Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(4):681-689. doi:10.7150/ijms.90814 This issue Cite
Research Paper
Rosmarinic Acid Protects Skin Keratinocytes from Particulate Matter 2.5-Induced Apoptosis
Herath Mudiyanselage Udari Lakmini Herath, Mei Jing Piao, Kyoung Ah Kang, Pincha Devage Sameera Madushan Fernando, Jin Won Hyun ![]()
Department of Biochemistry, College of Medicine and Jeju Research Center for Natural Medicine, Jeju National University, Jeju 63243, Republic of Korea.
Received 2023-10-6; Accepted 2024-1-27; Published 2024-2-4
Abstract

Background: The exposure of the human skin to particulate matter 2.5 (PM2.5) results in adverse health outcomes, such as skin aging, wrinkle formation, pigment spots, and atopic dermatitis. It has previously been shown that rosmarinic acid (RA) can protect keratinocytes from ultraviolet B radiation by enhancing cellular antioxidant systems and reducing oxidative damage; however, its protective action against the adverse effects of PM2.5 on skin cells remains unclear. Therefore, in this study, we explored the mechanism underlying the protective effects of RA against PM2.5-mediated oxidative stress in HaCaT keratinocytes.
Methods: HaCaT keratinocytes were pretreated with RA and exposed to PM2.5. Thereafter, reactive oxygen species (ROS) production, protein carbonylation, lipid peroxidation, DNA damage, and cellular apoptosis were investigated using various methods, including confocal microscopy, western blot analysis, and flow cytometry.
Results: RA significantly inhibited PM2.5-induced lipid peroxidation, protein carbonylation, DNA damage, increases in intracellular Ca2+ level, and mitochondrial depolarization. It also significantly attenuated PM2.5-induced apoptosis by downregulating Bcl-2-associated X, cleaved caspase-9, and cleaved caspase-3 protein levels, while upregulating B-cell lymphoma 2 protein level. Further, our results indicated that PM2.5-induced apoptosis was associated with the activation of the mitogen-activated protein kinase (MAPK) signaling pathway and that MAPK inhibitors as well as RA exhibited protective effects against PM2.5-induced apoptosis.
Conclusion: RA protected HaCaT cells from PM2.5-induced apoptosis by lowering oxidative stress.
Keywords: Rosmarinic acid, PM2.5, Oxidative stress, Apoptosis
Introduction
Globally, urban air pollution is a serious threat to public health. Fine particulate matter with aerodynamic diameter < 2.5 μm (PM2.5) is a constituent of airborne particulate matter that is primarily derived from industrial soot [1]. The components of PM2.5 differ depending on its source, and PM2.5 emitted by diesel exhaust predominantly contains polycyclic aromatic hydrocarbons (PAHs), black carbon, and hydrocarbons (C14-C35) and their derivatives. PAHs in PM2.5 have a high mutagenic potential and can easily penetrate the skin via the appendageal route and the stratum corneum, thereby causing PM2.5-induced skin injury [2,3]. Numerous studies have demonstrated that PM2.5 exposure increases the risk of cardiovascular and respiratory damage as well as neurotoxicity [4,5]. Further, PM2.5 has been linked to various skin disorders, including acne, atopic dermatitis, and skin aging [6,7]. It has also been shown that exposure to PM2.5 induces reactive oxygen species (ROS) generation in keratinocytes, and this excessive intracellular ROS generation induces cell damage owing to oxidative stress [3,8], which can considerably harm nucleic acids, proteins, lipids, cell membranes, and organelles, such as the mitochondria, and even induce apoptosis [9].
Most phenolic compounds can function as antioxidants or free-radical scavengers. Specifically, rosmarinic acid (RA), a naturally occurring hydroxylated polyphenolic compound that is commonly found in Rosmarinus officinalis, exhibits antimicrobial, anti-inflammatory, antioxidative, antiapoptotic, and antitumor activities [10-12]. Our previous study demonstrated that it exerts cytoprotective effects against ultraviolet B radiation by modulating cellular antioxidant systems in keratinocytes and ameliorating oxidative damage [11,12]. However, the cytoprotective action of RA against PM2.5 remains unclear. Therefore, in this study, we aimed to explore the cytoprotective effect of RA on PM2.5-induced skin damage.
Materials and Methods
Reagents
RA (100% purity), primary antibodies for Bax, Bcl-2, PARP, ERK, phospho-ERK, and p38 were procured from Santa Cruz Biotechnology (Dallas, TX, USA); diesel particulate matter NIST 1650b (PM2.5), NAC (antioxidant), Hoechst 33342, Z-VAD-FMK (caspase inhibitor), SB203580 (p38 inhibitor), trypan blue solution and avidin-TRITC were procured from Sigma-Aldrich Inc. (St. Louis, MO, USA). MTT and DMSO were purchased from Amresco LLC (Solon, OH, USA). H2DCFDA, Fluo-4 AM and DPPP were purchased from Molecular Probes (Eugene, OR, USA). JC-1 was purchased from Invitrogen (Carlsbad, CA, USA). SP600125 (JNK inhibitor) and U0126 (MEK inhibitor) were obtained from Tocris (Bristol, UK) and Calbiochem (La Jolla, CA, USA), respectively. Actin, caspase-3, caspase-9, phospho-p38, JNK and phospho-JNK primary antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA).
Preparation of RA and PM2.5
Stock solutions of RA and PM2.5 were prepared using DMSO. Specifically, the prepared PM2.5 stock solution (25 mg/mL) was subjected to sonication for 30 min to prevent the particles from clustering [13].
Cell culture
The human skin HaCaT cell line was procured from Cell Lines Service (Eppelheim, Germany). The cells were cultured according to standard procedures at 37 °C in an incubator with 5% CO2 and full humidity. Further, the culture medium, DMEM, was supplemented with 10% heat-inactivated FBS and an antibiotic-antimycotic mix [14].
MTT assay
The cells were incubated with MAPK pathway-targeted chemical inhibitors (50 nM U0126; 10 µM SB203580; and 5 µM SP600125) with or without 2.5 μM RA for 30 min and then exposed to 50 μg/mL PM2.5 for 24 h at 37 °C. Thereafter, MTT solution was added to each well after which incubation was further performed at 37 °C for 4 h. The purple precipitate thus obtained was dissolved by adding DMSO and cellular metabolic activity measurements were performed at 540 nm using a VersaMax ELISA microplate reader (Molecular Devices, Sunnyvale, CA, USA) [14].
Trypan blue assay
Cells were exposed to 30 μM Z-VAD-FMK with or without 2.5 μM RA for 30 min and then exposed to PM2.5 at 50 μg/mL for 24 h. After this treatment, the cells were stained using 0.1% trypan blue solution and observed using a fluorescence microscope [15].
ROS measurement
Cells were treated with 2.5 μM RA or 1 mM NAC for 30 min and then exposed to 50 μg/mL PM2.5 for 24 h. Then, for total ROS and superoxide anion detection, 25 μM H2DCFDA and 10 μM DHE, respectively, were added to the culture medium and fluorescent cells were analyzed using a flow cytometer (Becton Dickinson, Mountain View, CA, USA) [3,14]. Further, cellular H2O2 level was measured via the ROS-Glo™ H2O2 assay (Promega, Madison, WI, USA) according to the manufacturer's protocols [16].
Lipid peroxidation
Cells were stained with the fluorescent probe DPPP (5 μM), and stained cells were observed under a confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany) [8].
Protein carbonylation
Protein carbonylation level was assessed using the OxiSelect™ protein carbonyl ELISA kit (Cell Biolabs, San Diego, CA, USA) following the guidelines provided by the manufacturer [8].
Comet assay
Cells were collected on a slide, electrophoresed (25 V, 300 mA), stained with ethidium bromide, and observed under a fluorescence microscope equipped with image analysis software (Kinetic Imaging, Komet 5.5, UK). The percentage of total fluorescence in the comet tail and the length of the tail were measured in 50 cells/slide [17].
8-Oxoguanine (8-oxoG)
The avidin-TRITC conjugate was used to detect 8-oxoG, a biomarker of oxidative stress-induced DNA damage. Stained cells were observed using a confocal microscope [8].
Intracellular Ca2+ level
To detect intracellular Ca2+ level, cells were stained with 10 μM Fluo-4 AM and fluorescence intensity measurements were performed via flow cytometry and confocal microscopy [3].
Mitochondrial membrane potential (Δψm)
The cells were first labeled with JC-1, a lipophilic cationic fluorescent dye [3]. Thereafter, mitochondrial membrane potential (Δψm) was measured via confocal microscopy.
Western blot analysis
Extracted cell lysates were electrophoresed on SDS-polyacrylamide gel to separate proteins. Thereafter, the separated proteins were electroblotted to nitrocellulose membranes, which were then incubated with the appropriate primary antibodies (1:1,000), followed by incubation with a secondary antibody. Protein expression levels were then detected using an enhanced chemiluminescence plus western blot detection system (GE Healthcare Life Sciences, Buckinghamshire, UK) [14].
Hoechst 33342 staining
Cells were stained with a DNA-specific fluorescent dye, Hoechst 33342 and examined using a fluorescence microscope (Media Cybernetics, Rockville, MD, USA). Finally, the proportion of apoptotic cells was quantified [14].
Annexin V/PI staining assay
Apoptotic cells were quantified via flow cytometry using the Alexa FluorTM 488 annexin V/dead cell apoptosis kit (Invitrogen, Thermo Fisher Scientific, Inc.) in accordance with the guidelines provided by the manufacturer [18].
Statistical analysis
Statistical analyses were performed using sigmaStat version 3.5 (Systat SoftwareInc., San Jose, CA, USA). p < 0.05 indicated significance.
Results
RA protected skin cells by lowering PM2.5-induced increases in ROS levels
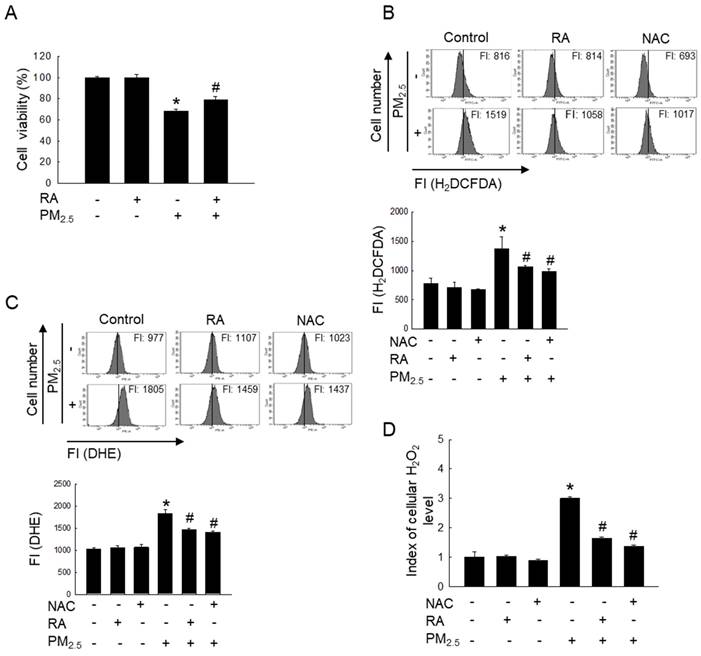
Reportedly, RA at 2.5 μM does not induce any form of cytotoxicity in HaCaT cells; it maintains cell viability at approximately > 95% [11]. Therefore, 2.5 μM RA was used as the optimal RA concentration in this study. The exposure of HaCaT cells to PM2.5 significantly reduced cell viability; however, treatment with RA significantly reversed the PM2.5-induced loss of cell viability (Figure 1A). Further, RA pretreatment significantly lowered the PM2.5-induced increase in the levels of ROS, including superoxide anion and hydrogen peroxide (Figures 1B-D).
RA ameliorated PM2.5-mediated cell death and ROS generation. (A) Cell viability measured via the MTT assay. (B) Cellular ROS amount detected via H2DCFDA staining. (C) Superoxide anion detected via DHE staining. (D) Cellular H2O2 level measured via the ROS-Glo™ H2O2 assay. (B, C) FI means fluorescence intensity. (A-D) *p < 0.05 vs. control; #p < 0.05 vs. PM2.5.
RA attenuated PM2.5-induced macromolecular damage
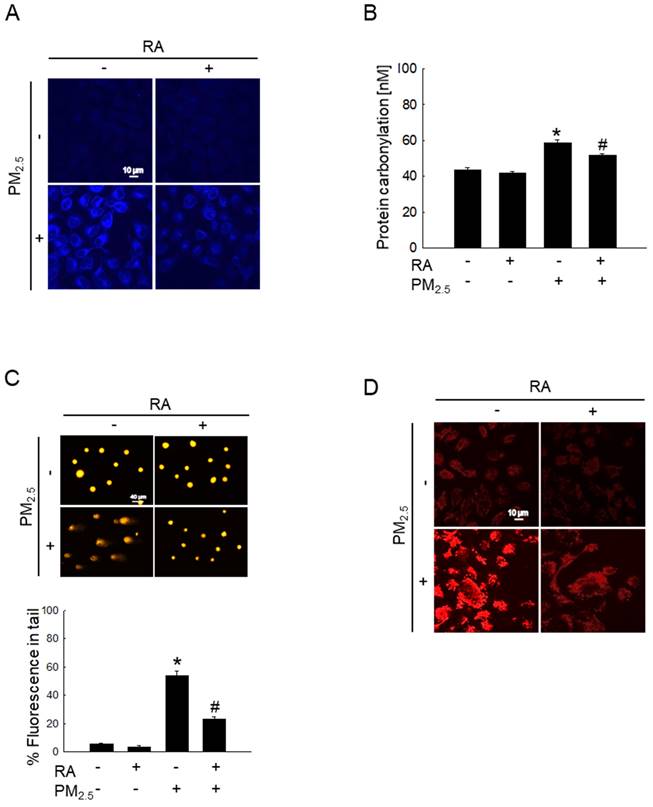
Oxidative stress significantly increases intracellular macromolecular damage [19]. In this study, we examined the protective properties of RA against macromolecular damage caused by PM2.5. The examination of PM2.5-induced lipid peroxidation via DPPP staining showed the significant enhancement of fluorescence intensity in the PM2.5-treated group; however, pretreatment with RA significantly decreased this fluorescence intensity (Figure 2A). Further, PM2.5-mediated oxidative stress resulted in a significant increase in protein carbonylation level, while RA pretreatment prevented this phenomenon (Figure 2B). Additionally, the examination of PM2.5-induced DNA damage via the comet assay showed that PM2.5-induced oxidative stress increased comet tail length and tail fluorescence percentage; however, these observations were reversed following RA pretreatment (Figure 2C). 8-Oxoguanine, a major form of oxidative DNA damage, was examined using an avidin-conjugated TRITC reagent [20]. The confocal microscopy images obtained thereafter revealed severe DNA lesions in the PM2.5-exposed cells; however, pretreatment with RA ameliorated this effect (Figure 2D).
RA prevented oxidative damage to intracellular molecules caused by PM2.5. (A) DPPP staining for the detection of lipid oxidation performed. (B) Protein carbonylation measured using the protein carbonyl ELISA kit. (C, D) DNA damage observed via (C) a comet assay and (D) avidin-TRITC staining. (B, C) *p < 0.05 vs. control; #p < 0.05 vs. PM2.5.
RA decreased PM2.5-induced apoptotic cell death
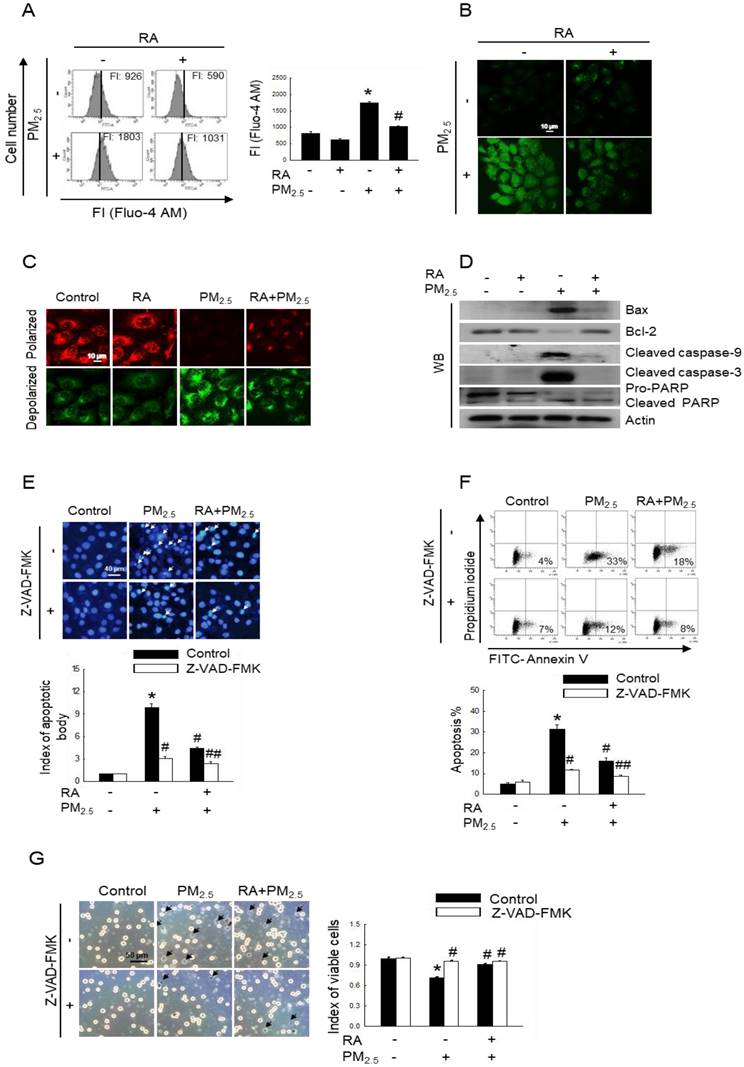
Oxidative stress results in increased cytoplasmic Ca2+ concentration, and such elevated Ca2+ level can damage mitochondria and nuclei, leading to apoptosis [21]. After labelling cells with Fluo-4 AM, we measured intracellular Ca2+ level via flow cytometry and confocal imaging. Thus, we observed an increased fluorescence intensity for cells exposed to PM2.5, indicating increased Ca2+ level; however, pretreatment with RA resulted in a decrease in Ca2+ level, as depicted in Figure 3A and B. Mitochondria act as apoptosis-regulating centers. PM2.5-induced excessive ROS generation can induce mitochondrial oxidative damage, and reportedly, severe mitochondrial damage is associated with mitochondrial malfunction and apoptosis [22]. Our results showed that PM2.5 enhanced mitochondrial depolarization, which was reversed by pretreatment with RA (Figure 3C). Previous studies have shown that PM2.5 enhances keratinocyte apoptosis by activating the caspase signaling pathway [15,23]. As shown in Figure 3D, PM2.5 exposure upregulated Bax, cleaved caspase-9, cleaved caspase-3, and cleaved PARP protein levels, while pretreatment with RA downregulated these proteins. In contrast, the expression level of antiapoptotic protein, Bcl-2 was decreased following PM2.5 exposure; however, it was enhanced owing to RA pretreatment. To verify the effect of caspase activation on apoptosis, HaCaT cells were pretreated with a caspase inhibitor (Z-VAD-FMK) with or without RA, both of which are associated with decreases in the amounts of apoptotic bodies (Figure 3E). As was observed for Hoechst 33342-staining, the percentage of annexin V-stained cells in the PM2.5 group increased and this effect was ameliorated by Z-VAD-FMK or/with RA pretreatment (Figure 3F). Trypan blue staining further revealed that Z-VAD-FMK and RA pretreatment ameliorated PM2.5-induced decreases in cell viability (Figure 3G).
RA reduced apoptosis triggered by PM2.5. (A, B) Ca2+ level after Fluo-4 AM staining detected via (A) flow cytometry and (B) confocal microscopy. (C) Δψm following JC-1 staining detected using a confocal microscope. (D) Bax, Bcl-2, cleaved caspase-9, cleaved caspase-3, and PARP protein expression levels detected via western blot analysis. Actin used as the loading control. (E, F) Apoptotic levels observed via (E) Hoechst 33342 staining and (F) annexin V/PI staining. Apoptotic bodies indicated using arrows. (G) Cell viability detected using trypan blue staining. (A) FI means fluorescence intensity. (A, E-G) *p < 0.05 vs. control; #p < 0.05 vs. PM2.5; ## p < 0.05 vs. RA+PM2.5.
RA downregulated the PM2.5-activated MAPK signaling pathway
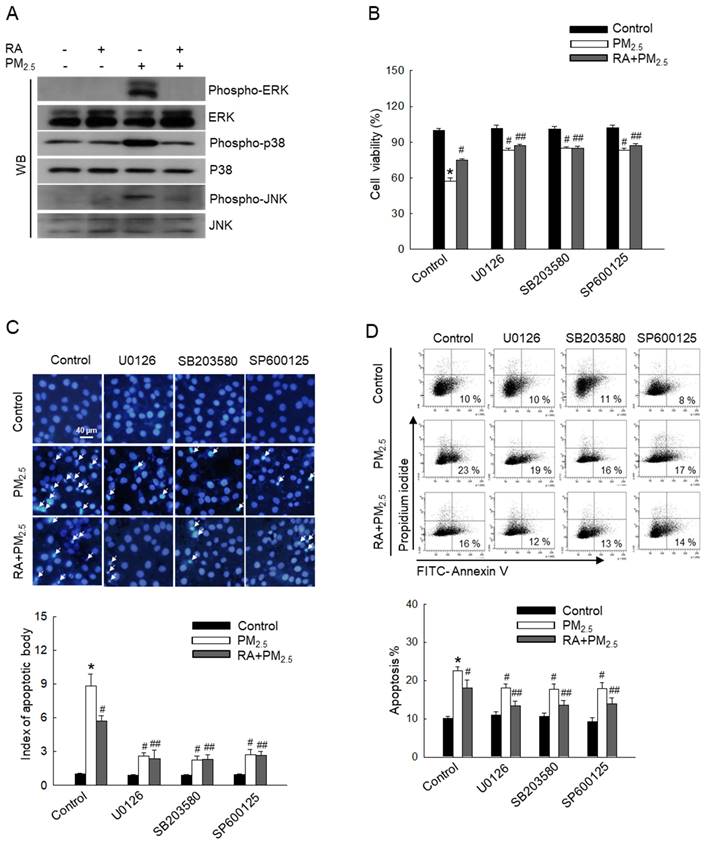
MAPK signaling pathways regulate various biological processes, such as apoptosis [24]. To further investigate the effect of PM2.5 on MAPK signaling pathway-mediated apoptosis, MAPK signaling cascade-associated proteins, ERK, p38, and JNK were detected via western blot analysis (Figure 4A). Thus, we observed that PM2.5 elevated the levels of phospho-ERK, phospho-p38, and phospho-JNK in comparison with the control treatment. However, RA pretreatment reversed this effect. Cells pretreated with each ERK, p38, and JNK inhibitor (U0126, SB203580, and SP600125) exhibited decreased PM2.5-induced cytotoxicity and apoptosis, similar to cells pretreated with RA (Figure 4B-D). These results demonstrated that RA ameliorates PM2.5-induced apoptosis by downregulating the PM2.5-induced MAPK signaling pathway.
RA lowered PM2.5-induced activation of the MAPK signaling pathway. (A) Western blot analysis performed for detection of ERK, p38, and JNK protein expression levels. Total ERK, p38 and JNK protein levels used as the loading control. (B) Cell viability detected via the MTT assay. Apoptotic levels observed via (C) Hoechst 33342 staining and (D) Annexin V/PI staining. Apoptosis bodies indicated using arrows. (B-D) *p < 0.05 vs. control; #p < 0.05 vs. PM2.5; ## p < 0.05 vs. RA + PM2.5.
Discussion
Skin keratinocytes constitute the primary barrier against environmental stressors and exposure of keratinocytes to PM2.5 could lead to increased ROS production [25]. Recent studies have demonstrated that PM2.5 may penetrate the skin and directly affect viable skin cells, including keratinocytes [9,25]. Therefore, in this study, we aimed to explore the protective effects of RA against PM2.5-induced damage in HaCaT cells. Our previous study showed that at concentrations up to 2.5 μM, RA does not exert any cytotoxic effects on HaCaT and that at 2.5 μM, RA scavenges up to 60% of intracellular ROS [11]. Thus, in this study, we used 2.5 μM as the optimal RA concentration. Further, based on our previous study, which showed that the optimal PM2.5 exposure concentration required to induce excessive ROS generation in HaCaT cells is 50 μg/mL [10]. In this study, we observed that RA significantly reversed PM2.5-induced decreases in the viability of HaCaT cells. RA also significantly reduced PM2.5-induced increases in ROS levels.
PM2.5-induced excessive ROS generation can trigger oxidative macromolecular damage, leading to apoptosis [26]. In the lipid peroxidation process, oxidants, such as free radicals or ROS attack several carbon-carbon double bonds in polyunsaturated fatty acids, initiating the removal of hydrogen from carbon to produce water and fatty acid radicals. The unstable fatty acid radicals thus obtained react with molecular oxygen to form lipid peroxyl radicals and hydroperoxides [27]. This membrane lipid peroxidation alters the physical properties of lipid bilayers, and consequently, affects membrane permeability, lipid-lipid interactions, ion gradients, and membrane fluidity [28]. Thus, lipid peroxidation negatively affects cellular functions. Additionally, oxidative stress caused by ROS overproduction results in protein carbonylation owing to protein oxidation or DNA modification [8]. In this study, we observed that RA exhibited a protective effect against PM2.5-induced macromolecular damage by diminishing ROS.
Owing to oxidative stress, Ca2+ flows into the cytoplasm from internal cellular stores, such as the sarcoplasmic reticulum/endoplasmic reticulum and can also be imported from extracellular spaces. As the Ca2+ concentration in the cytoplasm increases, Ca2+ flows into the mitochondria and nuclei. The accumulation of Ca2+ in mitochondria and nuclei as such disrupts normal cell metabolism, leading to apoptosis [21,29]. In this study, we observed that RA significantly attenuated oxidative stress-mediated cellular Ca2+ influx. Mitochondria play a major role in the onset of apoptosis. Moreover, PM2.5-induced oxidative stress leads to mitochondrial damage, including changes in Δψm [15,19,30]. In this study, we observed that RA regulated Δψm and facilitated the normal functioning of mitochondria. It has been shown that mitochondrial damage is associated with Bcl-2, which maintains the integrity of the mitochondrial membrane. The loss of Δψm induces the release of apoptotic factors into the cytosol. Further, the balance between the pro-apoptotic protein, Bax and the antiapoptotic proteins of the Bcl-2 family serves as the determining factor for the activation or inhibition of the caspase cascade. Specifically, caspase-3 initiates the intrinsic apoptotic pathway and cleaves nuclear proteins, such as PARP [31,32]. It has also been shown that PM2.5 upregulates apoptotic protein levels while downregulating the expression of antiapoptotic proteins [8,15]. In this study, we observed that RA reversed these effects.
The MAPK signaling cascade plays a crucial role in mediating cellular stress response. Previous reports have shown that PM2.5 activates MAPK pathway-associated proteins via ROS-mediated pathways [15,19]. The activation of p38 and JNK in response to PM2.5 is thought to induce apoptosis [33]. In contrast, the ERK pathway plays a dual role in cell survival and death. Further, recent studies have shown that continuous ERK activation can promote apoptosis [15,34]. Further exploration in this regard using ERK, p38, and JNK inhibitors revealed that RA pretreatment reduced the phosphorylation of ERK, p38, and JNK, similar to their respective inhibitors, leading to a decrease in the levels of apoptotic bodies.
Conclusion
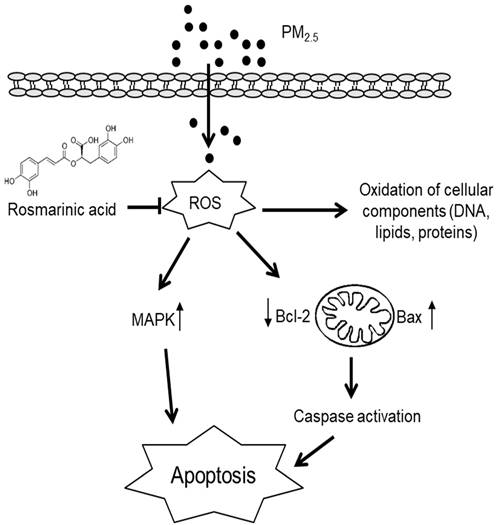
In this study, we investigated the effects of RA on PM2.5-induced damage in keratinocytes. Our results revealed that PM2.5 exacerbated skin cell damage by increasing ROS generation and activating apoptotic pathways; however, RA ameliorated these observed PM2.5-induced effects. Thus, it showed protective effects on skin cells against PM2.5-induced damage (Figure 5).
Summary of the protective effects of RA against PM2.5-induced skin cell damage. RA ameliorated PM2.5-induced intracellular ROS levels, hence attenuated oxidative damage to DNA, lipids, and proteins. Further, RA downregulated mitochondria-mediated caspase activation and the MAPK signaling pathway, and thus ameliorated apoptosis.
Abbreviations
PM2.5: particulate matter 2.5; RA: rosmarinic acid; ROS: reactive oxygen species; PAHs: polycyclic aromatic hydrocarbons; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; ERK: extracellular signal-regulated kinase; PARP: poly-ADP ribose polymerase; MAPK: mitogen-activated protein kinase; avidin-TRITC: avidin-tetramethylrhodamine isothiocyanate; MTT: thiazolyl blue tetrazolium bromide; DMSO: dimethyl sulfoxide; H2DCFDA: 2′,7′-dichlorodihydrofluorescein diacetate; DHE: dihydroethidium; DPPP: diphenyl-1-pyrenylphosphine; PI: propidium iodide; JC-1: 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; NAC: N-acetyl cysteine; JNK: c-Jun N-terminal kinase; DMEM: Dulbecco-modified Eagle medium; Fluo-4 AM: fluo-4-acetoxymethyl ester; MEK: mitogen-activated protein kinase kinase; FI: fluorescence intensity.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (RS-2023-00270936).
Author contributions
Conceptualization: Herath HMUL, Hyun JW; Investigation: Herath HMUL, Piao MJ, Fernando PDSM; Writing-original draft preparation: Herath HMUL, Hyun JW; Writing-review and editing: Herath HMUL, Piao MJ, Fernando PDSM, Kang KA, Hyun JW. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cui Y, Chen G, Yang Z. Mitochondrial superoxide mediates PM2.5-induced cytotoxicity in human pulmonary lymphatic endothelial cells. Environ Pollut. 2020;263:114423
2. Piao MJ, Kang KA, Zhen AX. et al. Particulate matter 2.5 mediates cutaneous cellular injury by inducing mitochondria-associated endoplasmic reticulum stress: protective effects of ginsenoside Rb1. Antioxidants. 2019;8:383
3. Zhen AX, Piao MJ, Kang KA. et al. Niacinamide protects skin cells from oxidative stress induced by particulate matter. Biomol Ther. 2019;27:562
4. Yang J, Chen Y, Yu Z. et al. The influence of PM2.5 on lung injury and cytokines in mice. Exp Ther Med. 2019;18:2503-2511
5. Li Q, Zheng J, Xu S. et al. The neurotoxicity induced by PM2.5 might be strongly related to changes of the hippocampal tissue structure and neurotransmitter levels. Toxicol Res. 2018;7:1144-1152
6. Kim YM, Kim J, Jung K. et al. The effects of particulate matter on atopic dermatitis symptoms are influenced by weather type: application of spatial synoptic classification (SSC). Int J Hyg Environ Health. 2018;221:823-829
7. Ding A, Yang Y, Zhao Z. et al. Indoor PM2. 5 exposure affects skin aging manifestation in a chinese population. Sci Rep. 2017;7:15329
8. Fernando PDSM, Piao MJ, Zhen AX. et al. Extract of Cornus officinalis protects keratinocytes from particulate matter-induced oxidative stress. Int J Med Sci. 2020;17:63-70
9. Piao MJ, Ahn MJ, Kang KA. et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch Toxicol. 2018;92:2077-2091
10. Gerogianni PS, Chatziathanasiadou MV, Diamantis DA. et al. Lipophilic ester and amide derivatives of rosmarinic acid protect cells against H2O2-induced DNA damage and apoptosis: The potential role of intracellular accumulation and labile iron chelation. Redox Biol. 2018;15:548-556
11. Fernando PMDJ, Piao MJ, Kang KA. et al. Rosmarinic acid attenuates cell damage against UVB radiation-induced oxidative stress via enhancing antioxidant effects in human HaCaT cells. Biomol Ther. 2016;24:75-84
12. Piao MJ, Fernando PMDJ, Kang KA. et al. Rosmarinic acid inhibits ultraviolet B-mediated oxidative damage via the AKT/ERK-NRF2-GSH pathway in vitro and in vivo. Biomol Ther. 2024;32:84-93
13. Herath HMUL, Piao MJ, Kang KA. et al. Hesperidin exhibits protective effects against PM2.5-mediated mitochondrial damage, cell cycle arrest, and cellular senescence in human HaCaT Keratinocytes. Molecules. 2022;27:4800
14. Fernando PDSM, Piao MJ, Kang KA. et al. Hesperidin protects human HaCaT keratinocytes from particulate matter 2.5-induced apoptosis via the inhibition of oxidative stress and autophagy. Antioxidants. 2022;11:1363
15. Zhen AX, Hyun YJ, Piao MJ. et al. Eckol inhibits particulate matter 2.5-induced skin keratinocyte damage via MAPK signaling pathway. Mar Drugs. 2019;17:444
16. Kim CW, Go RE, Hwang KA. et al. Effects of cigarette smoke extracts on apoptosis and oxidative stress in two models of ovarian cancer in vitro. Toxicol In Vitro. 2018;52:161-169
17. Zhen AX, Piao MJ, Kang KA. et al. 3-Bromo-4, 5-dihydroxybenzaldehyde protects keratinocytes from particulate matter 2.5-induced damages. Antioxidants. 2023;12:1307
18. Wang H, Paton JC, Herdman BP. et al. The B subunit of an AB5 toxin produced by Salmonella enterica serovar Typhi up-regulates chemokines, cytokines, and adhesion molecules in human macrophage, colonic epithelial, and brain microvascular endothelial cell lines. Infect Immun. 2013;81:673-683
19. Zhen AX, Piao MJ, Hyun YJ. et al. Purpurogallin protects keratinocytes from damage and apoptosis induced by ultraviolet B radiation and particulate matter 2.5. Biomol Ther. 2019;27:395
20. Kim KC, Lee IK, Kang KA. et al. 7,8-Dihydroxyflavone suppresses oxidative stress-induced base modification in DNA via induction of the repair enzyme 8-oxoguanine DNA glycosylase-1. Biomed Res Int. 2013;2013:863720
21. Xiao Y, Li Z, Bianco A. et al. Recent advances in calcium-based anticancer nanomaterials exploiting calcium overload to trigger cell apoptosis. Adv Funct Mater. 2023;33:2209291
22. Wei H, Yuan W, Yu H. et al. Cytotoxicity induced by fine particulate matter (PM 2.5) via mitochondria-mediated apoptosis pathway in rat alveolar macrophages. Environ Sci Pollut Res. 2021;28:25819-25829
23. Zhen AX, Piao MJ, Hyun YJ. et al. Diphlorethohydroxycarmalol attenuates fine particulate matter-induced subcellular skin dysfunction. Mar Drugs. 2019;17:95
24. Yue J, López JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. 2020;2:2346
25. Li Q, Kang Z, Jiang S. et al. Effects of ambient fine particles PM2.5 on human HaCaT cells. Int J Environ Res Public Health. 2017;14:72
26. Jin X, Xue B, Zhou Q. et al. Mitochondrial damage mediated by ROS incurs bronchial epithelial cell apoptosis upon ambient PM2.5 exposure. J Toxicol Sci. 2018;43:101-111
27. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438
28. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419-425
29. Kang P, Zhang W, Chen X. et al. TRPM2 mediates mitochondria-dependent apoptosis of melanocytes under oxidative stress. Free Radic Biol Med. 2018;126:259-268
30. Zhu S, Li X, Dang B. et al. Lycium Barbarum polysaccharide protects HaCaT cells from PM2.5-induced apoptosis via inhibiting oxidative stress, ER stress and autophagy. Redox Rep. 2022;27:32-44
31. Park C, Cha HJ, Hong SH. et al. Protective effect of phloroglucinol on oxidative stress-induced DNA damage and apoptosis through activation of the Nrf2/HO-1 signaling pathway in HaCaT human keratinocytes. Mar Drugs. 2019;17:225
32. Zhou F, Huang X, Pan Y. et al. Resveratrol protects HaCaT cells from ultraviolet B-induced photoaging via upregulation of HSP27 and modulation of mitochondrial caspase-dependent apoptotic pathway. Biochem Biophys Res Commun. 2018;499:662-668
33. Hou GR, Zeng K, Lan HM. et al. Juglanin ameliorates UVB-induced skin carcinogenesis via anti-inflammatory and proapoptotic effects in vivo and in vitro. Int J Mol Med. 2018;42:41-52
34. Tan BJ, Chiu GN. Role of oxidative stress, endoplasmic reticulum stress and ERK activation in triptolide-induced apoptosis. Int J Oncol. 2013;42:1605-1612
Author contact
![]() Corresponding author: Professor Jin Won Hyun, Department of Biochemistry, College of Medicine and Jeju Research Center for Natural Medicine, Jeju National University, Jeju 63243, Republic of Korea. Telephone: +82-64-754-3838; Fax: +82-64-702-2687; E-mail: jinwonhac.kr.
Corresponding author: Professor Jin Won Hyun, Department of Biochemistry, College of Medicine and Jeju Research Center for Natural Medicine, Jeju National University, Jeju 63243, Republic of Korea. Telephone: +82-64-754-3838; Fax: +82-64-702-2687; E-mail: jinwonhac.kr.