Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(4):664-673. doi:10.7150/ijms.90485 This issue Cite
Research Paper
METTL3 Promotes Osteogenic Differentiation of Human Periodontal Ligament Stem Cells through IGF2BP1-Mediated Regulation of Runx2 Stability
Xuefei Sun1,2† ![]() , Xiujiao Meng1,2†, Yu Piao1,2, Shaojie Dong1,2, Qianqian Dong1,2
, Xiujiao Meng1,2†, Yu Piao1,2, Shaojie Dong1,2, Qianqian Dong1,2 ![]()
1. Key Laboratory of Shaanxi Province for Craniofacial Precision Medicine Research, College of Stomatology, Xi'an Jiaotong University, Xi'an, China.
2. Clinical Research Center of Shaanxi Province for Dental and Maxillofacial Diseases, Department of Endodontics, College of Stomatology, Xi'an Jiaotong University, Xi'an, China.
†: Co-first authors.
Received 2023-9-25; Accepted 2024-1-23; Published 2024-2-4
Abstract

N6-Methyladenosine (m6A) has been reported to play a dynamic role in osteoporosis and bone metabolism. However, whether m6A is involved in the osteogenic differentiation of human periodontal ligament stem cells (hPDLSCs) remains unclear. Here, we found that methyltransferase-like 3 (METTL3) was up-regulated synchronously with m6A during the osteogenic differentiation of hPDLSCs. Functionally, lentivirus-mediated knockdown of METTL3 in hPDLSCs impaired osteogenic potential. Mechanistic analysis further showed that METTL3 knockdown decreased m6A methylation and reduced IGF2BP1-mediated stability of runt-related transcription factor 2 (Runx2) mRNA, which in turn inhibited osteogenic differentiation. Therefore, METTL3-based m6A modification favored osteogenic differentiation of hPDLSCs through IGF2BP1-mediated Runx2 mRNA stability. Our study shed light on the critical roles of m6A on regulation of osteogenic differentiation in hPDLSCs and served novel therapeutic approaches in vital periodontitis therapy.
Keywords: METTL3, hPDLSCs, Osteogenic differentiation, Runx2, m6A, IGF2BP1
1. Introduction
Periodontitis is the most prevalent oral infection, which destroys the periodontal supporting tissue and eventually leads to dentition loss[1, 2]. Repair and reconstruction of destructed periodontal supporting tissue rely on mechanical plaque removal and guided tissue regeneration techniques[3]. In recent years, human periodontal ligament stem cells (hPDLSCs) have brought great hope to periodontal tissue engineering due to their excellent osteogenic differentiation potential[4, 5]. Studies have shown that RUNX2 (runt-related transcription factor 2), as an osteoblast-specific transcription factor, plays a key role in the osteogenic differentiation of hPDLSCs[6, 7]. Therefore, endogenous regulation of RUNX2 is an important means to regulate the osteogenic differentiation of PDLSCs.
N6-methyladenosine is the most common modification of eukaryotic mRNA which regulates nearly every step of the RNA metabolic cycle, including splicing, export, decay, and translation[8, 9]. m6A modification is catalyzed by a methyltransferase complex including METTL3, METTL14 and their cofactors, and demethylated by ALKBH5 and FTO enzymes, forming a homeostasis[10]. Ultimately, m6A-binding proteins with YTH domains, including cytoplasmic proteins YTHDF1, YTHDF2, YTHDF3, IGF2BP1, and nuclear protein YTHDC1, mediate mRNA stability and translation to regulate downstream biological effects[11, 12]. Recently, the insulin-like growth factor-2 mRNA binding protein (IGF2BP) family has been reported as a novel m6A reading protein that directly recognizes m6A mRNA modifications, thereby recruiting cofactors including Human antigen R (HuR) and matrix protein 3 (MATR3). The IGF2BP protein family consists of three main members: IGF2BP1, IGF2BP2 and IGF2BP3. Functionally, IGF2BPs mainly recognizes and binds mRNA through KH domain, thus enhancing the stability of m6A-modified mRNA and improving the translation efficiency of mRNA, thus regulating various physiological and pathological functions. IGF2BP1 has been identified to play an important role in the regulation of mRNA targets such as CPT1A, E2F1, PEG10, c-MYC, and CCL5 in an m6A-dependent manner[13-16]. Zhou et al. reported that METTL3 promotes osteoblast differentiation of bone marrow mesenchymal stem cells (BMSCs) through IGF2BP1-mediated Runx2 mRNA stabilization to regulate osteogenic responses[17]. METTL14 promotes postmenopausal osteoporosis through m6A-guided epigenetic inheritance by regulating SMAD1 mRNA stability in an IGF2BP1 dependent manner[18]. Whether m6A methylation plays a role in regulating osteogenic differentiation of hPDLSCs and its molecular mechanism are still unclear.
In the current study, we revealed a significant increase of METTL3 and m6A levels in hPDLSCs undergoing osteogenic differentiation. Our results further verified that METTL3-mediated m6A RNA methylation induced hPDLSCs osteogenic differentiation via targeting Runx2 mRNA stability. Taken together, our findings elucidated the regulatory role of METTL3-mediated m6A RNA methylation in the osteogenic differentiation of hPDLSCs and afforded a novel potential molecule target for periodontitis therapy.
2. Materials and methods
2.1. Cell culture
The hPDLSCs were isolated and cultured according to the previous study[19, 20]. The healthy third molars or orthodontically treated premolars were extracted after obtaining informed consent from 18-22-year-old healthy donors. After adequate cleaning of the teeth, periodontal ligament tissue was scraped from the middle third of the root surface, digested with collagenase, and cultured in complete medium containing a-MEM (Hyclone, USA) and 10% fetal bovine serum (FBS) (GIBCO, Invitrogen, USA) at 37℃ in 5% CO2. The culture medium was changed every 3 days, and hPDLSCs between passages 3 and 6 were used for subsequent experiments. The medium was refreshed every 3 days, and hPDLSCs between passage 3 and 6 were used in the following experiment.
2.2. Overexpression and knockdown of genes
The METTL3 gene was transfected into cells for overexpression and knockdown by lentivirus (Genepharma, Hangzhou, China). Overexpression and knockdown of the indicated genes were transfected into hPDLSCs using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) with overexpression plasmids or siRNA, and analyzed 48-72 h later, followed by analysis 48-72 h later. The selected sequences for siRNA and shRNA knockdown as follow: sh-METTL3: 5′-GCTACCTGGACGTCAGTATCT-3′. sh-control: 5′-ACGTGACACGTTCGGAGAATT-3′. si-IGF2BP1: 5′-AUGAAACAUAACUUUCUUGUU-3′. si-control: 5′-TTCTCCGAACGTGTCACG A-3′.
2.3. RNA purification and quantitative real‑time PCR (qPCR)
Total RNA was isolated by using TRIzol reagent (Invitrogen, USA) from the samples according to the manufacturer's instructions. RNA was reverse transcribed into first‑strand cDNA using the PrimeScript™ RT reagent kit (Takara, Japan). Amplification was performed using 7500 Real-Time PCR System (Applied Biosystems) with SYBR Green PCR Master Mix (Takara, Japan). Amplification was performed as follows: denaturation at 95 °C for 60 s, followed by 40 cycles denaturation at 95 °C for 10 s, annealing at 60 °C for 30 s, and extension at 70 °C for 5 min. The qRT‑PCR was analyzed by using the 2-∆∆Ct method and normalized to GAPDH expression. Primers for qPCR are as follows (Table 1):
Primers for qPCR.
| Gene | Forward primer | Reverse primer | |
|---|---|---|---|
| METTL3 | 5′-CAAGCTGCACTTCAGACGAA-3′ | 5′-GCTTGGCGTGTG GTCTTT-3′ | |
| METTL14 | 5′-GTCTTAGTCTTCCCAGGATTGTTT-3′ | 5′-AATTGATGAGATTGCAGCACC-3′ | |
| ALKBH5 | 5′-ACTGAGCACAGTCACGCTTCC-3′ | 5′-GCCGTCATCAACGACTACCAG-3′ | |
| FTO | 5′-GACCTGTCCACCAGATTTTCA-3′ | 5′-AGCAGAGCAGCATACAACGTA-3′ | |
| Runx2 | 5′-GGGTAAGACTGGTCATAGGACC-3′ | 5′-CCCAGTATGAGAGTAGGTGTCC-3′ | |
| Osx | CCTCTGCGGGACTCAACAAC | AGCCCATTAGTGCTTGTAAAGG | |
| Ocn | 5′-GGGTAAGACTGGTCATAGGACC-3′ | 5′-CCCAGTATGAGAGTAGGTGTCC-3′ | |
| Alp | 5′-CATGCTGAGTGACACAGACAAGAA-3′ | 5′-ACAGCAGACTGCGCCTGGTA-3′ | |
| GAPDH | 5′-GGTCGGAGTCAACGGATTTG-3′ | 5′-GGAAGATGGTGATGGGATTTC-3′ | |
2.4. Western blot analysis
For western blot analysis, cells were washed with ice-cold PBS and lysed in lysis buffer (Beyotime Biotechnology, Shanghai, China). Then, the lysates were added to 5× loading dye and separated by 10% SDS-PAGE, and then transferred to PVDF membranes (Millipore, Germany). The PVDF membranes were blocked in 5% fat-free dry milk for 1 h and then incubated with primary antibodies (1:1,000; Abcam, UK) against METTL3 (ab195352), RUNX2 (ab192256), OSX (ab209484), METTL14 (ab220030), WTAP (ab195380), IGF2BP1 (ab290736), Tubulin (1:1,000; ab6046), and Lamin B (1:1,000; ab133741) overnight at 4 °C. The PVDF membranes were incubated with secondary antibodies (Proteintech, USA) at RT for 1 h, and detected by odyssey chemiluminescence, and quantified by using the Image Studio software (LI-COR Biosciences, USA).
2.5. Alizarin red staining
The osteogenic differentiation of hPDLSCs was induced in the osteogenic differentiation medium for 3 weeks. The medium of osteogenic differentiation was composed of α-MEM (Hyclone, USA), 10 mM β-glycerophosphate (Sigma, USA), 10% FBS (GIBCO, USA), 0.1 μM dexamethasone (Sigma, USA), and 50 μg/mL of ascorbic acid (Sigma, USA). The cells after osteogenic differentiation were fixed with 4% paraformaldehyde for 15 min and rinsed with ddH2O. Then the cells were stained with 0.1% alizarin red staining (ARS; pH 4.2) at RT for 10 min. Subsequently, cells were washed with deionized water to wash away unbound Alizarin Red. To quantify ARS, 10% cetylpyridinium chloride was added to the well plate to fully dissolve and quantitatively analyzed under a spectrophotometer at 562 nm.
2.6. Oil red O staining
The adipogenic differentiation of hPDLSCs was induced in the adipogenic differentiation medium for 4 weeks. The medium of adipogenic differentiation was composed of α-MEM (Hyclone, USA), 10% FBS (GIBCO, USA), 0.5 mM IBMX, 200 μM indomethacin, 1 μM dexamethasone, and 10 μg/mL of insulin (Sigma, USA). The cells after adipogenic differentiation were fixed with 4% paraformaldehyde for 15 min and rinsed with ddH2O. Then the cells were stained with 0.1% oil red O staining for 30 min at room temperature.
2.7. Flow cytometry analysis
The immunophenotype of hPDLSCs was detected by flow cytometry according to the manufacturer's instructions (BD Bioscience, USA). The following antibodies were used: MSC positive markers (CD105-PE, 146-PE, and CD29-PE), the hematopoietic marker (CD34-PE) and the leukocyte marker (CD45-PE). hPDLSCs were digested by trypsin and the cell density was adjusted to 1*107 in PBS solution. Then incubate with the antibodies for 30 min at 37°C in the dark. Unbound antibodies were washed with PBS and detected by flow cytometry.
2.8. Cell Counting Kit-8 (CCK-8) and Clonal Formation Unit
Cell proliferation was assessed by CCK-8 following the manufacturer's instructions. Briefly, we adjusted the cell density to 3000 cells/well seeded in 96-well dishes. The 96-well dishes were then incubated in a 37°C, 5% CO2 incubator. Cell samples were collected every day, and then 100 µl of CCK-8 solution was added to each well, and the absorbance at 450 nm was measured with a microplate reader.
HPDLSCs were limitedly diluted and seeded into 10 cm dishes at 200 cells/well. The cell culture medium was refreshed every 3 days. After 14 days, the stem cells were washed with PBS, fixed with 4% paraformaldehyde for 15 minutes, and then stained with 0.1% crystal violet to calculate the formation of cell clone-forming units.
2.9. m6A Dot Blot
Total RNAs were extracted by using TRIzol reagent (Invitrogen, USA) and then extracted and purified by an mRNA purification kit (TOYOBO, Japan) according to the manufacturer's protocol. The extracted mRNAs were directly spotted onto a Hybond-N+ membrane (Beyotime, Shanghai, China) and then UV-crosslinked to the Hybond-N+ membrane by a Stratalinker 2400 UV crosslinker (UVP, CA, USA). The membrane was washed with Tris-buffered saline with Tween 20 (TBST) for three time to remove unbound mRNAs and then blocked with 5% fat-free dry milk for 1 h at room temperature. Next, the membrane was incubated with N6-mA antibody (1:500, Synaptic Systems, Germany) at 4℃ overnight followed by HRP-conjugated anti-rabbit IgG (1:500, Synaptic Systems, Germany) for 1 h at room temperature. Finally, the membrane was mixed with Thermo ECL Signal Western Blotting Detection Reagent (Thermo Fisher Scientific, Waltham, USA) and analyzed with Image Studio software (LI-COR Biosciences, USA).
2.10. m6A Quantification
Quantification of m6A was measured by an m6A RNA methylation assay kit (Abcam, Cambridge, UK) according to the manufacturer's instructions. Briefly, positive control (PC), negative control (NC), and 200 ng isolated mRNA were added to each well with the capture antibody and then detected the percentage of m6A in the sample. After the RNA reacted with the substrate, the absorbance was determined with an enzyme-linked instrument at 450 nm. The formula for calculating the percentage of m6A in RNA is as follows: (S=200ng, P=1ng)

S: amount of input of RNA sample (200ng), P: amount of input of positive control (1ng), PC: positive control.
2.11. RNA stability assay
Cells were plated in 6-well plates and transfected with desired plasmid, siRNA or virus as described above. Cells were treated with 5 μg/ml actinomycin D (Sigma, USA) to inhibit RNA transcription and collected at indicated time points (0, 3, 6 h) to assess mRNA degradation. The total RNA was extracted and analyzed by qRT-PCR.
2.12. RNA agarose gel electrophoresis
Agarose (1g) was added to an erlenmeyer flask containing 90 mL of DEPC water, and heated it completely dissolve before adding 10 ml of 10x TAE buffer. Then pour the slightly cooled gel into the glue tank to solidify for later experiments. 4 μl of total RNA, 5 μl of 1×TAE buffer, and 1 μl of 10X Gel Red loading buffer were mixed and added to the gel well. RNA electrophoresis was performed from the negative electrode to the positive electrode at a voltage of 100V for about 30 minutes. The RNA gel was then placed on an Odyssey detector to observe the electrophoresis results.
2.13. Methylated RNA immunoprecipitation (MeRIP)-PCR
The Magna MeRIP Kit (Millipore, Massachusetts, USA) was used following the manufacturer's protocol to detect m6A modification on genes of interest. Briefly, 100 μg of total RNA was mixed with 100 μL RIP lysis buffer on ice for 5 min and then incubated with anti-m6A antibody (Synaptic Systems, Germany)- or IgG (CS200621, Millipore)-conjugated beads in 500 μl IP buffer supplemented with RNase inhibitors at 4 °C overnight. Methylated RNA was immunoprecipitated with beads, and purified with RNeasy kit (Qiagen, Germany). Then, RNA samples were subjected to reverse transcription and RT-PCR as previously described.
2.14. Luciferase Reporter Assay
Luciferase assay was performed using luciferase assay reagent reporter and lysis buffer (Promega, USA) according to the manufacturer's protocol. Briefly, the pmirGLO-YAP luciferase reporter by ligating YAP CDS to the mutiple cloning site (MCS) site. The wild-type and sh-METTL3 cells were transfected with YAP-luciferase reporter plasmid with Lipofectamine 2000 reagent (Invitrogen, USA). After transfection for 24 h, cells were analyzed with the Dual Luciferase Assay system (Promega, USA). Firefly luciferase (F-luc) activity was normalized to Renilla Luciferase (R-luc) to appraise reporter translation efficiency.
2.15. Protein co-immunoprecipitation (Co-IP)
Protein co-immunoprecipitation was performed using Dynabeads™ Co-Immunoprecipitation Kit (Thermo Fisher Scientific, Waltham, USA) according to the manufacturer's instructions and previous study[21]. Briefly, cells were washed twice with ice old PBS and then lysed with NP40 lysis buffer supplement with protease inhibitor on ice. After the protein was sonicated and centrifuged at 4 °C, 10% supernatant was collected as input. Remaining supernatant was incubated with indicated IP antibody or IgG-conjugated protein A/G Magnetic Beads in IP buffe supplemented at 4 °C overnight. Bound protein was immunoprecipitated with magnetic beads and treated with elution buffer. The recovered proteins were washed 3 times and analyzed by Western blotting.
2.16. Immunofluorescent staining
Cells were seeded on glass slides and fixed in 4% paraformaldehyde for 15 minutes, then permeabilized in 0.5% Triton X-100 for 10 minutes. The samples were then treated with 2N HCl for 15 minutes and neutralized with 0.1 M sodium borate buffer pH 8.5 for 15 minutes. Then, the cell slides were blocked in blocking solution (100 mg/ml RNaseA in PBS with 5% serum) for 2 h, and then N6-mA antibody (1:200, Synaptic Systems, Germany), RUNX2 (1:200, Abcam, ab192256), METTL3 (1:200, Abcam, ab195352) was added dropwise to the samples for incubation. Samples containing primary antibodies were incubated at 4°C overnight. The next day, samples were incubated with species appropriate secondary antibodies conjugated to Alexa Fluor 488 or Cy3 (1:200; Proteintech, USA) for 1 h in a humidified chamber at 37°C. Finally, slides were rinsed with PBS, nuclei were stained with DAPI, and image capture was observed under a fluorescence microscope (Olympus, Japan).
2.17. Statistical Analysis
Statistics were performed using Graphpad Prizm 8 (GraphPad Software Inc.,San Diego, CA, USA). Statistically significant differences were determined using a two-tailed Student's t-test or ANOVA. Data represented the mean ± SD of at least n=3 independent experiments. *p<0.05, ns, not significant.
3. Results
3.1. Identification of hPDLSCs
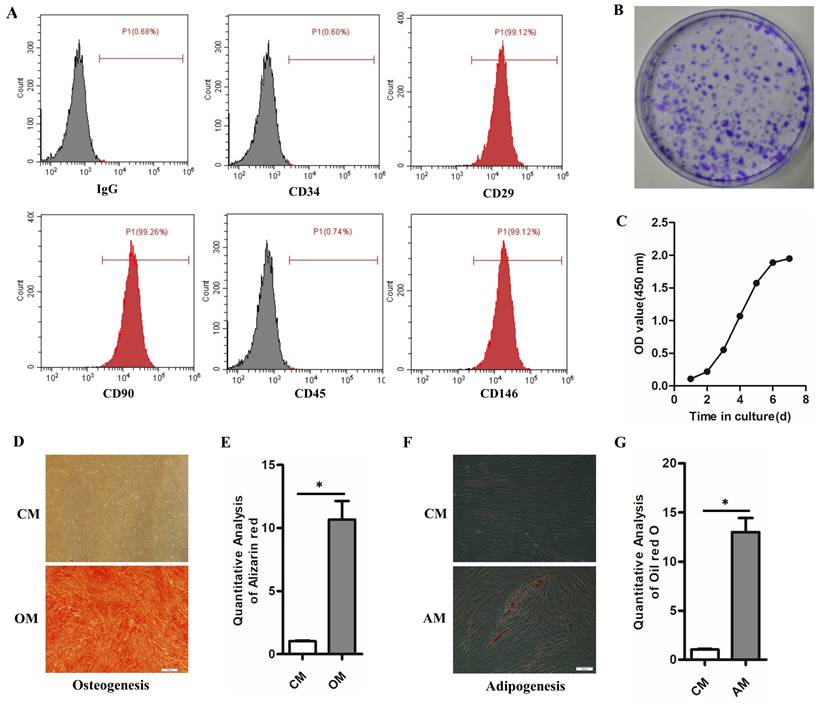
hPDLSCs express stem cell surface molecules, and possess high proliferation rate, self-renewal and multilineage differentiation capabilities. Flow cytometry results showed that hPDLSCs highly expressed CD29, CD90, and CD146 (mesenchymal stem cell markers) but scarcely expressed CD34 (hematopoietic marker) and CD45 (leukocyte marker) (Fig. 1A). Colony formation staining and cell growth curve verified the self-renewal ability and high proliferation ability of hPDLSCs (Figs. 1B and C). The multi-differentiation ability of hPDLSCs was verified by the Alizarin Red and Oil Red O staining. The Alizarin Red S staining indicated that mineralized calcium deposits were formed in hPDLSCs after culturing for 3 weeks in OM (Figs. 1D, E). Oil red O staining result demonstrated that hPDLSCs formed lipid droplets after 4 weeks of adipogenic induction (Figs. 1F, G). By contrast, few mineralized calcium deposits and lipid droplets were observed in the control groups (Figs. 1D-F). These data manifested that the isolated hPDLSCs hold the abilities of self-renewal and multi-differentiation.
Identification of hPDLSCs (A) Cell markers CD29, CD34, CD45, CD90 and CD146 expression was measured by flow cytometry. (B) The self-renewal ability of hPDLSCs was detected by colony-forming unit staining. (C) The proliferation curve was detected by spectrophotometry. (D) The osteogenesis capabilities of hPDLSCs were detected by alizarin red. Scale bars, 100 μm. (E) The quantitative analysis of alizarin red staining. (F) The adipogenesis capabilities were detected by oil red O staining. Scale bars, 20 μm. (G) The quantitative analysis of alizarin red staining and oil red O staining. OM, osteogenic medium; CM, control medium; AM, adipogenic medium. *P< 0.05.
3.2. The m6A methylase METTL3 is upregulated with the osteogenic differentiation of hPDLSCs
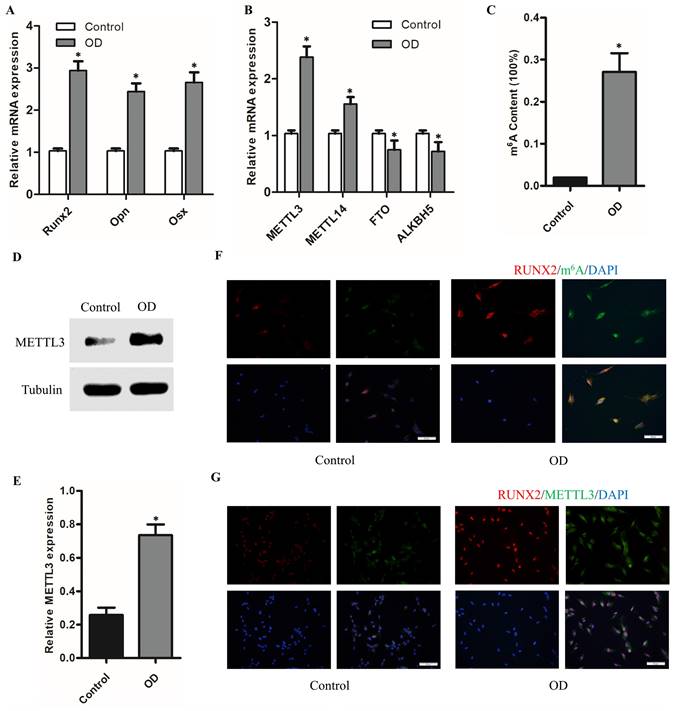
To understand the role of m6A in the osteogenic differentiation of hPDLSCs, we evaluated the expression of methylase (METTL3 and METTL14), demethylase (FTO and ALKBH5) and total methylated m6A RNA levels. As shown in the Figure 2A, the osteogenic-related mRNA level was remarkedly upregulated with the osteogenic differentiation of hPDLSCs. However, among the methyltransferase and demethylases, METTL3 level was remarkedly elevated with the osteogenic differentiation (Fig. 2B). Notably, the total m6A methylated RNA level was also increased with the osteogenic differentiation (Fig. 2C). The increased expression of METTL3 protein in hPDLSCs was further confirmed by western blotting (Figs. 2D, E). In accordance with the above results, the immunofluorescence (IF) assays results showed that m6A contents increased significantly with the up-regulation of RUNX2 in osteogenic-differentiation hPDLSCs (Fig. 2F). Moreover, there was a significant increase in methylase METTL3 correlated with m6A contents in OD group as compared with control group (Fig. 2G). Intriguingly, we also found that part of the METTL3 protein was transferred from the nucleus to the cytoplasm (Fig. 2G). These results evidenced that METTL3 may be involved in the osteogenic differentiation of hPDLSCs via m6A modification.
Increased m6A modification level together with the elevated expression of methylase METTL3 during the osteogenic differentiation of hPDLSCs. (A) The osteogenic-related genes level was detected by qPCR after osteogenic differentiation of hPDLSCs. (B) The expression of methylase (METTL3 and METTL14) and demethylase (FTO and ALKBH5) was detected by qPCR. (C) Quantification of m6A RNA methylation was determined by m6A ELISA kit. (D) The expression of methylase METTL3 was detected by Western Blotting. (E) The quantitative analysis of METTL3 expression. (F) The RUNX2 and m6A level was measured by immunofluorescent staining. Scale bars, 50 μm. (G) Immunofluorescent staining assay indicated that the protein levels of RUNX2 and METTL3 were elevated during the osteogenic differentiation of hPDLSCs. Scale bars, 50 μm. OD, osteogenic differentiation; *P< 0.05.
3.3 METTL3 promote osteogenic differentiation of hPDLSCs by modulating Runx2 mRNA stability
To verify the function of METTL3 in osteogenic differentiation of hPDLSCs, we use lentivirus to conduct gain-of-function and loss-of-function studies in hPDLSCs. As shown in Fig. 3A-B, compared to the NC group, the protein level of METTL3 was dramatically decreased in sh-METTL3 groups, and obviously increased in ov-METTL3 groups. Spontaneously, the m6A methylated mRNA level was repressed in sh-METTL3 groups, and elevated in ov-METTL3 groups (Fig. 3C). Moreover, alizarin red staining (ARS) was performed to investigate the osteogenic function of METTL3 in hPDLSCs. The results revealed that METTL3 knockdown markedly inhibited the osteogenic differentiation of hPDLSCs, while overexpression of METTL3 induced the opposite effects (Fig. 3D, E). In addition, the qPCR results showed that knocking down METTL3 impaired the expression of the osteogenic biomarkers (Runx2 and Opn), and vice versa (Fig. 3F). The protein level of osteogenic biomarkers was detected by Western Blotting, which was consistent with the results of qPCR and Alizarin Red (Fig. 3G, H). Taken together, these findings suggest that METTL3 promotes osteogenic differentiation of hPDLSCs.
METTL3 promotes osteogenic differentiation of hPDLSCs via increasing Runx2 RNA stability. (A) Western blot analysis of the expression of METTL3 in hPDLSCs after transfection with negative control (NC), METTL3 shRNA or METTL3. (B) Quantitative analysis of the expression of METTL3 in A. (C) The m6A mRNA methylation was determined by m6A dot blot. (D) The results of alizarin red staining in hPDLSCs that were treated as described above. (E) Quantitation of ARS absorbance measurement was performed. (F) The osteogenic biomarkers expression was detected by qPCR. (G) The expression of METTL3 and osteogenic biomarkers was detected by Western Blotting. (H) The quantitative analysis of Western Blotting in G. (I) The osteogenic biomarkers methylated m6A level was measured by MeRIP RT-PCR. (J) The Runx2 methylated m6A level was detected by MeRIP-PCR. (K) The RNA stability assay was initiated by adding actinomycin D (an RNA polymerase II inhibitor, 5 µg/mL). The mRNA level of Runx2 was analyzed by qPCR in Actinomycin D treated the indicated time points (0, 3, 6 h). *P< 0.05.
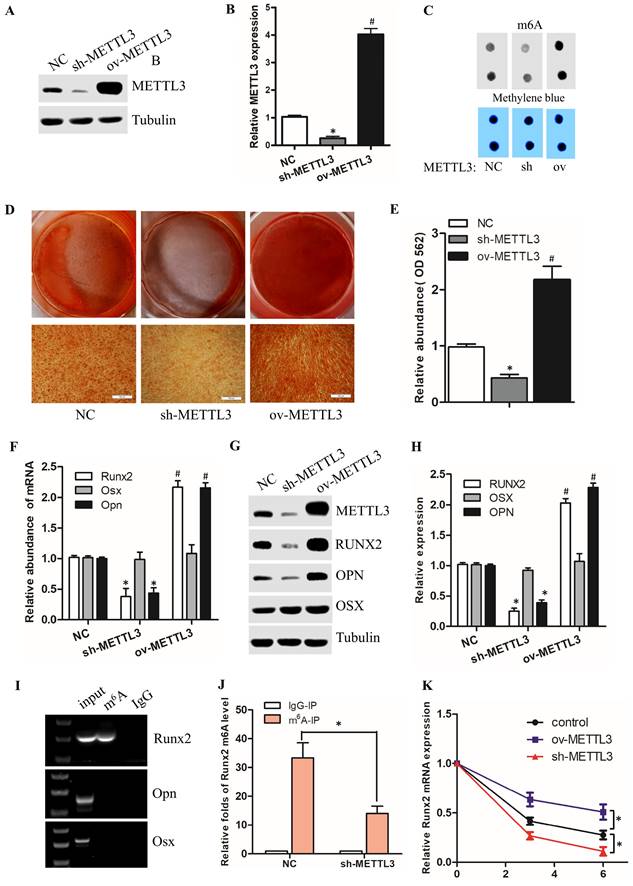
To identify the underlying mechanisms of METTL3 modulate osteogenic differentiation, we analyzed the methylated m6A level of osteogenic markers (Runx2, Opn, and Osx). The m6A-RIP RT-PCR revealed that among the above 3 osteogenic biomarkers, only Runx2 had m6A methylation modification in hPDLSCs (Fig. 3I). Moreover, we also found that the Runx2 methylated m6A level was inhibited in sh-METTL3 group compared to the control group (Fig. 3J). The above result showed that the mRNA and protein levels of Runx2 was both dramatically up-regulated in METTL3 overexpressed hPDLSCs (Fig. 3G-H). Based on the positive correlation between Runx2 and METTL3, we hypothesize that METTL3 promotes the osteogenic differentiation of hPDLSCs due to the increased stability of Runx2 mRNA. To accurately evaluate the stability of Runx2 mRNA associated with METTL3, we determined mRNA decay in actinomycin D (Act D)-treated hPDLSCs by qPCR. We observed that the Runx2 mRNA was decreased or increased in ActD-treated hPDLSCs after knock down or overexpression of METTL3, respectively, compared with the control vectors. Taken together, the data suggested that METTL3 promotes osteogenic differentiation of hPDLSCs via increasing Runx2 RNA stability.
3.4. METTL3 regulate Runx2 mRNA stability via IGF2BP1 involvement
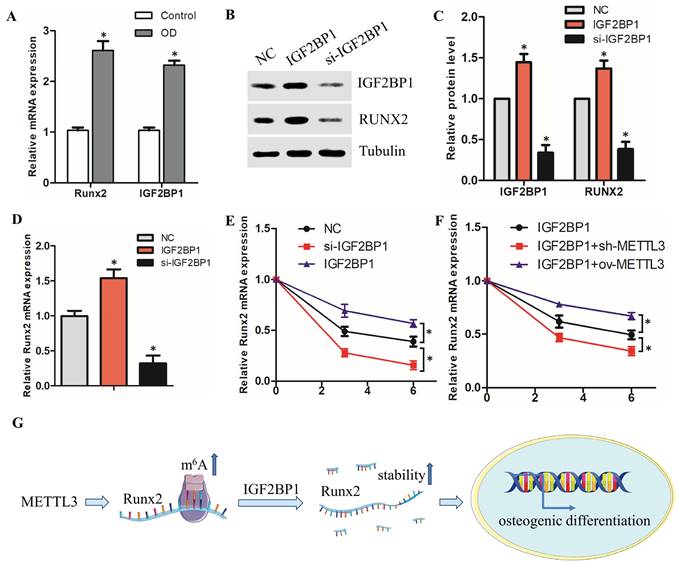
Recent studies have shown that m6A mediates mRNA fates by promoting the stability of mRNA via IGF2BP1[14-17]. We therefore investigated whether and how the IGF2BP1 protein in hPDLSCs, which recognize the m6A modification to regulate Runx2 mRNA stability. Firstly, we found that the IGF2BP1 mRNA level was remarkedly upregulated with the osteogenic differentiation of hPDLSCs (Fig. 4A). The positive correlation between Runx2 and IGF2BP1 suggests that IGF2BP1 may mediate the decay of Runx2 mRNA. To investigate the function of IGF2BP1 in regulating Runx2 mRNA stability, we knocked down IGF2BP1 using si-IGF2BP1 or overexpressed the protein with a plasmid (Fig. 4B). IGF2BP1 overexpression increased, while knocking down of IGF2BP1 decreased, the mRNA and protein levels of Runx2 in hPDLSCs, as analyzed by qPCR and Western Blotting (Fig. 4B-D). The RNA stability assay revealed that the Runx2 RNA degradation rates after transcription inhibition was dramatically decreased or increased after overexpression or knock down of IGF2BP1, respectively (Fig. 4E). Furthermore, IGF2BP1-associated Runx2 mRNA stability was decreased with sh-METTL3 cotransfection in hPDLSCs, but stability of Runx2 mRNA was increased with METTL3 overexpression (Fig. 4F). These observations revealed that IGF2BP1-facilitated Runx2 mRNA stability is regulated by METTL3-mediated m6A modification (Fig. 4G).
METTL3 regulate Runx2 mRNA stability via IGF2BP1 involvement. (A) The expression of Runx2 and IGF2BP1 was detected by qPCR after osteogenic differentiation of hPDLSCs. (B, C) Western blot analysis of the expression of IGF2BP1 and RUNX2 in hPDLSCs after transfection with negative control (NC), IGF2BP1 siRNA or IGF2BP1 plasmid. (D) The expression of Runx2 was detected by qPCR in hPDLSCs that were treated as described above. (E) The mRNA level of Runx2 was analyzed by qPCR in Actinomycin D (5 µg/mL) treated the indicated time points (0, 3, 6 h). (F) The RNA stability assay was performed to analyze the mRNA level of Runx2 by qPCR in Actinomycin D (5 µg/mL) treated cells with transfected with indicated genes. (G) Schematic illustration of this study. METTL3 promotes osteogenic differentiation of human periodontal ligament stem cells through mediating Runx2 stability via IGF2BP1-mediated m6A modification. OD, osteogenic differentiation; *P< 0.05.
4. Discussion
The treatment of periodontitis aims at promoting the regeneration of alveolar bone tissue as much as possible after plaque removal[22]. Osteogenic differentiation of hPDLSCs is an important process of alveolar bone formation[23]. However, the amount of hPDLSCs in periodontal tissue is very limited. Identifying novel methods to improve the osteogenic differentiation ability of hPDLSCs is a key issue in periodontal tissue engineering[24]. In the current study, we clarified the molecular regulatory mechanism of m6A modification and function of Runx2, regulated by METTL3 and YTHDFs proteins, in the regulation of hPDLSCs osteogenic differentiation. The presented findings indicate that m6A modification in hPDLSCs is a novel target for a potential periodontitis therapy.
m6A is a conserved post-transcriptional modification with broad functional effects on homeostasis, accounts for more than 60% of all RNA modifications in eukaryotes, and its disturbance may lead to dysfunction or disease[25, 26]. The dynamic modification of m6A is regulated by methylases and demethylases. Knockout of the methylase Mettl3 has been reported to lead to increased osteoporosis in mouse[27]. However, prior to the present study, it was unclear whether the m6A methylase METTL3 exerted effects on the osteogenic differentiation of hPDLSCs. For the first time, we found that the methylases METTL3 and m6A increased synchronously with the osteogenic differentiation of hPDLSCs. Furthermore, overexpression of METTL3 upregulates Runx2 levels by methylating the transcript through m6A modification and promotes osteogenic differentiation of hPDLSCs, and vice versa (Figure 3). It is worth mentioning that we also silenced FTO or ALKBH5 by siRNA, both of which could slightly promote Runx2 expression (data not shown). Given that FTO, ALKBH5 and METTL3, METTL14 are the two ends of the m6A "seesaw", they further verified the function of METTL3 in promoting osteogenic differentiation of hPDLSCs.
The m6A modification requires interaction with binding proteins to exert biological functions[28]. It has been previously reported that YTHDF1 binding proteins promotes FZD5 mRNA translation in an m6A-dependent manner to facilitates the progression of hepatocellular carcinoma[29]. Zhang et al. revealed that silencing METTL3 inhibits osteoblast differentiation of MC3T3-E1 cells by stabilizing Smurf1 and Smad7 mRNA transcripts via YTHDF2 involvement[30]. However, it also remains elusive whether these binding proteins function in the osteogenic differentiation of hPDLSCs. As a core transcription factor of osteoblasts, RUNX2 plays an indispensable role in osteogenic differentiation[31]. In present study, we found that METTL3 promote osteogenic differentiation of hPDLSCs via increasing Runx2 RNA stability, further observations revealed IGF2BP1-reduced Runx2 mRNA degradation is regulated by METTL3-mediated m6A modification. We also found that the methylation of Opn and Osx was not detected in periodontal ligament stem cells, suggesting that the changes in Opn expression after METTL3 knockdown may be regulated by the RUNX2 transcription factor. Our observations indicate that METTL3 regulates the degradation efficiency of Runx2 mRNA through IGF2BP1, thereby clarifying the regulatory molecular mechanism of Runx2 m6A modification involved in the osteogenic differentiation of hPDLSCs from a novel perspective.
Collectively, our study clarified that METTL3 promotes the osteogenic differentiation of hPDLSCs in an m6A-dependent manner, acts at least partially by facilitating Runx2 expression through mechanisms with IGF2BP1-mediated Runx2 mRNA stability (Fig. 4G). These findings provide a novel perspective for effective therapeutic strategies for periodontitis.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (82102221) and Natural Science Basic Research Program of Shaanxi Province (212275386003).
Author contributions
Xuefei Sun: Designed the experiments and writing original draft; Funding acquisition. Xiujiao Meng: Methodology; Visualization and Performed the experiments. Yu Piao: Advised on the experimental design and edited the manuscript. Shaojie Dong: Conceived the experiments and Funding acquisition. Qianqian Dong: Conceptualization, Project administration. All authors have read and agreed to the published version of the manuscript.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang C, Xue P, Ke J, Cai Q. Development of Ferroptosis-Associated ceRNA Network in Periodontitis. Int Dent J. 2023;73:186-94
2. Slots J. Periodontitis: facts, fallacies and the future. Periodontol 2000. 2017;75:7-23
3. Hajishengallis G. New developments in neutrophil biology and periodontitis. Periodontol 2000. 2020;82:78-92
4. Xiang L, Zhang X, Yu H, Wang B, Lin Z, Gong P. Overexpression of αCGRP promotes osteogenesis of periodontal ligament cells by regulation of YAP signaling. J Cell Physiol. 2019;234:5077-85
5. Purwaningrum M, Giachelli CM, Osathanon T, Rattanapuchpong S, Sawangmake C. Dissecting specific Wnt components governing osteogenic differentiation potential by human periodontal ligament stem cells through interleukin-6. Sci Rep. 2023;13:9055
6. Zhao Y, Zhai Q, Liu H, Xi X, Chen S, Liu D. TRIM16 Promotes Osteogenic Differentiation of Human Periodontal Ligament Stem Cells by Modulating CHIP-Mediated Degradation of RUNX2. Frontiers in cell and developmental biology. 2020;8:625105
7. Dong T, Sun X, Jin H. Role of YAP1 gene in proliferation, osteogenic differentiation, and apoptosis of human periodontal ligament stem cells induced by TNF-α. J Periodontol. 2021;92:1192-200
8. Pan Y, Liu Y, Cui D, Yu S, Zhou Y, Zhou X. et al. METTL3 enhances dentinogenesis differentiation of dental pulp stem cells via increasing GDF6 and STC1 mRNA stability. BMC Oral Health. 2023;23:209
9. Wu S, He G, Liu S, Cao Y, Geng C, Pan H. Identification and validation of the N6-methyladenosine RNA methylation regulator ZC3H13 as a novel prognostic marker and potential target for hepatocellular carcinoma. Int J Med Sci. 2022;19:618-30
10. Xu J, Qi Y, Ju Q. Promotion of the resistance of human oral epithelial cells to herpes simplex virus type I infection via N6-methyladenosine modification. BMC Oral Health. 2023;23:121
11. Garbo S, Zwergel C, Battistelli C. m6A RNA methylation and beyond - The epigenetic machinery and potential treatment options. Drug discovery today. 2021;26:2559-74
12. Sendinc E, Shi Y. RNA m6A methylation across the transcriptome. Mol Cell. 2023;83:428-41
13. Liu Y, Guo Q, Yang H, Zhang XW, Feng N, Wang JK. et al. Allosteric Regulation of IGF2BP1 as a Novel Strategy for the Activation of Tumor Immune Microenvironment. ACS central science. 2022;8:1102-15
14. Shi J, Zhang Q, Yin X, Ye J, Gao S, Chen C. et al. Stabilization of IGF2BP1 by USP10 promotes breast cancer metastasis via CPT1A in an m6A-dependent manner. Int J Biol Sci. 2023;19:449-64
15. Mao Y, Jiang F, Xu XJ, Zhou LB, Jin R, Zhuang LL. et al. Inhibition of IGF2BP1 attenuates renal injury and inflammation by alleviating m6A modifications and E2F1/MIF pathway. Int J Biol Sci. 2023;19:593-609
16. Zhang L, Wan Y, Zhang Z, Jiang Y, Gu Z, Ma X. et al. IGF2BP1 overexpression stabilizes PEG10 mRNA in an m6A-dependent manner and promotes endometrial cancer progression. Theranostics. 2021;11:1100-14
17. Zhou S, Zhang G, Wang K, Yang Z, Tan Y. METTL3 potentiates osteogenic differentiation of bone marrow mesenchymal stem cells via IGF2BP1/m6A/RUNX2. Oral Dis. 2023
18. Huang C, Wang Y. Downregulation of METTL14 improves postmenopausal osteoporosis via IGF2BP1 dependent posttranscriptional silencing of SMAD1. Cell Death Dis. 2022;13:919
19. Sun X, Li M, Ban J, Li Z. miR-23b mediates TNF-α-Inhibited Osteogenic Differentiation of Human Periodontal Ligament Stem Cells by Targeting Runx2. Int J Med Sci. 2021;18:3674-83
20. Sun X, Li Z, Dong S, Dong Q. Long non-coding RNA SNHG5 promotes osteogenic differentiation of human periodontal ligament stem cells via mediating miR-23b-3p/Runx2 axis. Int J Med Sci. 2023;20:958-68
21. Lin X, Chai G, Wu Y, Li J, Chen F, Liu J. et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nature communications. 2019;10:2065-78
22. Chen X, Arias Z, Omori K, Yamamoto T, Shinoda-Ito Y, Takashiba S. Autophagy as a potential mechanism underlying the biological effect of 1,25-Dihydroxyvitamin D3 on periodontitis: a narrative review. BMC Oral Health. 2023;23:90
23. Zhang Z, Deng M, Hao M, Tang J. Periodontal ligament stem cells in the periodontitis niche: inseparable interactions and mechanisms. J Leukoc Biol. 2021;110:565-76
24. Calabrese EJ. Human periodontal ligament stem cells and hormesis: Enhancing cell renewal and cell differentiation. Pharmacol Res. 2021;173:105914
25. Cai W, Ji Y, Han L, Zhang J, Ni Y, Cheng Y. et al. METTL3-Dependent Glycolysis Regulates Dental Pulp Stem Cell Differentiation. J Dent Res. 2021: doi:10.1177/00220345211051594.
26. Zhuang H, Yu B, Tao D, Xu X, Xu Y, Wang J. et al. The role of m6A methylation in therapy resistance in cancer. Mol Cancer. 2023;22:91
27. Wu Y, Xie L, Wang M, Xiong Q, Guo Y, Liang Y. et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nature communications. 2018;9:4772-84
28. Zaccara S, Jaffrey SR. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell. 2020;181:1582-95
29. Liu X, Qin J, Gao T, Li C, He B, Pan B. et al. YTHDF1 Facilitates the Progression of Hepatocellular Carcinoma by Promoting FZD5 mRNA Translation in an m6A-Dependent Manner. Molecular therapy Nucleic acids. 2020;22:750-65
30. Zhang Y, Gu X, Li D, Cai L, Xu Q. METTL3 Regulates Osteoblast Differentiation and Inflammatory Response via Smad Signaling and MAPK Signaling. Int J Mol Sci. 2019;21:199-214
31. Komori T. Molecular Mechanism of Runx2-Dependent Bone Development. Mol Cells. 2020;43:168-75
Author contact
![]() Corresponding author: sunxuefeiedu.cn; +86 029-87283389. qianqian2232com; +86 029 87283389.
Corresponding author: sunxuefeiedu.cn; +86 029-87283389. qianqian2232com; +86 029 87283389.