced expression levels of the p53 protein and increased Bax/Bcl-2 ratio [101,102]. This suggested that PASMC senescence was a critical factor in the process of pulmonary vascular remodeling during PAH. Therefore, targeting p53 is a potential therapeutic strategy to prevent PASMC senescence and subsequent alleviation of PAH.
PAEC senescence plays a significant role in the development of PAH. The levels of senescence markers in the PAECs are significantly higher in the patients with idiopathic PAH (IPAH) compared to those in the healthy subjects. The expression of TWIST1 and PDGFB is elevated in the senescent cells and mediates the progression of PAH [103]. DNA damage and senescence is induced by shear stress in the pulmonary microvascular endothelial cells (MVECs) of patients with idiopathic PAH [104]. Furthermore, telomere erosion is associated with a higher proportion of senescent lung endothelial cells in patients with chronic obstructive pulmonary disease (COPD) [105]. Hypoxic mice with endothelial FXN deficiency exhibit cell-specific senescence, elevated expression of the senescence-associated secretory phenotype (SASP) markers, accumulation of perivascular monocytes, and increased vascular collagen deposition. This suggested that endothelial FXN deficiency exacerbated endothelial cell senescence and worsened the severity of PAH [106,107]. Collectively, these findings suggested a close association between endothelial cell senescence and the development of PAH. Therefore, targeting senescence and its associated proteins are potential therapeutic targets for PAH in the future.
Further investigations are necessary to unravel the role of cellular senescence in the pathophysiology of PAH and for preclinical trials regarding the efficacy of senescence-targeting drugs. Although there is a great deal of evidence regarding the close association of senescence in the PASMC and PAECs with PAH, it is not clear whether these cells influence the development of PAH together or independently. Secondly, the occurrence of PAH is multifactorial, and the relationship between senescence and other pathogenic factors is not completely clear. The clinical importance of senescence is a treatment target requires in-depth study. Furthermore, the study on the molecular mechanisms underlying the role of cell senescence in PAH is still in the preliminary stages. Therefore, further basic and clinical research is necessary. It is plausible that once the integral role of cell senescence has been confirmed in PAH, inhibition of senescence or the removal of senescent cells will provide a novel avenue for treating patients with PAH.
Summary and Future Perspectives
In the pathogenesis of pulmonary arterial hypertension (PAH), the cells of the pulmonary arterial system undergo programmed cell death through different pathways. However, there is a significant lack of information regarding the relationship between different mechanisms of programmed cell death and PAH pathogenesis. Therefore, significant investigations are necessary to elucidate the mechanisms by which different cell death mechanisms are triggered in vivo. Moreover, further research is necessary to determine how these survival and death signals are balanced to determine specific cell fate. It is also not clear as to how these complex signals in the dying cells influence PAH development and progression. This remains a promising area of research in the field. The cell death mechanisms and signaling pathways play a critical essential role in growth, development, homeostasis, and disease. Deciphering these mechanisms will unravel novel therapeutic targets for the prevention and alleviation of PAH.
Abbreviations
AIF: Apoptosis inducing factor; CI: Cardiac index; COPD: Chronic obstructive pulmonary disease; CTEPH: Chronic thromboembolic pulmonary hypertension; DAMP: Damage-associated molecular patterns; DEG: Differentially expressed genes; EPC: Endothelial progenitor cells; IPAH: Idiopathic pulmonary hypertension; MAL: Myelin and lymphocyte; PAEC: Pulmonary artery endothelial cells; PAH: Pulmonary arterial hypertension; PAMP: Pathogen-associated molecular patterns; PAP: Pulmonary artery pressure; PAR: Poly-ADP ribose; PASMC: Pulmonary artery smooth muscle cells; PDK: Pyruvate dehydrogenase kinase; PFD: Pore-forming domain; PRR: Pattern-recognition receptors; PVR: Pulmonary vascular resistance; RD: Repressor domain; ROS: Reactive oxygen species; SASP: Senescence-associated secretory phenotype; VEGF: Vascular endothelial growth factor.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant number 82305214]; Natural Science Foundation of Hunan Province [grant number 2023JJ40401]; Hunan Administration of Traditional Chinese Medicine [grant number B2023024]; the Hunan Provincial Department of Education Scientific Research Outstanding Youth Project [grant number 22B0394]; and State Key Laboratory Project of Chinese Medicine powder and Innovative drugs [grant number 21PTKF1002].
Author Contributions
Xia Li searched the literature and drafted the manuscript; Ai-Guo Dai conceived and designed the review; Jun-lan Tan, Jia-jing Wan, and Bei-bei Cheng examined the literature and constructed the figures; Yu-hong Wang, Ai-Guo Dai made critical revisions of the review. All the authors contributed to the article and approved the final version for submission.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hou Wenwen, Qu Nini. Research Overview of Traditional Chinese Medicine Intervention in Pulmonary Arterial Hypertensio. Journal of Practical Traditional Chinese Internal Medicine. 2022;36(02):129-131
2. Dubroff J, Melendres L, Lin Y. et al. High geographic prevalence of pulmonary artery hypertension: associations with ethnicity, drug use, and altitude. Pulm Circ. 2020;10(1):2045894019894534
3. Jones PL, Rabinovitch M. Tenascin-C is induced with progressive pulmonary vascular disease in rats and is functionally related to increased smooth muscle cell proliferation. Circ Res. 1996;79(6):1131-1142
4. Marsboom G, Pokreisz P, Gheysens O. et al. Sustained endothelial progenitor cell dysfunction after chronic hypoxia-induced pulmonary hypertension. Stem Cells. 2008;26(4):1017-1026
5. Lee SJ, Smith A, Guo L. et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2011;183(5):649-658
6. Chen HY, Pan L, Yang HL. et al. Integrin alpha5beta1 suppresses rBMSCs anoikis and promotes nitric oxide production. Biomed Pharmacother. 2018;99:1-8
7. Zhang M, Xin W, Yu Y. et al. Programmed death-ligand 1 triggers PASMCs pyroptosis and pulmonary vascular fibrosis in pulmonary hypertension. J Mol Cell Cardiol. 2020;138:23-33
8. Zhang F, Liu H. Identification of ferroptosis-associated genes exhibiting altered expression in pulmonary arterial hypertension. Math Biosci Eng. 2021;18(6):7619-7630
9. Lv ZC, Li F, Wang L. et al. Impact of Parthanatos on the Increased Risk of Onset and Mortality in Male Patients With Pulmonary Hypertension. Am J Mens Health. 2021;15(3):15579883211029458
10. Poch D, Mandel J. Pulmonary Hypertension. Ann Intern Med. 2021;174(4):ITC49-ITC64
11. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147(4):742-758
12. Yuan J, Najafov A, Py BF. Roles of Caspases in Necrotic Cell Death. Cell. 2016;167(7):1693-1704
13. Deng C, Zhong Z, Wu D. et al. Role of FoxO1 and apoptosis in pulmonary vascular remolding in a rat model of chronic thromboembolic pulmonary hypertension. Sci Rep. 2017;7(1):2270
14. Duan H, Zhang Q, Liu J. et al. Suppression of apoptosis in vascular endothelial cell, the promising way for natural medicines to treat atherosclerosis. Pharmacol Res. 2021;168:105599
15. Nickel N, Jonigk D, Kempf T. et al. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir Res. 2011;12(1):62
16. Lévy M, Maurey C, Celermajer DS. et al. Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. J Am Coll Cardiol. 2007;49(7):803-810
17. Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10(1):95
18. Zhang W, Li Y, Xi X. et al. MicroRNA-15a-5p induces pulmonary artery smooth muscle cell apoptosis in a pulmonary arterial hypertension model via the VEGF/p38/MMP-2 signaling pathway. Int J Mol Med. 2020;45(2):461-474
19. Hong YM, Kwon JH, Choi S. et al. Apoptosis and inflammation associated gene expressions in monocrotaline-induced pulmonary hypertensive rats after bosentan treatment. Korean Circ J. 2014;44(2):97-104
20. Tajsic T, Morrell NW. Smooth muscle cell hypertrophy, proliferation, migration and apoptosis in pulmonary hypertension. Compr Physiol. 2011;1(1):295-317
21. Veith C, Vartürk-Özcan I, Wujak M. et al. SPARC, a Novel Regulator of Vascular Cell Function in Pulmonary Hypertension. Circulation. 2022;145(12):916-933
22. Rai N, Sydykov A, Kojonazarov B. et al. Targeting peptidyl-prolyl isomerase 1 in experimental pulmonary arterial hypertension. Eur Respir J. 2022;60(2):2101698
23. Perini GF, Ribeiro GN, Pinto Neto JV. et al. BCL-2 as therapeutic target for hematological malignancies. J Hematol Oncol. 2018;11(1):65
24. Liu Jinjun, Li Qingqing, Wu Shili. et al. Role of myelin and lymphocyte protein in regulating pulmonary artery smooth muscle cell proliferation and apoptosis in pulmonary hypertension. Journal of Southern Medical University. 2022;42(10):1572-1577
25. Cai C, Xiang Y, Wu Y. et al. Formononetin attenuates monocrotaline-induced pulmonary arterial hypertension via inhibiting pulmonary vascular remodeling in rats. Mol Med Rep. 2019;20(6):4984-4992
26. Rybka V, Suzuki YJ, Shults NV. Effects of Bcl-2/Bcl-xL Inhibitors on Pulmonary Artery Smooth Muscle Cells. Antioxidants (Basel). 2018;7(11):150
27. Ferrian S, Cao A, McCaffrey EF. et al. Single-Cell Imaging Maps Inflammatory Cell Subsets to Pulmonary Arterial Hypertension Vasculopathy. Am J Respir Crit Care Med. 2024;209(2):206-218
28. Chen D, Zhang HF, Yuan TY. et al. Puerarin-V prevents the progression of hypoxia- and monocrotaline-induced pulmonary hypertension in rodent models. Acta Pharmacol Sin. 2022;43(9):2325-2339
29. Wang J, Liu W, Lu W. et al. Sodium tanshinone IIA sulfonate enhances the BMP9-BMPR2-Smad1/5/9 signaling pathway in rat pulmonary microvascular endothelial cells and human embryonic stem cell-derived endothelial cells. Biochem Pharmacol. 2022;199:114986
30. Nie X, Shen C, Tan J. et al. Andrographolide Attenuates Established Pulmonary Hypertension via Rescue of Vascular Remodeling. Biomolecules. 2021;11(12):1801
31. Long L, Ormiston ML, Yang X. et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension[J]. Nat Med. 2015;21(7):777-785
32. Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet. 2009;43:95-118
33. Otera H, Mihara K. Mitochondrial dynamics: functional link with apoptosis. Int J Cell Biol. 2012;2012:821676
34. Chen KH, Dasgupta A, Lin J. et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation. 2018;138(3):287-304
35. Dasgupta A, Wu D, Tian L. et al. Mitochondria in the Pulmonary Vasculature in Health and Disease: Oxygen-Sensing, Metabolism, and Dynamics. Compr Physiol. 2020;10(2):713-765
36. Colpman P, Dasgupta A, Archer SL. The Role of Mitochondrial Dynamics and Mitotic Fission in Regulating the Cell Cycle in Cancer and Pulmonary Arterial Hypertension: Implications for Dynamin-Related Protein 1 and Mitofusin2 in Hyperproliferative Diseases. Cells. 2023;12(14):1897
37. Harvey LD, Chan SY. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. J Clin Med. 2017;6(4):43
38. Guo Y, Liu X, Zhang Y. et al. 3-Bromopyruvate ameliorates pulmonary arterial hypertension by improving mitochondrial metabolism. Life Sci. 2020;256:118009
39. Liu J, Wang W, Wang L. et al. 3-Bromopyruvate alleviates the development of monocrotaline-induced rat pulmonary arterial hypertension by decreasing aerobic glycolysis, inducing apoptosis, and suppressing inflammation. Chin Med J (Engl). 2020;133(1):49-60
40. Sauler M, Bazan IS, Lee PJ. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu Rev Physiol. 2019;81:375-402
41. Tian Q, Fan X, Ma J. et al. Critical role of VGLL4 in the regulation of chronic normobaric hypoxia-induced pulmonary hypertension in mice. FASEB J. 2021;35(8):e21822
42. Yan J, Wang A, Cao J. et al. Apelin/APJ system: an emerging therapeutic target for respiratory diseases. Cell Mol Life Sci. 2020;77(15):2919-2930
43. Siques P, Pena E, Brito J. et al. Oxidative Stress, Kinase Activation, and Inflammatory Pathways Involved in Effects on Smooth Muscle Cells During Pulmonary Artery Hypertension Under Hypobaric Hypoxia Exposure. Front Physiol. 2021;12:690341
44. Berghausen EM, Janssen W, Vantler M. et al. Disrupted PI3K subunit p110α signaling protects against pulmonary hypertension and reverses established disease in rodents. J Clin Invest. 2021;131(19):e136939
45. Savai R, Al-Tamari HM, Sedding D. et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20(11):1289-1300
46. Song S, Wang S, Ma J. et al. Biliverdin reductase/bilirubin mediates the anti-apoptotic effect of hypoxia in pulmonary arterial smooth muscle cells through ERK1/2 pathway. Exp Cell Res. 2013;319(13):1973-1987
47. Xue Z, Zhou M, Liu Y. et al. A modified Fangji Huangqi decoction ameliorates pulmonary artery hypertension via phosphatidylinositide 3-kinases/protein kinase B-mediated regulation of proliferation and apoptosis of smooth muscle cells in vitro and in vivo. J Ethnopharmacol. 2023;314:116544
48. Zhang Q, Fan K, Wang P. et al. Carvacrol induces the apoptosis of pulmonary artery smooth muscle cells under hypoxia. Eur J Pharmacol. 2016;770:134-146
49. Li W, He P, Huang Y. et al. Selective autophagy of intracellular organelles: recent research advances. Theranostics. 2021;11(1):222-256
50. Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176(1-2):11-42
51. Feng W, Wang J, Yan X. et al. ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. 2021;54(6):e13048
52. Hassoun PM. Pulmonary Arterial Hypertension. N Engl J Med. 2021;385(25):2361-2376
53. Lahm T, Frump AL, Albrecht ME. et al. 17β-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;311(2):L375-388
54. Teng RJ, Du J, Welak S. et al. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302(7):L651-663
55. Dalvi P, Sharma H, Chinnappan M. et al. Enhanced autophagy in pulmonary endothelial cells on exposure to HIV-Tat and morphine: Role in HIV-related pulmonary arterial hypertension. Autophagy. 2016;12(12):2420-2438
56. Ma C, Xu Q, Huang S. et al. The HIF-1α/miR-26a-5p/PFKFB3/ULK1/2 axis regulates vascular remodeling in hypoxia-induced pulmonary hypertension by modulation of autophagy. FASEB J. 2023;37(5):e22906
57. Zhai C, Shi W, Feng W. et al. Activation of AMPK prevents monocrotaline-induced pulmonary arterial hypertension by suppression of NF-κB-mediated autophagy activation. Life Sci. 2018;208:87-95
58. Zhang X, Zheng Y, Chen Z. Autophagy and Mitochondrial Encephalomyopathies. Adv Exp Med Biol. 2020;1207:103-110
59. Chen Xinling, Wang Shenglan. Cell autophagy, pathway, regulation and its multiple correlations with pulmonary hypertension. Chinese Journal of Tissue Engineering Research. 2021;25(02):311-316
60. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42(4):245-254
61. Wu Y, Pan B, Zhang Z. et al. Caspase-4/11-Mediated Pulmonary Artery Endothelial Cell Pyroptosis Contributes to Pulmonary Arterial Hypertension. Hypertension. 2022;79(3):536-548
62. Rong W, Liu C, Li X. et al. Caspase-8 Promotes Pulmonary Hypertension by Activating Macrophage-Associated Inflammation and IL-1β (Interleukin 1β) Production. Arterioscler Thromb Vasc Biol. 2022;42(5):613-631
63. Zhong Z, Liang S, Sanchez-Lopez E. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198-203
64. Sun Y, Lu M, Sun T. et al. Astragaloside IV attenuates inflammatory response mediated by NLRP-3/calpain-1 is involved in the development of pulmonary hypertension. J Cell Mol Med. 2021;25(1):586-590
65. Jiang Y, Liu H, Yu H. et al. Circular RNA Calm4 Regulates Hypoxia-Induced Pulmonary Arterial Smooth Muscle Cells Pyroptosis via the Circ-Calm4/miR-124-3p/PDCD6 Axis. Arterioscler Thromb Vasc Biol. 2021;41(5):1675-1693
66. Feng W, Xu X, Zhao G. et al. EETs and CYP2J2 inhibit TNF-α-induced apoptosis in pulmonary artery endothelial cells and TGF-β1-induced migration in pulmonary artery smooth muscle cells. Int J Mol Med. 2013;32(3):685-693
67. Xie SS, Deng Y, Guo SL. et al. Endothelial cell ferroptosis mediates monocrotaline-induced pulmonary hypertension in rats by modulating NLRP3 inflammasome activation. Sci Rep. 2022;12(1):3056
68. Dixon SJ, Lemberg KM, Lamprecht MR. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death[J]. Cell. 2012;149(5):1060-1072
69. Liao J, Xie SS, Deng Y. et al. PRDX6-mediated pulmonary artery endothelial cell ferroptosis contributes to monocrotaline-induced pulmonary hypertension. Microvasc Res. 2023;146:104471
70. Hu P, Xu Y, Jiang Y. et al. The mechanism of the imbalance between proliferation and ferroptosis in pulmonary artery smooth muscle cells based on the activation of SLC7A11. Eur J Pharmacol. 2022;928:175093
71. Vogel NT, Annis J, Prisco SZ. et al. Ferroptosis Promotes Pulmonary Hypertension. bioRxiv. 2023
72. Wang Y, An R, Umanah GK. et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016;354(6308):aad6872
73. Robinson N, Ganesan R, Hegedűs C. et al. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019;26:101239
74. Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171(8):2000-2016
75. Huang P, Chen G, Jin W. et al. Molecular Mechanisms of Parthanatos and Its Role in Diverse Diseases. Int J Mol Sci. 2022;23(13):7292
76. David KK, Andrabi SA, Dawson TM. et al. Parthanatos, a messenger of death. Front Biosci (Landmark Ed). 2009;14(3):1116-1128
77. Neto-Neves EM, Frump AL, Vayl A. et al. Isolated heart model demonstrates evidence of contractile and diastolic dysfunction in right ventricles from rats with sugen/hypoxia-induced pulmonary hypertension. Physiol Rep. 2017;5(19):e13438
78. Taddei ML, Giannoni E, Fiaschi T. et al. Anoikis: an emerging hallmark in health and diseases. J Pathol. 2012;226(2):380-393
79. Silginer M, Weller M, Ziegler U. et al. Integrin inhibition promotes atypical anoikis in glioma cells. Cell Death Dis. 2014;5:e1012
80. Xiaowei C, Jia M, Xiaowei W. et al. Overexpression of CXCL12 chemokine up-regulates connexin and integrin expression in mesenchymal stem cells through PI3K/Akt pathway. Cell Commun Adhes. 2013;20(3-4):67-72
81. Silva NA, Cooke MJ, Tam RY. et al. The effects of peptide modified gellan gum and olfactory ensheathing glia cells on neural stem/progenitor cell fate. Biomaterials. 2012;33(27):6345-6354
82. Lo CY, Weil BR, Palka BA. et al. Cell surface glycoengineering improves selectin-mediated adhesion of mesenchymal stem cells (MSCs) and cardiosphere-derived cells (CDCs): Pilot validation in porcine ischemia-reperfusion model. Biomaterials. 2016;74:19-30
83. Lee S, Choi E, Cha MJ. et al. Cell adhesion and long-term survival of transplanted mesenchymal stem cells: a prerequisite for cell therapy. Oxid Med Cell Longev. 2015;2015:632902
84. Burst VR, Gillis M, Pütsch F. et al. Poor cell survival limits the beneficial impact of mesenchymal stem cell transplantation on acute kidney injury. Nephron Exp Nephrol. 2010;114(3):e107-116
85. He N, Xu Y, Du W. et al. Extracellular Matrix can Recover the Downregulation of Adhesion Molecules after Cell Detachment and Enhance Endothelial Cell Engraftment. Sci Rep. 2015;5:10902
86. Umesh A, Paudel O, Cao YN. et al. Alteration of pulmonary artery integrin levels in chronic hypoxia and monocrotaline-induced pulmonary hypertension. J Vasc Res. 2011;48(6):525-537
87. Lu X, Kassab GS. Integrins mediate mechanical compression-induced endothelium-dependent vasodilation through endothelial nitric oxide pathway. J Gen Physiol. 2015;146(3):221-232
88. Abe S, Ishida K, Masuda M. et al. A prospective, randomized study of inhaled prostacyclin versus nitric oxide in patients with residual pulmonary hypertension after pulmonary endarterectomy[J]. Gen Thorac Cardiovasc Surg. 2017;65(3):153-159
89. Kozij NK, Granton JT, Silkoff PE. et al. Exhaled Nitric Oxide in Systemic Sclerosis Lung Disease. Can Respir J. 2017;2017:6736239
90. Chanvorachote P, Pongrakhananon V, Chunhacha P. Prolonged nitric oxide exposure enhances anoikis resistance and migration through epithelial-mesenchymal transition and caveolin-1 upregulation. Biomed Res Int. 2014;2014:941359
91. Childs BG, Gluscevic M, Baker DJ. et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16(10):718-735
92. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482-496
93. Hou Y, Dan X, Babbar M. et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565-581
94. Pignatelli P, Menichelli D, Pastori D. et al. Oxidative stress and cardiovascular disease: new insights. Kardiol Pol. 2018;76(4):713-722
95. Ashraf S, Cha BH, Kim JS. et al. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis Cartilage. 2016;24(2):196-205
96. Aoshiba K, Nagai A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6(7):596-601
97. Noureddine H, Gary-Bobo G, Alifano M. et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res. 2011;109(5):543-553
98. Roger I, Milara J, Belhadj N. et al. Senescence Alterations in Pulmonary Hypertension. Cells. 2021;10(12):3456
99. Liu L, Wei Y, Giunta S. et al. Potential role of cellular senescence in pulmonary arterial hypertension. Clin Exp Pharmacol Physiol. 2022;49(10):1042-1049
100. Wang Z, Yang K, Zheng Q. et al. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2019;316(1):L216-L228
101. van der Feen DE, Berger R, Bartelds B. Converging Paths of Pulmonary Arterial Hypertension and Cellular Senescence. Am J Respir Cell Mol Biol. 2019;61(1):11-20
102. Semen KO, Bast A. Senescence in pulmonary arterial hypertension: is there a link? Curr Opin Pulm Med. 2022;28(4):303-306
103. Kyi P, Hendee K, Hunyenyiwa T. et al. Endothelial senescence mediates hypoxia-induced vascular remodeling by modulating PDGFB expression. Front Med (Lausanne). 2022;9:908639
104. Culley MK, Chan SY. Endothelial Senescence: A New Age in Pulmonary Hypertension[J]. Circ Res. 2022;130(6):928-941
105. Amsellem V, Gary-Bobo G, Marcos E. et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(12):1358-1366
106. Culley MK, Zhao J, Tai YY. et al. Frataxin deficiency promotes endothelial senescence in pulmonary hypertension[J]. J Clin Invest. 2021;131(11):e136459
107. Culley MK, Rao RJ, Mehta M. et al. Frataxin deficiency disrupts mitochondrial respiration and pulmonary endothelial cell function. Vascul Pharmacol. 2023;151:107181
Author contact
![]() Corresponding author: Aiguo Dai, Hunan Provincial Key Laboratory of Vascular Biology and Translational Medicine, Changsha, China. Email: Daiaiguo2023com.
Corresponding author: Aiguo Dai, Hunan Provincial Key Laboratory of Vascular Biology and Translational Medicine, Changsha, China. Email: Daiaiguo2023com.