Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(6):1129-1143. doi:10.7150/ijms.94570 This issue Cite
Review
The role of immune cells in different stages of atherosclerosis
Cong He3#, Hyo In Kim4#, Jinbong Park5 ![]() , Junli Guo2
, Junli Guo2 ![]() , Wei Huang1,2
, Wei Huang1,2 ![]()
1. Key Laboratory of Tropical Translational Medicine of Ministry of Education, School of Basic Medicine and Life Sciences, Hainan Medical University, Haikou 571199, PR China.
2. Key Laboratory of Tropical Translational Medicine of Ministry of Education & Key Laboratory of Tropical Cardiovascular Diseases Research of Hainan Province, School of Public Health, Hainan Medical University, Haikou 571199, PR China.
3. Department of Pharmacology, Harbin Medical University-Daqing, Daqing 163319, PR China.
4. Department of Surgery, Beth Israel Deaconess Medical Center, Boston, MA 02215, United States.
5. Department of Pharmacology, College of Korean Medicine, Kyung Hee University, Seoul 02447, Republic of Korea.
#Cong He and Hyo In Kim contributed equally and share first authorship.
Received 2024-1-22; Accepted 2024-4-17; Published 2024-4-22
Abstract

Atherosclerosis is a chronic inflammatory disease characterized by the accumulation of immune cells in the intima of arteries. Experimental and clinical evidence shows that both innate and adaptive immunity orchestrate the progression of atherosclerosis. The heterogeneous nature of immune cells within atherosclerosis lesions is important. Studies utilizing high-dimensional mass spectrometry and single-cell RNA sequencing of leukocytes from atherosclerotic lesions show the diversity and adaptability of these immune cell subtypes. Their migration, compositional changes, phenotypic alterations, and adaptive responses are key features throughout atherosclerosis progression. Understanding how these immune cells and their subtypes affect atherogenesis would help to develop novel therapeutic approaches that control atherosclerosis progression. Precise targeting of specific immune system components involved in atherosclerosis, rather than broad suppression of the immune system with anti-inflammatory agents, can more accurately regulate the progress of atherosclerosis with fewer side effects. In this review, we cover the most recent advances in the field of atherosclerosis to understand the role of various immune cells on its development. We focus on the complex network of immune cells and the interaction between the innate immune system and adaptive immune system.
Keywords: atherosclerosis, inflammation, innate immunity, adaptive immunity
Introduction
Cardiovascular diseases (CVDs) include myocardial infarction, heart failure, ischemic stroke, and sudden cardiac death. CVDs are the main causes of highly morbidity and mortality in both developed and developing countries (1). Atherosclerosis is the main cause of cardiovascular diseases. It is a chronic inflammatory disease which occurs in the intima of middle and large arteries (2). Both innate and adaptive immunity play important roles in the progression of atherosclerosis. Infiltrated low-density lipoprotein (LDL) can activate endothelial cells, and subsequently induce the recruitment of monocytes (3). Macrophages derived from monocytes absorb lipoproteins to form lipid rich foam cells, leading to the formation of necrotic cores and eventually forming atherosclerosis plaques (4). In addition, macrophages in plaques recognize LDL derived epitopes and release various pro-inflammatory mediators to drive the initiation of vascular inflammation (4). Furthermore, macrophages, along with dendritic cells (DCs), mediate antigen presentation in secondary lymphoid organs which accelerates excessive plaque growth through triggering an adaptive immune response (5). Advanced plaques are characterized by large necrotic cores containing activated immune cells, extracellular lipids and lipoproteins derived cholesterol crystals. These plaques are located under the fibrous cap composed of smooth muscle cells (SMCs) and collagen rich matrix, preventing them from interacting with platelets (6). Rupture or erosion of plaques may trigger thrombosis in the coronary arteries which directly cause myocardial infarction (7). In this review, we summarize literature on the role of the immune system in atherosclerosis progression, with particular attention to the interaction between innate and adaptive immunity in atherogenesis.
Innate immunity
In the artery wall, LDL undergoes modification and acquires immunogenic properties triggering the initiation of innate immunity that attracts innate and adaptive immune cells (4). Monocytes and macrophages can polarize to pro-inflammatory phenotypes upon certain stimuli, then possess potentially pro-atherogenic roles (8). Dendritic cells and neutrophils aggregation, and cause arterial inflammation which drives the expansion of atherosclerotic lesions (9, 10). The role of other innate immune cells including mast cells and natural killer T (NKT) cells in atherosclerosis have been previously reviewed (11).
Monocytes
Monocytes are one of the key cells of the innate immune system. Monocytes are rarely seen in healthy coronary arteries. Atherosclerosis-related inflammation and hypercholesterolemia destroy the homeostasis of monocytes in blood, bone marrow and spleen (12). This promotes the maturation of hematopoietic stem cells and progenitor cells into monocytes (13). Circulating monocytes are positively correlated with the severity and plaque size of atherosclerosis (14), thus is a promising target for treatment. Inhibiting monocyte accumulation in the arterial intima almost eliminates atherosclerosis in mice (15). Monocytes are usually be divided into two categories: classical monocytes (Ly6Chigh in mice and CD14+ CD16- in humans) and non-classical monocytes (Ly6Clow in mice and CD14low CD16+ in humans) (14, 16). Classical monocytes accumulate in the intima of artery wall and are necessary for the initiation and progression of innate immunity (4). In particular, the C-C chemokine receptor type 2 (CCR2, CCR5) and C-X-(3)-C motif chemokine receptor 1(CX3CR1) of classical monocytes play an important role in development of atherosclerotic plaques (15). Plaque monocytes not only transform into macrophages but also produce DCs (Fig. 1), which together trigger local inflammation and thus accelerate the progression of atherosclerosis (17). Non-classical monocytes, which accounts for ~10% of the total monocyte population (18), function to maintain the vascular homeostasis without transmigration through the endothelium. Unlike classical monocytes, most non-classical monocytes have been widely viewed as anti-inflammatory, as maintain vascular homeostasis that patrol and crawl along arterial endothelium to resolve arterial inflammation (16, 19).
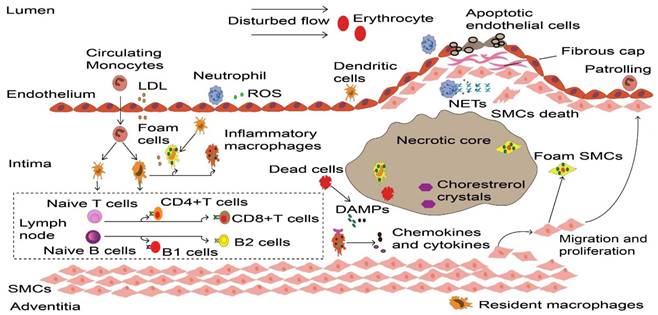
Dynamic process of inflammatory cells in atherosclerosis. Upon the vascular endothelial cell dysfunction induced by disturbed flow, erythrocytes and LDL enter the intima that drives the initiation of atherosclerosis. These atherogenic particles trigger vascular inflammation, thereby accelerate monocyte and neutrophil recruitment. Neutrophils release ROS and various granule proteins increase chemotactic molecules expression in activated endothelial cells, in turn attract more immune cells into intima. Monocyte-derived macrophages, partly intimal DCs and SMCs that acquire macrophage properties together take up LDL and become foam cells in the onset of atherosclerosis. Persistence of accumulated LDL leads to foam cells stress and death, these dead cells form necrotic cores and eventually develop atherosclerotic plaques. Inflammatory macrophages recognize ROS-induced oxLDL and DAMPs provided by dead cells, releasing various cytokines and chemokines to reconstruct the microenvironment in atherosclerosis. More importantly, DCs along with macrophages present LDL-derived antigen to naive B and T cells in artery tertiary lymphoid organs that located in the surrounding of diseased arteries such as the adventitia, lead to autoreactive B cells and T cells. These adaptive immune cells start proliferation and differentiation, producing cytokines and antibodies to accelerate or dampen atherosclerosis progression. To prevent plaque components interacting with platelets, SMCs transmigrates and their derived collagen matrix together constitute a fibrous cap that overlaps advanced plaque in sub-endothelial space. Meanwhile, neutrophils and their secondary products such as NETs promote the death of endothelial cells and SMCs, which elicit plaque rupture or erosion, respectively. LDL, low-density lipoprotein. ROS, Reactive oxygen species. DCs, dendritic cells. SMCs, smooth muscle cells. oxLDL, Oxidized low-density lipoprotein. DAMPs, damage-associated molecular patterns. NETs, neutrophil extracellular traps.
Macrophages
Macrophages are the most abundant innate immune cells found in atherosclerosis (20). Macrophages predominate in all stages of atherosclerosis, including lesion initiation, foam cell formation, necrotic core expansion, plaque rupture or erosion (21), lesion regression and inflammation (22). Macrophages exist in adventitia and intima of healthy arteries (23). These resident macrophages originate from embryonic hematopoiesis, infiltrate before or shortly after birth, and are maintained through local proliferation (24). In general, resident macrophages can prevent arteriosclerosis by maintaining vascular homeostasis through interaction with vascular smooth muscle cells (25) and by eliminating apoptotic cells and damaged mitochondria (26). The outer membrane macrophages with high expression of lymphatic vessel endothelial hyaluronic acid receptor 1 (LYVE1) limit inadaptable cardiac remodeling after myocardial infarction (27). On the other hand, resident macrophages in the aortic intima can promote atherosclerosis during the early stage by acting as the earliest foam cell in the lesions (23).
Resident macrophages and monocyte-derived macrophages are bound by CCR2 on cell surface (26), which reconstructs macrophages in lesions and form the majority of macrophages through continuous infiltration of classical monocytes (15). Macrophages are the pivotal factor in the conversion of pathological intimal thickening into early atherosclerotic plaques by promoting necrotic core formation (28). Macrophages also capture and phagocytose modified LDL through scavenger receptors such as scavenger receptor type A (SR-A) and cluster of differentiation 36 (CD36) (29). Continuous engulfment of lipoprotein particles matures macrophage into foam cells (21). Even more, SMCs accelerate plaque formation after turning into macrophage-like cells (Fig. 1) (30).
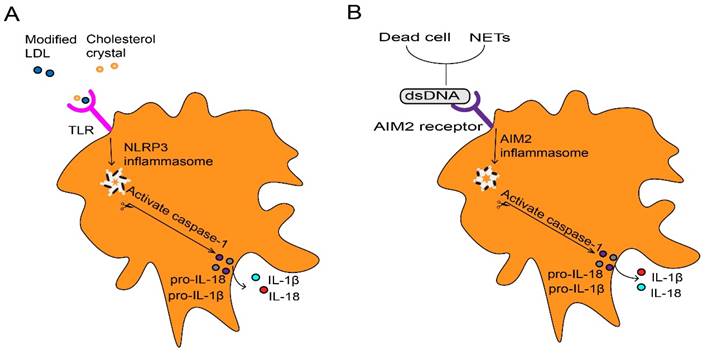
These contribute as the majority of foam cells in atherosclerosis (31). The formation of foam cells is a key event, and they are trapped within the injured endothelium to form the first sign structure of atherosclerosis called fatty streak (21). At first, foam macrophages rather inhibit inflammatory responses (32). However, accumulation of modified lipoproteins result in oxidative stress then disrupts cellular metabolism and eventually leads to the death of foam cells (33). Dead foam cells are initially engulfed and cleared by macrophages through effervescence (34), but insufficient clearance results in the formation of a necrotic core of dead cells (21). Cell debris and modified LDL serve as danger signals that further promote the recruitment of atherogenic immune cells in plaques (21). Furthermore, inflammatory macrophages abundantly promote the NOD-like receptor protein 3 (NLRP3) inflammasome formation in response to modified LDL and cholesterol crystals in atherosclerotic lesions (35). This can further stimulate caspase-1 expression and drive immature interleukin 1β (IL-1β) and IL-18 into pro-inflammatory isoforms with strong pro-atherosclerotic effects (Fig. 2A) (33). IL-1 family cytokines not only increase oxidative stress in macrophages, but also promotes proliferation of them during atherosclerosis (36). The dead cells that release nuclear double-stranded (ds) DNA, partly derived from neutrophil extracellular traps (NETs) (37), act as cytosolic DNA that recognized by the absent in melanoma 2 (AIM2) receptor (38), which is restricted in macrophages (37). The recognition stimulates the activation of AIM2 inflammasome in macrophages, leading to the production of IL-1β and IL-18 in atherosclerosis (Fig. 2B). In the later stage of atherosclerosis, AIM2 macrophages are found surrounding the necrotic core (39). Targeting depletion of AIM2 significantly inhibited inflammatory macrophage proliferation and necrotic core formation (36) while promoting plaque stability by attenuating the death of SMCs (37). Additionally, macrophages within the plaque contribute to the expansion of the necrotic core, thereby increasing plaque instability. They achieve this by degrading extracellular matrix components through the release of Matrix metalloproteinases (MMPs), consequently thinning the fibrous cap (40). Macrophages contribute to plaque instability by providing tissue factors that induce bleeding within the plaque (41). Furthermore, the IL-17A/IL-17RA axis also increases aortic arch inflammation during atherogenesis through the induction of aortic chemokines, and the acceleration of neutrophil and monocyte recruitment to this site (42).
Inflammasome activation in macrophage. (A) Extracellular inflammatory stimuli such as modified LDL and cholesterol crystals, which are recognized by macrophages through TLR, thus promote transcription of pro-IL-1β and pro-IL-18 as well as NLRP3. Assembly of the NLRP3 inflammasome stimulates caspase-1 activation, which processes pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. (B) Dead cells and NETs release dsDNA as danger signals which lead to the AIM2 receptor activation. Activated AIM2 inflammasome also contributes to the maturation and production of IL-1β and Il-18 through activating protease caspase-1. LDL, low-density lipoproteins. IL, interleukin. TLR, toll-like receptor. NLRP3, NOD-like receptor protein 3. NETs, neutrophils extracellular traps. dsDNA, double-stranded DNA. AIM2, absent in melanoma.
Different macrophage subsets exhibit distinct phenotypes and effects within atherosclerotic plaques Their plasticity is contingent upon the local microenvironment of the lesion, the macrophage origin, and the activation of intracellular signaling pathways (43). Single-cell sequencing of atherosclerotic aortas reveals that pathological macrophages cannot be simply classified into M1 and M2 (44). Recent characterization of human coronary atherosclerotic lesions using single-cell RNA sequencing unveiled three distinct subpopulations of myeloid macrophages (My.0, My.1, and My.2) were present within plaques associated with major adverse cardiovascular diseases (45). Consistent with a previous study on macrophages in carotid plaques (46), My.0 and My.2 macrophages release various pro-inflammatory cytokines, detecting danger-associated molecular patterns (DAMPs) through toll-like receptors (TLR) (45). These inflammatory macrophages are major drivers of atherosclerosis and do not exist in healthy arteries (43). My.0 selectively expresses TNF, whereas My.2 macrophages are characterized by a selectively high level of CXCL3 expression and a tendency to encode IL-1β (45). My.1 macrophages mainly consist of foamy macrophages without inflammatory cytokines. They significantly express triggering receptors encoded on myeloid cells 2 (TREM2), which regulate lipid metabolism and cholesterol efflux (20). Furthermore, My.1 macrophages promote plaque stability by inducing fibrosis (20). The number of these macrophages decreases in symptomatic patients compared to asymptomatic patients (46).
Neutrophils
Neutrophils are the first white blood cells recruited to the site of arterial damage (47). They induce strong inflammatory responses through various killing mechanisms (9). Neutrophils play a crucial role in the onset and progression of atherosclerosis (9), and their recruitment is necessary for the atherogenic effects caused by endothelium breaches (48). Significant accumulation of neutrophils is found in the intima of human coronary arteries (9), and neutralizing neutrophils can reduce arterial intimal thickening in mice under flow disturbance (48). Infiltrated neutrophils aggravate endothelium damage by secreting reactive oxygen species (ROS) and proteases (Fig. 3A), which contribute to the foundation of necrotic core formation by increasing the accumulation of leukocytes and LDL in the sub-endothelial region (9). A recent study showed that the association between neutrophil count and microvascular obstruction has been eliminated in AMI (acute myocardial infarction) patients treated with metoprolol and metoprolol inhibits neutrophil migration as a potential target for the therapeutic reduction of infarct size (49). In addition, neutrophils release various chemotactic proteins such as CCL2, Cathepsin G and α-Defensin. These chemokines accelerate the infiltration of monocytes (12), thus paving the way for macrophage expansion (15). Furthermore, the alarmins produced by neutrophils increase circulating monocytes by inducing the activation of bone marrow monocytes (50).
Neutrophil and neutrophil extracellular traps (NETs) in atherosclerosis. (A) During atherosclerosis progression, activated neutrophils release ROS and protease promotes endothelial cell stress and apoptosis, thus paving the way for myeloid cell accumulation and LDL extravasation. Neutrophils release various chemotactic proteins that contribute to monocyte recruitment. Consequently, infiltrated monocytes give rise to macrophage, neutrophil-derived ROS and MPO, which mediate modification of LDL, thereby promoting foam cell formation. Neutrophils and NETs also have an important role in the priming and activation of inflammatory macrophage, and stimulating the initiation of B cells and the differentiation of T cells, which triggers the generation and release of IL-1β, IL-18 and IFN-α. NETs protein-DNA complex serves as autoantigens to promote a strong IFN-α signal in dendritic cells. NETs are also involved in anti-inflammatory by activating macrophages to phagocyte dead cells. (B) Plaque destabilization can be divided into plaque rupture and plaque erosion, and neutrophil is the key element in the process. NETs containing cytotoxic histone H4 lead to death and lysis in SMCs, and neutrophils implicit in collagen degradation, thus provoking plaque rupture. Interestingly, when plaques are not at risk of rupture, neutrophils and NETs cause endothelial cell death and desquamation that contribute to plaque erosion. ROS, reactive oxygen species. LDL, low-density lipoproteins. MPO, myeloperoxidase. IL, interleukin. IFN-α, interferon-alpha. NETs, neutrophils extracellular traps. SMCs, smooth muscle cells.
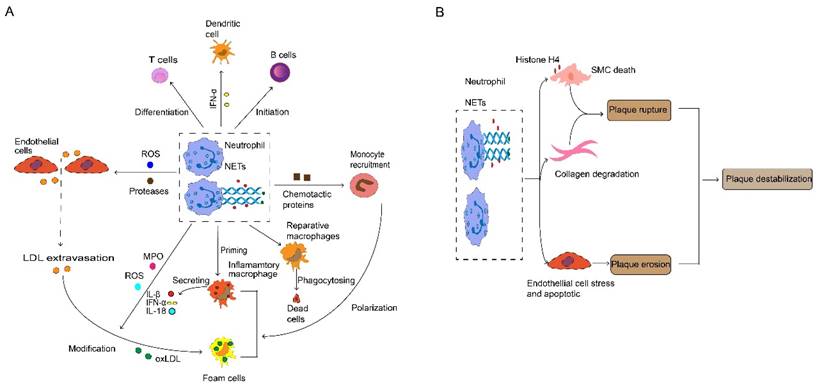
Neutrophils and their secreted products stimulate macrophages and promote the formation of foam cells. Catherine and α-defensin drives macrophages to polarize into an inflammatory state (9). Neutrophils produce reactive oxygen species (ROS) and myeloperoxidase (MPO), contributing to LDL modification, thereby accelerating the transition of macrophages into foam cells (9). Moreover, neutrophils inhibit the increase in macrophage effervescence, thereby exacerbating pathological inflammation (51). NETs released by neutrophils are characterized by reticular structure and contain de-concentrated chromatin and granular protein. These proteins are related to pathogen eradication, autoimmune diseases and vascular disease (52). An imbalanced immune response may lead to dysfunction of NETs and leakage of antimicrobial components, thereby exacerbating inflammation and host tissue damage (53). The contribution of neutrophils was demonstrated by targeted depletion using antibodies. Depletion of neutrophils results in decreased size of atherosclerotic lesions in mice (54). After sensing cholesterol crystals, NETs activate macrophages to activate NLRP3 inflammasome which eventually increases IL-1β generation (55). Conversely, the activation of NLRP3 in macrophages increases the accumulation of neutrophils and the formation of NETs in plaques (56). As mentioned above, NETs stimulate AIM2 inflammasome and increases caspase-1 expression in macrophages, thereby increasing IL-1β secretion (38). On the other hand, NETs can also directly enhance the TLR9/NF-kB (nuclear factor-k-gene binding) pathway in macrophages, promoting the production of IL-8 (57) and Tumor Necrosis Factor-α (TNF-α) (58).
In addition to their effect on innate immune responses, neutrophils also have the ability to mediate adaptive immunity in atherosclerosis (Fig. 3A). Neutrophils can act as APCs that express major histocompatibility complex Ⅱ and costimulatory molecules in the cell surface after being stimulated by interferon-γ (IFN-γ). IFN-γ originate from various immune cells including itself (59), which induces T cell differentiation (47). For example, neutrophils facilitate the differentiation of CD4 T cells into Th1 and Th17 cells (59). Particularly, NETs can directly activate immature T cells, inducing Th17 differentiation (60), and further recruit more immune cells into the plaque via by promoting the activation of Th17 cells (55). Also, neutrophils play an important role in antigen presentation and the development of effector T cells through IFN-γ secretion (47). Neutrophil-derived B cell activating factor and a proliferation-inducing ligand affect the survival and activation of B cells (61). During myocardial infarction, recruited neutrophils release various alarmins which induce granulocyte formation to aggravate damage (62). They also promote mature B cells into plasma cells in the blood, which in turn accelerates atherosclerosis (63). The presence of long-lived antibody secreting cells may explain the risk of recurrence of myocardial infarction and stroke following the initial myocardial infarction. In addition, NETs stimulate the production of IFN-α in plasmacytoid dendritic cells to promote plaque growth (64).
The destruction of atherosclerotic plaque is a major driving factor of cardiovascular disease (65). Such destruction can be categorized into two different ways: plaque rupture, caused by the expansion of necrotic core, and plaque erosion, which means the thinning of fibrous cap (66). Neutrophils play an indispensable role in all forms of plaque instability (Fig. 3B). Compared to plaque erosion, plaque rupture usually accompanies a more severe inflammatory state, characterized by extremely thin fiber caps and higher lipid load (66). Neutrophils not only contribute to necrotic core expansion (52) but also increase lipid infiltration by disrupting the artery wall (9). Moreover, pathological SMCs attract NETs which release histone H4, thus thinning the fibrous cap by driving the death of SMCs (67). Neutrophils can also promote collagen degradation and accelerate plaque rupture (68). Due to the progress in lifestyle modifications and medical interventions, the incidence of plaque rupture and plaque superficial erosion-induced acute coronary artery diseases are rising (69). Plaque erosion is characterized by the preservation of vascular integrity and platelet rich thrombus. This mainly occurs in atherosclerosis lacking a lipid core or a compromised fiber cap, representing lower risk (70). Eroded plaques remain intact and are seen in sites of the vasculature where endothelial cells are deficient and disturbed flow occurs (71), facilitated by neutrophils and NETs that accelerate endothelial cell death and detachment (72). Furthermore, NETs not only promote platelet activation when binding to the injured endothelium (73) but also interact with locally activated platelets, initiating the coagulation cascade and accelerating the formation of wall thrombus, which consequently contributes to cardiovascular diseases (74).
Neutrophils mainly exert a pro-inflammatory response, but can also be anti-inflammatory (75-78). Neutrophils expressed high pro-inflammatory markers at Day 1 and high anti-inflammatory markers at Days 5 and 7 post-MI (myocardial infarction). Pro-inflammatory N1 neutrophils always dominate and can be mediated by DAMPs, and the percentage of anti-inflammatory N2 neutrophils gradually increased post-MI from Day 1 to Day 7 in mice (75). Neutrophils are also involved in the modulation of the healing and remodeling response: the protein S100A8/A9 (S100 calcium-binding protein A8/A9) in NETs activated macrophages to phagocyte dead cells (76). The transformation of neutrophils from pro-inflammatory to anti-inflammatory initiates the reparative process mostly by dedifferentiating cardiomyocytes and promoting the accumulation of reparative macrophages (77, 78).
Dendritic cells
Dendritic cells (DCs) are another important coordination factor of immune-inflammatory response in atherosclerosis (10). DCs originate from hematopoietic progenitor cells, possess various pathogen recognition receptors (PPRs) and establish a network of sentinel cells on both the outer and inner surfaces of most tissues (10). While only a small number of DCs are present in the heart of healthy individuals, arteries susceptible to atherosclerosis harbor myeloid cells displaying characteristics and markers associated with DCs (79). The intimal resident DCs contribute to the retention of lipids in the endothelium during the initiation of atherosclerosis (80). Additionally, DCs and macrophages share similar functional characteristics (8), contributing to foam cell formation and accelerating necrotic core expansion (80, 81). Moreover, as the most potent and versatile antigen-presenting cells (APCs), DCs serve as an important bridge linking innate and adaptive immune responses (10). These cells absorb their self and foreign antigens through specialized surface receptors (10) and subsequently present antigens on major histocompatibility complex (MHC) molecules in the cell surface following danger signals (82). Upon activation, DCs migrate into the lymph node located below advanced plaques. There, the activated DCs physically interact with immature T cells, presenting antigens to initiate adaptive immunity (5). Such process is also observed in plaques (83).
DCs exhibit similar transcriptional characteristics and developmental potential in humans and mice (84). They can be widely divided into two main subtypes: conventional DCs (cDCs) and plasmacytoid DCs (pDCs) (10). pDCs, usually derived from lymphocytes and partly from monocytes, are characterized by large amounts of IFNs (64). These IFNs, for instance IFN-α, mediate the formation of atherogenesis through promoting maturation of cDCs (85), expansion of CD8+ cytotoxic T cells (CTLs) (86) and polarization of CD4+ T cells into T-helper 1 (Th1) cells (87). Moreover, pDCs enhance CD4+ T cells immunity through MHC class Ⅱ-restricted antigen presentation without IFN-α production (88). Typically found in the shoulder regions of human atherosclerotic plaques, increased plaque burden ultimately contributes to plaque rupture (89). Interestingly, pDCs exhibit both pro-atherogenic (90) and anti-atherogenic effects in mice (91). Such contradictory results may be due to the interaction with T cells and variations in animal models of atherosclerosis. DCs consist of type 1 conventional dendritic cells (cDC1s) and type 2 conventional dendritic cells (cDC2s) (10), serving as the main activator of the naive T cell pool (92). cDC1s mainly present exogenous antigens through MHC class Ⅰ, a process called cross-presentation, which is critical to the initiation of CD8+ CTLs (93). On the other hand, cDC2s are involved in the initiation of T cell responses (94). There are significant interactions between cDCs and CD4+ T cells in the aortic wall, promoting T cell activation, proliferation, and the production of IFN-γ and TNF-α cytokines (95). These proinflammatory cytokines increase the uptake of LDL by macrophages and the infiltration of immune cells (96), indicating that cDCs accelerate atherosclerosis by increasing chronic inflammation and formation of foam cells (95). Moreover, these cDCs exhibit interactions with antigen-experienced effector memory T cells within plaques, suggesting a recall reaction in the progression of atherosclerosis (95). The maturation and migration of DCs play a crucial role in facilitating interactions between T cells and DCs, forming the core of adaptive immunity. In the presence of CCR7, the receptor for CCL19 and CCL21, DCs migrate from peripheral tissues to draining lymph nodes (97). Additionally, circulating DCs exhibit a preference for migrating from the bloodstream to developing plaques or lymphoid tissues (10). Thus, decreased number of circulating pDCs can be used as a predictor of coronary artery disease (98). During atherosclerosis regression in mice, DCs migrate from atherosclerotic lesions into lymph nodes and circulation system (99). MyD88 is an adapter for downstream signal transduction through TLRs in mature DCs (100), and its depletion reduces the homing of effector T cells and Treg cells to plaque in atherosclerotic mice (101). Additionally, mediators derived from DCs play a significant role in adaptive immunity. For example, DCs release CCL17, inhibiting the expansion of regulatory T (Treg) cells in atherosclerosis and thereby promoting the recruitment of inflammatory monocytes (102). Also, the expression of CD40 on the cell surface by DCs contributes to Th1 polarization (103). Anti- and pro-atherosclerotic outcomes depend on interactions between T cells and DCs, followed by subsequent adaptive immune responses.
Adaptive immunity
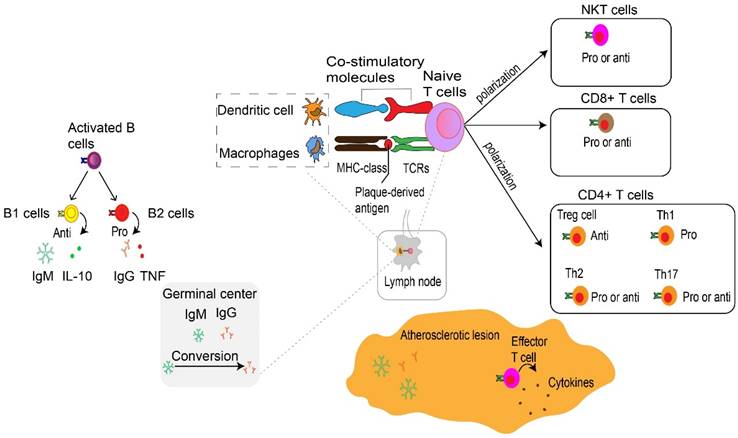
T cells, B cells, and their secondary products are the main components of adaptive immunity. The adaptive immune system coordinates the formation of atherosclerosis and the stability of plaque (Fig. 4 and 5). Adaptive immunity is primarily controlled by antigen presentation. In atherosclerosis, antigen-presenting cells (APCs) such as macrophages within atherosclerotic plaques, adventitial B cells, and DCs present peptide antigens. These antigens are recognized by T cells through specific T cell receptors (TCRs) (96). During this process, APCs present plaque-derived peptides on their MHC (major histocompatibility complex) class Ⅰ or MHC class Ⅱ molecules to immature CD4+ or CD8+ T cells, respectively (97). One finding indicate that the PD-1/PD-L (programmed death-1, Programmed death-ligand 1) pathway plays an important role in down-regulation of pro-atherogenic T cell response and atherosclerosis by limiting APC-dependent T cell activation (104). APCs released co-stimulatory molecules simultaneously bind to TCR, inducing not only clonal proliferation of T cells (105) but also occupying a dominant position in differentiation into effector T cells (96). The cytokines secreted by APCs also affect T cell differentiation (106). For example, IL-12 released by APCs contributes to the maturation of pro-inflammatory T helper type 1 (Th1) cells (107), TGF-β promotes transformation of CD4+ T cells into CD4+ regulatory T cells (108). IL-2 induced immature CD4+ T cells to transform into regulatory T cells (Tregs) (109). MHC class I and II molecules, along with co-stimulatory molecules provided by APCs, establish the basis of adaptive immunity in atherosclerosis (110). Results from TCR sequencing have demonstrated oligoclonal amplification of activated T cells, indicating their status as antigen-experienced effector T cells (111). These effector cells, with specific antigen memory, migrate from draining lymph nodes to the systemic circulation and accumulate in atherosclerotic plaques (5). Notably, T cells experiencing antigen show memory response upon re-stimulation (43). In contrast to innate immunity, adaptive immunity is a rather slower process, but is highly specific and persistent. Such characteristic is largely due to the development of memory effector cells. Similar to macrophages, T cells are found in all stages of atherosclerosis (112), with activated T cells as a sign of atherogenesis (5). Atherosclerotic lesions containing T cells and are often enriched in LDL-specific antibodies (92). Blocking the co-stimulatory pathway of T cells may be a therapeutic target for CVDs. On the contrary, inhibiting the T cell co-inhibitory pathway and stimulating the co-stimulatory pathway may pose a serious risk of cardiovascular events (113).
Adaptive immune cells in atherosclerosis. At the immune synapse, macrophages, dendritic cells and B cells, present lipid antigens to natural killer T (NKT) cells and peptide antigens to T cells, the latter engaging adaptive T cell and B cell responses. These antigens are macrophages and dendritic cells present plaque-derived antigens through MHC class molecules recognized by T cells through TCRs to naive CD4+ and CD8+ T cells in lymph nodes and atherosclerotic plaques, co-stimulatory molecules are also involved in the process which prime naive T cells and promote their proliferation and polarization. These matured effector T cells including CD8+ cells, CD4+ T cells such as Th1 cells, Th2 cells and Th17 cells as well as Treg cells together into the systemic circulation migrate to atherosclerotic lesions where they release various cytokines to promote atherogenesis or depress atherogenesis. CD8+ T cells in atherosclerotic lesions have also been found to have dual functions, with pro-atherogenic effects mediated by IFNγ production and macrophage activation, and atheroprotective effects via limiting the accumulation of macrophages and Th1 cells and B cell modulation. Th1 cells produce TNF-α and IFN-γ, indicating a pro-inflammatory and pro-atherogenic role of Th1 cells. Th2 and Th17 cells play a controversial role in atherosclerosis. Treg cells promote inflammatory resolution and dampen atherosclerosis progression via the production of IL-10 and TGFβ. The effects of NKT cells on the development of atherosclerotic plaques seem to be conflicting and need to be further investigated. Activated B cells mature into antibody-secreting plasma cells, and produce LDL-specific IgM and IgG antibodies that are enriched in atherosclerotic lesions. Conversion of IgM to IgG occurs in the germinal center located in the lymph node and form by B cells and Tfh cells. In addition, they secret IL-10 and TNF to regulate atherosclerosis progression. NKT, natural killer T cells. MHC, major histocompatibility complex. TCRs, specific T cell receptors. Th, T helper cells. Treg, regulatory T cells. IFNγ, interferon-gamma. TNF-α, Tumor Necrosis Factor-α. IL, interleukin. TGFβ, transforming growth factor β. LDL, low-density lipoproteins. Ig, immunoglobulin. Tfh, T follicular cells. Pro, Pro-inflammatory. Anti, Anti-inflammatory.
The main cytokines released by each effector T cell in atherosclerosis. The main cytokines released by each effector T cell are described, together with their roles in atherosclerosis. Especially, the Treg cells play an immunosuppressive effect by depressing the antigen-presentation process. The Th1 cells secrete TNF and IFN-γ, which increase inflammation by contributing to the transformation of naive CD4+ T cells to Th1 cells. NKT cells have a pro-atherogenic effect due to producing a high amount of pro-inflammatory cytokines such as IFNγ. Treg, regulatory T cells. Th, T helper cells. IFNγ, interferon-gamma. TNF, Tumor Necrosis Factor. NKT, natural killer T cells.
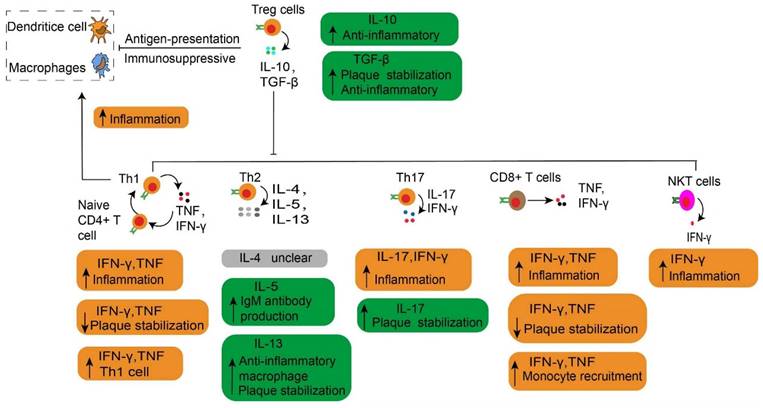
Both CD4+ and CD8+ T cells are found in human aortas (46), and particularly CD8+ T cells are prevalent in humans (114) and murine plaques (115). On the other hand, CD4+ T cells are the key regulator of adaptive immunity in atherosclerosis. They facilitate B cells to produce antibodies (97) and support CD8+ T cells to exert the full potential of their cytotoxic effects (43). After activation, CD4+ T cells mainly differentiate into distinct T-helper (Th) subtypes of Th-1, -2, -17, follicular helper T cells or Treg cells (97). These Th cells or Treg cells, along with their products, respectively contribute to immune activation or immunosuppression in atherosclerosis. Upon activation, these effectors use CCR5 (116) and CXCR6 (117) to home into atherosclerotic lesions. Then they target the primary ligand of CCR5 provided by activated platelets to deplete CCL5 (118) or block the interaction of CCL5 and CCR5, thereby reducing the infiltration of CD4+ T cells into advanced plaques (116). However, CCR5 deficiency does not affect the early stages of atherosclerosis (119). This observation suggests that Treg cells might neutralize the pro-atherosclerotic effects of Th cells. It is generally accepted that Treg cells play a dominant role in the initial stage of atherosclerosis by limiting the formation of inflammation and the development of atherosclerosis. Here, we discuss the most prevalent subtypes of CD4+ T cells and their pivotal role during the progression of atherosclerosis. The role of CD8+ T cells and B cells related to atherosclerosis is also discussed.
Th1 cells
Atherosclerosis is known to be a Th1-related disease. Th1 cells are the most abundant and predominant CD4+ effector T cell subtypes in atherosclerotic plaques (97, 120). Th1 cells are characterized by expression of the defining T box transcription factor (T-bet) (121). They also release pro-inflammatory cytokines IFN-γ and TNF within lesions (122), which can induce the activation of macrophages and T cells, amplifying atherogenic inflammation (96). Several experimental studies have demonstrated that Th1 inhibition via genetic deficiency of T-bet or IFN-γ, or by deleting its receptor, significantly protects mice from the development of atherosclerosis (96). Th1-derived IFN-γ induces plaque instability by restraining the proliferation of vascular smooth muscle cells (VSMCs) (123) and accelerating the degradation of collagen matrix (124). TNF-α, mainly released by T cells, contributes to increased lesion size by stimulating the infiltration of leukocytes (125) and LDL (126). Surprisingly, the elevated level of IFN-γ produced by Th1 cells, which synergize with IL-12 to promote CD4+ T cells to skew towards Th1 (127). TNF-α is not just an effector but also an initiator of inflammatory Th differentiation (128).
Th2 cells
Th2 cells play a controversial role in atherosclerosis (43). CD4+ T cells express the transcription factor GATA binding protein 3 (GATA3) within the Th2 subtype, which is important for humoral immunity (129). Th2 cells predominantly produce IL-4, IL-5, and IL-13 in plaques (130). IL-4 promotes atherogenesis (131), therefore targeted depletion of IL-4 inhibits atherogenesis in mice (132). However, IL-4 levels are also negatively correlated with clinical atherosclerosis in humans (133). Even more, exogenous IL-4 does not drive the expansion of lesions in mice (134). In contrast to IL-4, Th2 -derived IL-5 and IL-13 antagonize the Th1 response and show a protective effect in atherosclerosis (135). IL-5 promotes the production of oxLDL-targeted antibodies by inducing B1 cell maturation, thereby suppressing inflammation and the formation of necrotic cores through the elimination of dead cells (136). Administration of IL-13 in atherosclerosis models has been shown to stabilize atherosclerotic plaques via inducing collagen expression (97) and inhibiting monocyte/macrophage infiltration (137).
Th17 cells
Th17 cells are the third largest subpopulation of CD4+ T cells in atherosclerosis (97). Th17 cells are characterized by the expression of transcription factor RAR-related orphan receptor gamma (RORγ) and are activated by IL-23 (138), acting as the main producer of IL-17 (139). The plasticity of Th17 after recognizing its own antigen is similar to Tregs (97). It depends on local inflammatory environment (140) and the synergistic regulation of Th17 differentiation by IL-6 and transforming growth factor-β (TGF-β) (138). In the presence of IL-6 and IL-β, Th17 cells release IL-17 and IFN-γ to accelerate atherosclerosis progression (141). In addition, pathogenic Th17 cells secrete IL-6 and granulocyte-macrophage colony-stimulating factors (GM-CSFs) (139), stimulating leukocyte accumulation in plaques (142). Pro-inflammatory Th17 cells aggravate the development of atherosclerotic plaque in mice (143). Inhibition of IL-17 is shown to alleviate the development of atherosclerosis (142). On the other hand, Th17 enhances collagen production released by SMCs which increases plaque stability (144), so blockade of IL-17 leads to plaque fragility (145). This potentially reduces the TGF-β generation, which is dependent on the IL-17 pathway (146).
Treg cells
The Treg cell subset is CD25 positive and expresses the defined forkhead box protein P3 (Foxp3) transcription factor as a specific marker for human Treg cells, used to distinguish Treg from non-regulatory T cells (97, 147). Foxp3 controls Treg differentiation and plays a vital role in both the function and activation of Foxp3-expressing T cells (147). Treg cells exhibit inhibitory activity against various immune cells, including the proliferation and functional activity of CD4+ and CD8+ T cells, B cells, APCs, NKT, and cytokine production (147). Treg cells release anti-inflammatory cytokines such as TGF-β and IL-10, and thus protects from inflammation and subsequent atherosclerosis in humans and mice (43). Treg cells are important immunosuppressive modulators which collaborate with TGF-β and IL-10 to inhibit the proliferation and activation of pro-inflammatory effector T cells (148). They also suppress antigen presentation and reduce plaque inflammation (43). Notably, Treg depletion leads to a significant increase in lesions which is independent of vascular inflammation in atherosclerosis models (149). In addition, TGF-β derived from CD25+ Foxp3+ Treg cells contributes to plaque stability by increasing collagen contents (5). The progression of atherosclerosis is mainly determined by the balance between Treg cells and pro-inflammatory T cells, especially Th1 cells (Fig. 6). In the early stages of atherosclerosis, Th1 cells and monocyte-derived macrophages form atherosclerotic lesions (5). Treg cells counteract against these inflammatory cells and suppress the growth of lesions (150). However, inflammatory CD4+ effector T cells increase and Treg cells decrease in lesions, and thus disrupt the balance of these two, accelerating the progression of atherosclerosis (150).
The imbalance between pro-inflammatory and anti-inflammatory signals drives atherosclerosis. The prevailing notion is that Treg predominates before disease onset. However, the imbalance of the pro-atherogenic effect (yellow) and anti-atherogenic effect (green) promotes atherogenesis. It should be noted Treg cell plasticity and instability mainly converts to Th1 cells in plaque formation. The pro-resolving macrophage also can polarize inflammatory macrophage to accelerate atherosclerosis progression. An increased anti-atherogenic effect may contribute to atherosclerosis regression. Treg, regulatory T cells. Th1, T helper 1 cells.
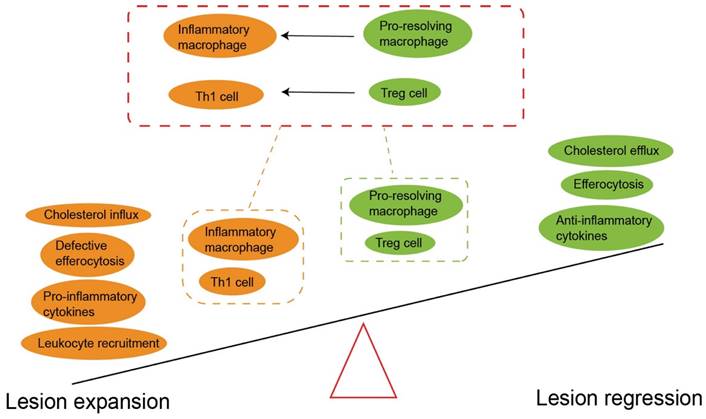
Treg cells are important linkers between immunity and lipoprotein metabolism (43). Reduced Treg cells lead to an impaired ability to regulate very low-density lipoprotein (VLDL) and LDL in the blood, and partly aggravates hypercholesterolemia (149). Conversely, long-term hypercholesterolemia inhibits the tolerance and accumulation of Treg cells, and also enhances the potential of inflammatory T cells to counteract Treg cells (151). Additionally, accumulation of cholesterol crystals amplifies the activation of immune cells and therefore induces complex inflammatory responses (55). Surprisingly, under atherosclerosis-related inflammation, Treg cells lose the expression of CD25 and Foxp3. Instead, they show high plasticity (Fig. 6), expressing other effector T-specific transcription factors, chemokine receptors and cytokines (97). Single cell RNA sequencing studies have demonstrated that Foxp3+ Treg cells transform into Th1 cells and begin expressing IFN-γ in advanced plaques (152). In addition, a subset of unstable Treg cells acquire the characteristics of T-follicle helper cells (Tfh) or convert into ex-Treg cells, both phenotypes losing the immunosuppressive ability during the progression of atherosclerosis (153).
In addition to suppressing the progression of atherosclerosis, Treg cells promote plaque regression by resolving inflammation and repairing damaged tissues (22). In the presence of an LDL reduction strategy, the amount of Treg cells in plaques should not decrease (150). Expansion of Treg cells is considered as a sign of regression of atherosclerosis (22), and these Treg cells originate from peripheral immature T cells rather than thymocytes (154). Treg cells not only inhibit continuously activated macrophages and inflammatory effector T cells, but also promote macrophage polarization into M2 macrophages in degraded plaques (22). This M2 polarization increases the clearance of dead cells, reduces necrotic cores and lead to regression of inflammation (22). Activated M2 macrophages express IL-10 and TGF-β, which in turn promote Treg cell expansion (155).
CD8+ T cells
MHC class I molecules present apolipoprotein B-derived epitopes to CD8+ T cells (97). Following stimulation, CD8+ T cells polarize into CD8+ cytotoxic T lymphocytes (CTLs), possessing the ability to eliminate viral infections and other abnormal cells through various cytotoxic pathways (97). CD8+ T cells are reported to exert dual roles in atherosclerosis: they can both promote atherosclerosis or protect from its development in murine models (115, 156). CD8+ CTLs utilize cytotoxic granules, particularly perforin-1 and granzymes, to promote the formation of necrotic cores by inducing apoptosis of macrophages, VSMCs and endothelial cells. Additionally, CD8+ T cells aggravate inflammation and monocytosis in the bone marrow through TNF-α (115) and IFN-γ production (157). Of note, perforin-1 and granzymes released by CD8+ CTLs exhibit inhibitory actions against APCs and other effector T cells, potentially impeding the onset of atherogenesis. A subset of CD8+ T cells shows regulatory functions that actively inhibit atherosclerosis by limiting the accumulation of macrophages and Th1 cells (158) and B cell-mediated atherogenic antibody production (156). In advanced human plaques, CD8+ T cells account for the majority of effector T cells (159) and are localized in predominantly fibrous cap sites (160). The increased blood CD8+ T cells is a commonly seen in patients who recently suffered from coronary artery diseases (161). Although CD8+ T cells can recognize apolipoprotein B-derived epitopes, their effect on atherosclerosis is not fully understood.
Natural Killer T cells
Natural killer T (NKT) cells are another subset of T cells that express unique invariant T cell receptors and natural killer cell surface molecules, such as CD161 (also known as NK1.1 in mice) and killer cell immunoglobulin like receptors (similar to the Ly49 family in mice) (162). NKT cells have the ability to respond to lipid derived antigens presented by CD1d on antigen-presenting cells. Symptomatic plaques of carotid atherosclerosis are associated with increased NK cell infiltration and elevated serum levels of NK activated receptor ligands (163). In mice, NKT cells are considered atherogenic because they produce a large number of proinflammatory cytokines, such as IFNγ (164). In humans, the number of NKT cells in rupture-prone plaques is higher compared to stable plaques (164), but the exact mechanism of this process is still unclear. However, a recent study showed that the high activation or gene deletion of NKT cells would not affect the progression of atherosclerosis in the mouse model (165). In general, the effect of NKT cells on the development of atherosclerotic plaque seems to need further study. The adaptive immune system coordinates the formation of atherosclerosis and the stability of plaque (Fig. 4 and 5).
B cells
B cells can accelerate or inhibit atherosclerosis, mainly by producing antibodies to induce regulatory T cell responses (92). Like T cells, B cells have specific B cell receptors (BCR) which recognize foreign and autoantigens. BCRs mediate the activation, development and differentiation of B cells in draining lymph nodes (92). Generally, the number of B cells present in atherosclerosis plaque is low, with most of them stay in the outer membrane with lymphatic structure (166). Upon activation, B cells mature into plasma cells and secrete antibodies; in particular LDL-specific antibodies. These antibodies obtain high affinity in the germinal center of the lymphoid nodes, and are maintained by B cells and Tfh cells (167). B cells can be subcategorized into B1 and B2 cells (92). B1 cells can independently produce antibodies without T cells (92), while B2 cells require Tfh cells for activation and transformation into B2 plasma cells that generate soluble antibodies in the blood (96). B1 cells can suppress the development of atherosclerosis by secreting natural immunoglobulin M (IgM) antibodies directly counteracting ox-LDL, thereby limiting the formation and activation of foam cells (92). B1 cells also have a regulatory effect by releasing IL-10 to modulate inflammation responses (168). On the other hand, IgG antibodies produced by B2 cells amplify inflammation reaction in atherosclerosis and contribute to the activation and polarization of innate immune cells (92). In addition, B cells can transform IgM antibodies into other isotypes such as IgG, further promoting the progression of atherosclerosis (167). The developing B cells can also polarize into other phenotypes such as innate response-activated B cells and cytokine-producing B cells, which increase Th1 cell immune response (169) and pro-inflammatory cytokines TNF-α (170), respectively.
Conclusion
Atherosclerosis is a chronic inflammatory disease mainly driven by a maladaptive immune response. The immune system participates in all stages of atherosclerosis, from the initiation to progression, and also in atherosclerosis-associated complications. Emerging evidence suggests that combining LDL reduction strategies with anti-inflammatory drugs can successfully prevent major cardiovascular adverse events. However, systemic immune suppression can result in increased susceptibility of fatal infections. Therefore, greater attention should be directed toward safer and more precise therapeutic interventions targeting inflammation related to atherosclerosis. Especially, the subtypes of immune cells within plaques undergo phenotypic transformations under complex inflammatory conditions. Understanding the mechanisms underlying immune cells and their secondary products that orchestrate inflammation and atherogenesis would facilitate the development of a novel strategy. Such attempts should focus on maintaining the protective effect of the immune system while controlling maladaptive immune responses against the host.
Acknowledgements
This work was supported in part by Natural Science Foundation of China (82360056 to WH), Project supported by the Education Department of Hainan Province (Hnky2023ZD-10 to WH), Hainan Medical University Talent Research Launch Fund (2022026 to WH) and a grant from Kyung Hee University in 2022 (KHU-20220785 to JP).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fox KAA, Metra M, Morais J, Atar D. The myth of 'stable' coronary artery disease. Nat Rev Cardiol. 2020;17:9-21
2. Kobiyama K, Ley K. Atherosclerosis. Circ Res. 2018;123:1118-20
3. Skålén K, Gustafsson M, Rydberg EK. et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750-4
4. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341-55
5. Gisterå A, Hansson GK. The immunology of atherosclerosis. Nature Reviews Nephrology. 2017;13:368-80
6. Virmani R, Kolodgie FD, Burke AP. et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262-75
7. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852-66
8. Cybulsky MI, Cheong C, Robbins CS. Macrophages and Dendritic Cells: Partners in Atherogenesis. Circ Res. 2016;118:637-52
9. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. 2020;17:327-40
10. Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17:30-48
11. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092-104
12. Engelen SE, Robinson AJB, Zurke YX, Monaco C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol. 2022 10.1038/s41569-021-00668-4
13. Robbins CS, Chudnovskiy A, Rauch PJ. et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364-74
14. Hamers AAJ, Dinh HQ, Thomas GD. et al. Human Monocyte Heterogeneity as Revealed by High-Dimensional Mass Cytometry. Arterioscler Thromb Vasc Biol. 2019;39:25-36
15. Combadière C, Potteaux S, Rodero M. et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649-57
16. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical Monocytes in Health and Disease. Annu Rev Immunol. 2019;37:439-56
17. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349-62
18. Berg KE, Ljungcrantz I, Andersson L. et al. Elevated CD14++CD16- monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012;5:122-31
19. Marcovecchio PM, Thomas GD, Mikulski Z. et al. Scavenger Receptor CD36 Directs Nonclassical Monocyte Patrolling Along the Endothelium During Early Atherogenesis. Arterioscler Thromb Vasc Biol. 2017;37:2043-52
20. Cochain C, Vafadarnejad E, Arampatzi P. et al. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res. 2018;122:1661-74
21. Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res. 2016;118:653-67
22. Sharma M, Schlegel MP, Afonso MS. et al. Regulatory T Cells License Macrophage Pro-Resolving Functions During Atherosclerosis Regression. Circ Res. 2020;127:335-53
23. Williams JW, Zaitsev K, Kim KW. et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol. 2020;21:1194-204
24. Ensan S, Li A, Besla R. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol. 2016;17:159-68
25. Lim HY, Lim SY, Tan CK. et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity. 2018;49:326-41.e7
26. Lavine KJ, Liu Y. The dynamic cardiac cellular landscape: visualization by molecular imaging. Nat Rev Cardiol. 2022;19:345-7
27. Dick SA, Macklin JA, Nejat S. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20:29-39
28. Rayner KJ. Cell Death in the Vessel Wall: The Good, the Bad, the Ugly. Arterioscler Thromb Vasc Biol. 2017;37:e75-e81
29. Lusis AJ. Atherosclerosis. Nature. 2000;407:233-41
30. Shankman LS, Gomez D, Cherepanova OA. et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628-37
31. Wang Y, Dubland JA, Allahverdian S. et al. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:876-87
32. Kim K, Shim D, Lee JS. et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ Res. 2018;123:1127-42
33. Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. 2018;19:526-37
34. Kojima Y, Weissman IL, Leeper NJ. The Role of Efferocytosis in Atherosclerosis. Circulation. 2017;135:476-89
35. Duewell P, Kono H, Rayner KJ. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357-61
36. Fidler TP, Xue C, Yalcinkaya M. et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296-301
37. Paulin N, Viola JR, Maas SL. et al. Double-Strand DNA Sensing Aim2 Inflammasome Regulates Atherosclerotic Plaque Vulnerability. Circulation. 2018;138:321-3
38. Hornung V, Ablasser A, Charrel-Dennis M. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514-8
39. Hakimi M, Peters A, Becker A. et al. Inflammation-related induction of absent in melanoma 2 (AIM2) in vascular cells and atherosclerotic lesions suggests a role in vascular pathogenesis. J Vasc Surg. 2014;59:794-803
40. Deguchi JO, Aikawa M, Tung CH. et al. Inflammation in atherosclerosis: visualizing matrix metalloproteinase action in macrophages in vivo. Circulation. 2006;114:55-62
41. Badimon L, Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med. 2014;276:618-32
42. Butcher MJ, Gjurich BN, Phillips T, Galkina EV. The IL-17A/IL-17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ Res. 2012;110:675-87
43. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. 2022;22:251-65
44. Mills CD, Kincaid K, Alt JM. et al. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166-73
45. Emoto T, Yamamoto H, Yamashita T. et al. Single-Cell RNA Sequencing Reveals a Distinct Immune Landscape of Myeloid Cells in Coronary Culprit Plaques Causing Acute Coronary Syndrome. Circulation. 2022;145:1434-6
46. Depuydt MAC, Prange KHM, Slenders L. et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ Res. 2020;127:1437-55
47. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159-75
48. Franck G, Even G, Gautier A. et al. Haemodynamic stress-induced breaches of the arterial intima trigger inflammation and drive atherogenesis. Eur Heart J. 2019;40:928-37
49. García-Prieto J, Villena-Gutiérrez R, Gómez M. et al. Neutrophil stunning by metoprolol reduces infarct size. Nat Commun. 2017;8:14780
50. Nagareddy PR, Murphy AJ, Stirzaker RA. et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695-708
51. Caligiuri G. CD31 as a Therapeutic Target in Atherosclerosis. Circ Res. 2020;126:1178-89
52. Doring Y, Soehnlein O, Weber C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ Res. 2017;120:736-43
53. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. 2019;133:2178-85
54. Zernecke A, Bot I, Djalali-Talab Y. et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209-17
55. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316-20
56. Westerterp M, Fotakis P, Ouimet M. et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation. 2018;138:898-912
57. An Z, Li J, Yu J. et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-κB signaling in macrophages. Cell Cycle. 2019;18:2928-38
58. Hidalgo A, Libby P, Soehnlein O. et al. Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res. 2022;118:2737-53
59. Abi Abdallah DS, Egan CE, Butcher BA, Denkers EY. Mouse neutrophils are professional antigen-presenting cells programmed to instruct Th1 and Th17 T-cell differentiation. Int Immunol. 2011;23:317-26
60. Wilson AS, Randall KL, Pettitt JA. et al. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat Commun. 2022;13:528
61. Scapini P, Bazzoni F, Cassatella MA. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett. 2008;116:1-6
62. Sreejit G, Abdel-Latif A, Athmanathan B. et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation. 2020;141:1080-94
63. Kyaw T, Loveland P, Kanellakis P. et al. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur Heart J. 2021;42:938-47
64. Döring Y, Manthey HD, Drechsler M. et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation. 2012;125:1673-83
65. Anderson JL, Morrow DA. Acute Myocardial Infarction. N Engl J Med. 2017;376:2053-64
66. Nakajima A, Sugiyama T, Araki M. et al. Plaque Rupture, Compared With Plaque Erosion, Is Associated With a Higher Level of Pancoronary Inflammation. JACC Cardiovasc Imaging. 2022;15:828-39
67. Silvestre-Roig C, Braster Q, Wichapong K. et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. 2019;569:236-40
68. Mawhin MA, Tilly P, Zirka G. et al. Neutrophils recruited by leukotriene B4 induce features of plaque destabilization during endotoxaemia. Cardiovasc Res. 2018;114:1656-66
69. Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. The Lancet. 2017;389:197-210
70. Fahed AC, Jang IK. Plaque erosion and acute coronary syndromes: phenotype, molecular characteristics and future directions. Nat Rev Cardiol. 2021;18:724-34
71. Boren J, Chapman MJ, Krauss RM. et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41:2313-30
72. Quillard T, Araújo HA, Franck G. et al. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J. 2015;36:1394-404
73. Darbousset R, Thomas GM, Mezouar S. et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120:2133-43
74. Carminita E, Crescence L, Brouilly N. et al. DNAse-dependent, NET-independent pathway of thrombus formation in vivo. Proc Natl Acad Sci U S A. 2021 118
75. Ma Y, Yabluchanskiy A, Iyer RP. et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110:51-61
76. Marinković G, Koenis DS, de Camp L. et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ Res. 2020;127:664-76
77. Laera N, Malerba P, Vacanti G. et al. Impact of Immunity on Coronary Artery Disease: An Updated Pathogenic Interplay and Potential Therapeutic Strategies. Life (Basel). 2023 13
78. Lörchner H, Pöling J, Gajawada P. et al. Myocardial healing requires Reg3β-dependent accumulation of macrophages in the ischemic heart. Nat Med. 2015;21:353-62
79. Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res. 2014;114:1757-71
80. Paulson KE, Zhu SN, Chen M. et al. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383-90
81. Bobryshev YV, Watanabe T. Subset of vascular dendritic cells transforming into foam cells in human atherosclerotic lesions. Cardiovasc Pathol. 1997;6:321-31
82. Ait-Oufella H, Sage AP, Mallat Z, Tedgui A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ Res. 2014;114:1640-60
83. Llodrá J, Angeli V, Liu J. et al. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779-84
84. Anderson DA 3rd, Dutertre CA, Ginhoux F, Murphy KM. Genetic models of human and mouse dendritic cell development and function. Nat Rev Immunol. 2021;21:101-15
85. Ghislat G, Cheema AS, Baudoin E. et al. NF-κB-dependent IRF1 activation programs cDC1 dendritic cells to drive antitumor immunity. Sci Immunol. 2021 6
86. Schlecht G, Garcia S, Escriou N. et al. Murine plasmacytoid dendritic cells induce effector/memory CD8+ T-cell responses in vivo after viral stimulation. Blood. 2004;104:1808-15
87. Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. 2000;1:305-10
88. Sage AP, Murphy D, Maffia P. et al. MHC Class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation. 2014;130:1363-73
89. Yilmaz A, Lochno M, Traeg F. et al. Emergence of dendritic cells in rupture-prone regions of vulnerable carotid plaques. Atherosclerosis. 2004;176:101-10
90. Macritchie N, Grassia G, Sabir SR. et al. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:2569-79
91. Daissormont IT, Christ A, Temmerman L. et al. Plasmacytoid dendritic cells protect against atherosclerosis by tuning T-cell proliferation and activity. Circ Res. 2011;109:1387-95
92. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. 2019;16:180-96
93. Colbert JD, Cruz FM, Rock KL. Cross-presentation of exogenous antigens on MHC I molecules. Curr Opin Immunol. 2020;64:1-8
94. Merad M, Sathe P, Helft J. et al. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563-604
95. Koltsova EK, Garcia Z, Chodaczek G. et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. 2012;122:3114-26
96. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res. 2019;124:315-27
97. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17:387-401
98. Van Vré EA, Van Brussel I, de Beeck KO. et al. Changes in blood dendritic cell counts in relation to type of coronary artery disease and brachial endothelial cell function. Coron Artery Dis. 2010;21:87-96
99. Trogan E, Feig JE, Dogan S. et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781-6
100. Waisman A, Lukas D, Clausen BE, Yogev N. Dendritic cells as gatekeepers of tolerance. Semin Immunopathol. 2017;39:153-63
101. Subramanian M, Thorp E, Hansson GK, Tabas I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J Clin Invest. 2013;123:179-88
102. Weber C, Meiler S, Döring Y. et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest. 2011;121:2898-910
103. Lutgens E, Lievens D, Beckers L. et al. CD40 and its ligand in atherosclerosis. Trends Cardiovasc Med. 2007;17:118-23
104. Gotsman I, Grabie N, Dacosta R. et al. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. 2007;117:2974-82
105. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1-22
106. Zernecke A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arterioscler Thromb Vasc Biol. 2015;35:763-70
107. Hsieh CS, Macatonia SE, Tripp CS. et al. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547-9
108. Chen W, Jin W, Hardegen N. et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875-86
109. Dooms H, Abbas AK. Revisiting the role of IL-2 in autoimmunity. Eur J Immunol. 2010;40:1538-40
110. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204-12
111. Paulsson G, Zhou X, Törnquist E, Hansson GK. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:10-7
112. Zernecke A, Winkels H, Cochain C. et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ Res. 2020;127:402-26
113. Simons KH, de Jong A, Jukema JW. et al. T cell co-stimulation and co-inhibition in cardiovascular disease: a double-edged sword. Nat Rev Cardiol. 2019;16:325-43
114. Gewaltig J, Kummer M, Koella C. et al. Requirements for CD8 T-cell migration into the human arterial wall. Hum Pathol. 2008;39:1756-62
115. Kyaw T, Winship A, Tay C. et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. 2013;127:1028-39
116. Li J, McArdle S, Gholami A. et al. CCR5+T-bet+FoxP3+ Effector CD4 T Cells Drive Atherosclerosis. Circ Res. 2016;118:1540-52
117. Galkina E, Harry BL, Ludwig A. et al. CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation. 2007;116:1801-11
118. Huo Y, Schober A, Forlow SB. et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61-7
119. Kuziel WA, Dawson TC, Quinones M. et al. CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis. 2003;167:25-32
120. Buono C, Binder CJ, Stavrakis G. et al. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci U S A. 2005;102:1596-601
121. Szabo SJ, Kim ST, Costa GL. et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655-69
122. Frostegård J, Ulfgren AK, Nyberg P. et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999;145:33-43
123. Hansson GK, Hellstrand M, Rymo L. et al. Interferon gamma inhibits both proliferation and expression of differentiation-specific alpha-smooth muscle actin in arterial smooth muscle cells. J Exp Med. 1989;170:1595-608
124. Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb. 1991;11:1223-30
125. Edamitsu S, Matsukawa A, Ohkawara S. et al. Role of TNF alpha, IL-1, and IL-1ra in the mediation of leukocyte infiltration and increased vascular permeability in rabbits with LPS-induced pleurisy. Clin Immunol Immunopathol. 1995;75:68-74
126. Zhang Y, Yang X, Bian F. et al. TNF-α promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: crosstalk between NF-κB and PPAR-γ. J Mol Cell Cardiol. 2014;72:85-94
127. Uyemura K, Demer LL, Castle SC. et al. Cross-regulatory roles of interleukin (IL)-12 and IL-10 in atherosclerosis. J Clin Invest. 1996;97:2130-8
128. Alam MS, Otsuka S, Wong N. et al. TNF plays a crucial role in inflammation by signaling via T cell TNFR2. Proc Natl Acad Sci U S A. 2021 118
129. Nakayama T, Hirahara K, Onodera A. et al. Th2 Cells in Health and Disease. Annu Rev Immunol. 2017;35:53-84
130. Winkels H, Ehinger E, Vassallo M. et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ Res. 2018;122:1675-88
131. Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117-25
132. King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor-/- mice. Arterioscler Thromb Vasc Biol. 2002;22:456-61
133. Engelbertsen D, Andersson L, Ljungcrantz I. et al. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arterioscler Thromb Vasc Biol. 2013;33:637-44
134. King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am J Pathol. 2007;171:2040-7
135. Fernández-Gallego N, Castillo-González R, Méndez-Barbero N. et al. The impact of type 2 immunity and allergic diseases in atherosclerosis. Allergy. 2022;77:3249-66
136. Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res. 2014;114:1743-56
137. Cardilo-Reis L, Gruber S, Schreier SM. et al. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med. 2012;4:1072-86
138. Marks BR, Nowyhed HN, Choi JY. et al. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat Immunol. 2009;10:1125-32
139. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 Family of Cytokines in Health and Disease. Immunity. 2019;50:892-906
140. Hirota K, Duarte JH, Veldhoen M. et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255-63
141. Zielinski CE, Mele F, Aschenbrenner D. et al. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature. 2012;484:514-8
142. Smith E, Prasad KM, Butcher M. et al. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2010;121:1746-55
143. Gao Q, Jiang Y, Ma T. et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol. 2010;185:5820-7
144. Brauner S, Jiang X, Thorlacius GE. et al. Augmented Th17 differentiation in Trim21 deficiency promotes a stable phenotype of atherosclerotic plaques with high collagen content. Cardiovasc Res. 2018;114:158-67
145. Danzaki K, Matsui Y, Ikesue M. et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:273-80
146. Gisterå A, Robertson AK, Andersson J. et al. Transforming growth factor-β signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci Transl Med. 2013;5:196ra00
147. Churov AV, Chegodaev YS, Khotina VA. et al. Regulatory T Cells in Atherosclerosis: Is Adoptive Cell Therapy Possible? Life (Basel). 2023 13
148. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. 2015;35:280-7
149. Klingenberg R, Gerdes N, Badeau RM. et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest. 2013;123:1323-34
150. Maganto-García E, Tarrio ML, Grabie N. et al. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation. 2011;124:185-95
151. Mailer RKW, Gisterå A, Polyzos KA. et al. Hypercholesterolemia Enhances T Cell Receptor Signaling and Increases the Regulatory T Cell Population. Sci Rep. 2017;7:15655
152. Butcher MJ, Filipowicz AR, Waseem TC. et al. Atherosclerosis-Driven Treg Plasticity Results in Formation of a Dysfunctional Subset of Plastic IFNγ+ Th1/Tregs. Circ Res. 2016;119:1190-203
153. Gaddis DE, Padgett LE, Wu R. et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun. 2018;9:1095
154. Fernández-Ruiz I. T(reg) cells promote plaque regression. Nat Rev Cardiol. 2020;17:384
155. Schmidt A, Zhang XM, Joshi RN. et al. Human macrophages induce CD4(+)Foxp3(+) regulatory T cells via binding and re-release of TGF-β. Immunol Cell Biol. 2016;94:747-62
156. Clement M, Guedj K, Andreata F. et al. Control of the T follicular helper-germinal center B-cell axis by CD8⁺ regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation. 2015;131:560-70
157. Cochain C, Koch M, Chaudhari SM. et al. CD8+ T Cells Regulate Monopoiesis and Circulating Ly6C-high Monocyte Levels in Atherosclerosis in Mice. Circ Res. 2015;117:244-53
158. van Duijn J, Kritikou E, Benne N. et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc Res. 2019;115:729-38
159. Fernandez DM, Rahman AH, Fernandez NF. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576-88
160. Paul VS, Paul CM, Kuruvilla S. Quantification of Various Inflammatory Cells in Advanced Atherosclerotic Plaques. J Clin Diagn Res. 2016;10:Ec35-8
161. Hwang Y, Yu HT, Kim DH. et al. Expansion of CD8(+) T cells lacking the IL-6 receptor α chain in patients with coronary artery diseases (CAD). Atherosclerosis. 2016;249:44-51
162. Getz GS, Reardon CA. Natural killer T cells in atherosclerosis. Nat Rev Cardiol. 2017;14:304-14
163. Bonaccorsi I, Spinelli D, Cantoni C. et al. Symptomatic Carotid Atherosclerotic Plaques Are Associated With Increased Infiltration of Natural Killer (NK) Cells and Higher Serum Levels of NK Activating Receptor Ligands. Front Immunol. 2019;10:1503
164. Bobryshev YV, Lord RS. Co-accumulation of dendritic cells and natural killer T cells within rupture-prone regions in human atherosclerotic plaques. J Histochem Cytochem. 2005;53:781-5
165. Nour-Eldine W, Joffre J, Zibara K. et al. Genetic Depletion or Hyperresponsiveness of Natural Killer Cells Do Not Affect Atherosclerosis Development. Circ Res. 2018;122:47-57
166. Srikakulapu P, Hu D, Yin C. et al. Artery Tertiary Lymphoid Organs Control Multilayered Territorialized Atherosclerosis B-Cell Responses in Aged ApoE-/- Mice. Arterioscler Thromb Vasc Biol. 2016;36:1174-85
167. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity. 2019;50:1132-48
168. Kyaw T, Tipping P, Bobik A, Toh BH. Protective role of natural IgM-producing B1a cells in atherosclerosis. Trends Cardiovasc Med. 2012;22:48-53
169. Hilgendorf I, Theurl I, Gerhardt LM. et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation. 2014;129:1677-87
170. Lund FE. Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol. 2008;20:332-8
Author contact
![]() Corresponding authors: Wei Huang, Key Laboratory of Tropical Translational Medicine of Ministry of Education, School of Basic Medicine and Life Sciences, Hainan Medical University, Haikou 571199, PR China. E-mail: hy0206210edu.cn. Junli Guo, Key Laboratory of Tropical Translational Medicine of Ministry of Education & Key Laboratory of Tropical Cardiovascular Diseases Research of Hainan Province, School of Public Health, Hainan Medical University, Haikou 571199, PR China. E-mail: guojledu.cn. Jinbong Park, Department of Pharmacology, College of Korean Medicine, Kyung Hee University, Seoul, 02447, Republic of Korea. E-mail: thejinbongac.kr.
Corresponding authors: Wei Huang, Key Laboratory of Tropical Translational Medicine of Ministry of Education, School of Basic Medicine and Life Sciences, Hainan Medical University, Haikou 571199, PR China. E-mail: hy0206210edu.cn. Junli Guo, Key Laboratory of Tropical Translational Medicine of Ministry of Education & Key Laboratory of Tropical Cardiovascular Diseases Research of Hainan Province, School of Public Health, Hainan Medical University, Haikou 571199, PR China. E-mail: guojledu.cn. Jinbong Park, Department of Pharmacology, College of Korean Medicine, Kyung Hee University, Seoul, 02447, Republic of Korea. E-mail: thejinbongac.kr.