Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Molecular mechanism
3. Mitochondrial dysfunction
4. Inflammatory markers
5. Summary and outlook
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(2):284-298. doi:10.7150/ijms.88195 This issue Cite
Review
Exploring the Neuroprotective Effects of Lithium in Ischemic Stroke: A literature review
Weihua Wang1, Dunlin Lu1, Youkui Shi1, ![]() , Yanqiang Wang2,
, Yanqiang Wang2, ![]()
1. Department of Emergency, Affiliated Hospital of Weifang Medical University, Weifang, Shandong 261031, P.R. China.
2. Department of Neurology Ⅱ, Affiliated Hospital of Weifang Medical University, Weifang, Shandong 261031, P.R. China.
Received 2023-7-18; Accepted 2023-11-17; Published 2024-1-1
Abstract

Ischemic stroke ranks among the foremost clinical causes of mortality and disability, instigating neuronal degeneration, fatalities, and various sequelae. While standard treatments, such as intravenous thrombolysis and endovascular thrombectomy, prove effective, they come with limitations. Hence, there is a compelling need to develop neuroprotective agents capable of improving the functional outcomes of the nervous system. Numerous preclinical studies have demonstrated that lithium can act in multiple molecular pathways, including glycogen synthase kinase 3(GSK-3), the Wnt signaling pathway, the mitogen-activated protein kinase (MAPK)/ extracellular signal-regulated kinase (ERK) signaling pathway, brain-derived neurotrophic factor (BDNF), mammalian target of rapamycin (mTOR), and glutamate receptors. Through these pathways, lithium has been shown to affect inflammation, autophagy, apoptosis, ferroptosis, excitotoxicity, and other pathological processes, thereby improving central nervous system (CNS) damage caused by ischemic stroke. Despite these promising preclinical findings, the number of clinical trials exploring lithium's efficacy remains limited. Additional trials are imperative to thoroughly ascertain the effectiveness and safety of lithium in clinical settings. This review delineates the mechanisms underpinning lithium's neuroprotective capabilities in the context of ischemic stroke. It elucidates the intricate interplay between these mechanisms and sheds light on the involvement of mitochondrial dysfunction and inflammatory markers in the pathophysiology of ischemic stroke. Furthermore, the review offers directions for future research, thereby advancing the understanding of the potential therapeutic utility of lithium and establishing a theoretical foundation for its clinical application.
Keywords: Ischemic stroke, Neuroprotection, Lithium, Molecular mechanism, Mitochondrial dysfunction, Inflammatory marker
1. Introduction
Stroke is defined as a neurological deficit resulting from acute focal injury to the central nervous system of vascular origin [1]. According to the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD) 2019, stroke remained the second leading cause of death and the third leading cause of disability globally [2]. Stroke types are primarily classified as ischemic and hemorrhagic strokes, with diagnosis and differentiation based on clinical features and brain imaging [1]. In 2019, ischemic stroke constituted 62.4% of all newly diagnosed stroke cases, while intracerebral hemorrhage constituted 27.9% of cases worldwide [2]. Ischemic stroke, the predominant etiology among stroke types, accounts for about 87% of all cases [3].
Restoring blood flow (reperfusion) and preventing cell damage (neuroprotection) are two potential treatment strategies for ischemic stroke [4]. To restore blood perfusion, thrombolytic therapy and endovascular therapy are available [4]. Thrombolytic therapy is most effective when initiated within 4.5 hours of an acute stroke, given its narrow time window [5]. Patients with stroke episodes beyond 4.5 hours, proximal artery occlusion, and contraindications to thrombolysis are not eligible for intravenous thrombolysis [6]. In addition, intravenous tissue-type plasminogen activator (t-PA) carries a risk of symptomatic intracranial hemorrhage (sICH) [4, 7]. Endovascular therapy proves effective for acute occlusion of large vessels [8], encompassing direct injection of t-PA into the artery (arterial thrombolysis) and mechanical thrombectomy [6]. The clinical effect of arterial thrombolysis remains unclear. While thrombectomy has gained widespread use in clinical practice, it is associated with a higher rate of surgical complications [6, 9]. Mechanical thrombectomy is recommended within 6 hours from symptom onset in patients with large vessel occlusion (LVO), either in combination with intravenous thrombolysis within 4.5 hours of symptom onset or as a standalone procedure between 4.5 hours and 6 hours of symptom onset [10].
Progressive neurodegeneration and loss of function caused by stroke affect the quality of life of patients after stroke. Most patients suffer from sequelae such as cognitive impairment, movement disorders, depression, swallowing disorders, and language disorders [11]. Consequently, the quest for stroke neuroprotectors aimed at improving neurological outcomes has become a paramount focus, drawing attention from researchers worldwide.
Lithium was initially used for the treatment of urinary tract stones, but it later gained widespread acceptance for treating psychiatric disorders [12]. Lithium possesses antiviral properties [13, 14]. Preclinical evidence suggests its inhibitory effects extend to specific Coronaviridae viruses [15]. Lithium demonstrates the capacity to extend lifespan in various animal models and humans [16]. It proves beneficial for the cardiovascular system, renal function [17], bone metabolism, orthodontically induced root resorption (OIRR), and primordial follicle activation [18-20]. Furthermore, lithium shows promise in addressing conditions such as diabetes (including type 2 diabetes and the diabetogenic effects of chronic corticotherapy [21]), obesity, osteoporosis, and sarcopenia [19]. Additionally, lithium exhibits effects on microorganisms [22]. These interdisciplinary studies contribute to a more comprehensive understanding of lithium's potential applications.
Lithium exhibits its effects through rodent models [12], in vitro cellular models [23, 24], and in vivo substitution models such as Eisenia fetida [25]. Individuals with different genotypes may respond differently to lithium treatment [12, 26]. While experimental models have demonstrated the impact of lithium on ischemic stroke [27], its mechanism remains incompletely understood [12]. Furthermore, its clinical application is not well-established [27]. In this review, our emphasis is on elucidating the neuroprotective role of lithium against ischemic stroke (Figure 1). Additionally, we explore the potential of targeting mitochondria and inflammatory markers as viable therapeutic avenues for stroke.
Lithium exerts neuroprotective effects in ischemic stroke through multiple mechanisms. GSK-3: glycogen synthase kinase 3; mTOR: mammalian target of rapamycin; MAPK: mitogen-activated protein kinase; ERK: extracellular signal- regulated kinase; MMP: matrix metalloproteinase; NMDAR: N-methyl-d-aspartate receptor; FoxO3a: forkhead box O3; NLRP3: nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing protein 3; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B; CREB: cAMP-response element binding protein; BDNF: brain-derived neurotrophic factor; Trk: tropomyosin-related kinase; PSD-95: postsynaptic density protein 95; GAP-43: growth associated protein-43; BBB: blood-brain barrier. Lines with solid arrows represent stimulatory connections; lines with flattened ends represent inhibitory connections. Dashed lines represent pathways with reduced activity as a result of lithium treatment.
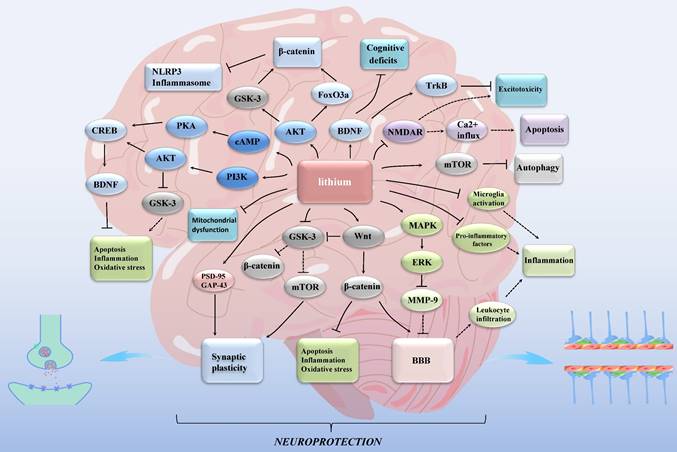
2. Molecular mechanism
2.1 GSK-3
Glycogen synthase kinase-3 (GSK-3), a serine/threonine kinase comprising α and β isoforms, represents a key target of lithium [28]. In contrast to the typical behavior of many protein kinases, GSK-3 is inherently active in unstimulated cells and experiences inhibition upon stimulation [28]. Its substrates often require an additional "trigger phosphorylation event," and GSK-3 demonstrates a broad spectrum of substrates [28], reflecting its involvement in diverse cellular processes, including cell proliferation and differentiation, cell cycle regulation, apoptosis, and autophagy [29, 30].
GSK-3 can be inhibited by lithium through direct binding to the adenosine triphosphate (ATP)-dependent magnesium-sensitive catalytic site of the enzyme. Additionally, lithium indirectly suppresses GSK-3 activity by promoting its serine phosphorylation through the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT), PI3K/protein kinase C (PKC), and cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signaling pathways [31]. Lithium enhances the serine phosphorylation of GSK-3 by disrupting the β-arrestin-2 (βArr2)-protein phosphatase 2A (PP2A)-AKT complex, which plays a role in dephosphorylating and deactivating AKT. Moreover, through the disinhibition of inhibitor-2 (I-2) inhibitory action on protein phosphatase-1 (PP-1), which is responsible for the dephosphorylation of GSK-3 at serine residues, lithium's direct inhibition of GSK-3 disrupts the auto-regulation of GSK-3 and further decreases its activity [31, 32].
Lithium inhibits IMPase to induce mTOR-independent autophagy. Simultaneously, it inhibits GSK-3β, which, in turn, reduces autophagy by activating mTOR [33, 34]. The differential effects of lithium on autophagy may be attributed to varying therapeutic concentrations [35].
The GSK-3 signaling pathway regulates autophagy by modulating downstream signaling molecules in both mTORC1-dependent and mTORC1-independent manners [36]. mTOR is a well-established major autophagy regulator, and GSK-3 interacts with mTORC1 to influence autophagy. On the one hand, GSK-3 inhibitors can down-regulate the expression of mammalian target of rapamycin complex (mTORC1), thereby inducing autophagy. On the other hand, mTORC1 regulates Foxk protein (Foxk1) through GSK-3, leading to autophagy inhibition [36, 37]. In the context of autophagy regulation, most studies have indicated that GSK-3 acts as a positive regulator of mTORC1 [28, 38], aligning with the earlier findings mentioned. This modulation of autophagy promotes the clearance of harmful substances favorable in treating neurological diseases [28]. However, some studies have suggested GSK-3 as a negative regulator of mTOR [39].
GSK-3β, the most extensively researched isoform of GSK-3 [40], is widely distributed throughout brain tissue [41]. It becomes activated during cerebral ischemia, and its activation is associated with adverse effects on post-ischemic neuronal survival [41]. The inhibition of GSK-3β plays a vital role in regulating processes such as inflammation [42], autophagy [43] (including mitophagy [44]), apoptosis [31], oxidative stress [45], excitotoxicity [31], and pyroptosis [46], thereby facilitating neuroprotection. Furthermore, the post-stroke administration of GSK-3β inhibitors enhances cognitive recovery and can mitigate recombinant tissue plasminogen activator (rt-PA)-induced hemorrhagic conversion [47, 48]. In the context of oxygen-glucose deprivation (OGD)-induced neuronal injury, GSK-3 inhibitors exhibit the potential to diminish the generation of mitochondrial reactive oxygen species (ROS), thus alleviating mitochondrial damage in neurons [31]. Consequently, GSK-3 emerges as a promising candidate for a therapeutic target in ischemic stroke.
Following hypoxia-ischemia (HI) in neonatal rats, lithium chloride induces axonal repair by inhibiting GSK-3β to activate the mTORC1 signaling pathway [49]. A recent study indicated that lithium chloride ameliorates ischemic brain injury and alleviates associated cognitive impairment by inhibiting nucleotide-binding oligomerization domain, leucine-rich repeat and nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing protein 3 (NLRP3) inflammasome activation through AKT/GSK-3β/β-catenin and AKT/forkhead box O3 (FoxO3a)/β-catenin pathways [12].
2.2 The Wnt signaling pathway
The Wnt signaling pathway is an intercellular signaling cascade activated by lipid-modifying proteins secreted by the Wnt family. The most basic form of the pathway consists of Wnt ligands from the secreting cell, their cognate receptors on the surface of the receiving cell, and signaling sensors within the receiving cell [50]. Subsequent responses elicited by the Wnt signaling pathway play an essential role in a variety of physiological aspects of an organism [51]. The Wnt signaling pathway is a complicated regulatory network consisting of two branches: canonical and non-canonical pathways [52]. The canonical Wnt signaling pathway is also known as the Wnt/β-catenin signaling pathway. The Wnt/β-catenin signaling pathway is involved in vascular development [53] and BBB formation in the brain [54]. In cerebrovascular endothelial cells, neurons, pericytes, astrocytes, microglia, and oligodendrocytes, the Wnt signaling pathway regulates cell survival and proliferation, even influencing their unique biological functions [55]. Lithium chloride activates the Wnt signaling pathway to reduce infarct volume and alleviate neurological deficits [56].
Wnt proteins can also activate signaling pathways independent of Wnt/β-catenin, mainly including Wnt/ planar cell polarity (PCP) and Wnt/Ca2+ signaling pathways, collectively referred to as non-canonical Wnt signaling pathways. Previous studies have demonstrated some synergistic [57] and antagonistic effects [58, 59] between canonical and non-canonical Wnt signaling pathways. After ischemic stroke, both canonical and non-canonical Wnt signaling pathways are down-regulated. However, the blood-brain barrier (BBB) is protected by the up-regulation of Wnt/β-catenin signaling from the interference of the non-canonical Wnt signaling pathway [60].
The activation of the typical Wnt signaling pathways leads to the inactivation of GSK-3β, resulting in the stabilization of β-catenin and facilitating its translocation to the nucleus [61]. The Wnt signaling pathways activate mTOR by inhibiting GSK-3 to regulate cell growth, independent of β-catenin-dependent transcription [39]. Furthermore, the Wnt pathways inhibit mTOR by suppressing the GSK-3/AMP-activated protein kinase (AMPK) pathway and inducing autophagy in hippocampal neurons [62]. Both of these mechanisms may be unrelated to β-catenin. However, the inhibition of GSK-3β activity by lithium chloride administration in the oxygen-glucose deprivation/reperfusion (OGD/R) model might increase the expression level of β-catenin [63, 39]. This study may elucidate the discrepancy, suggesting that the accumulation of β-catenin after Wnt signaling activation and the regulation of mTOR through GSK-3 by Wnt may involve two distinct pathways.
2.3 The MAPK/ERK signaling pathway
The MAPK pathway, playing a pivotal role in cellular processes like proliferation, differentiation, and transformation, represents an evolutionarily conserved signaling cascade [64, 65]. Comprising three primary subfamilies, including c-Jun-N-terminal kinase (JNK), p38, and ERK1/2 [66], with ERK1/2 being integral to the classical pathway within the MAPK cascade [65]. The MAPK/ERK1/2 signaling pathway is the most extensively studied among the MAPK pathways [67]. Activation of all MAPK pathways occurs in cerebral ischemia, where JNK and p38 activation can be detrimental, and ERK1/2 activation may exhibit both beneficial and harmful effects [68]. In light of these considerations, our focus now shifts to the neuroprotective role of the MAPK/ERK1/2 signaling pathway involved after ischemic stroke.
In experimental ischemic stroke studies, it is observed that lithium significantly improves the stability of the blood-brain barrier (BBB) through the activation of the MAPK/ERK1/2 pathway [69]. Inhibiting the MAPK/ERK1/2 pathway increases neuronal apoptosis and significantly decreases cellular activity after cerebral infarction in rats [70]. Additionally, both in vitro and in vivo experiments demonstrate that curcumin attenuates focal cerebral ischemia-reperfusion injury by positively regulating the MEK/ERK/cAMP-response element-binding protein (CREB) pathway [71].
The overexpression of ERK1/2 increased the infarct size and degree of neurological deficits in mice after transient middle cerebral artery occlusion (tMCAO). The stimulation of the MEK/ERK1/2 signaling proved detrimental to the functional outcome of ischemic stroke [72]. Inhibiting the MEK/ERK1/2/CREB signaling pathway may enhance BDNF expression and exert neuroprotective effects, thereby improving neurobehavioral function in rats after middle cerebral artery occlusion/reperfusion (MCAO/R) [73]. This result contradicts previous studies that suggested a neuroprotective effect of activating this pathway in ischemic stroke [69-71].
In conclusion, the MAPK/ERK1/2 pathway plays a dual role in cerebral ischemic stroke (Figure 2). On the one hand, activation of ERK1/2 inhibits apoptosis and exerts neuroprotective effects, yet also promotes inflammation. On the other hand, inhibition of ERK1/2 can alleviate post-ischemic inflammation and cellular injury by regulating downstream signaling pathways. This dual action may be related to whether the increase in ERK1/2 activity is due to injury such as stroke or caused by neuroprotective agents. Inhibition of the former-induced increase in endogenous ERK1/2 phosphorylation alleviated ischemic injury, and the increase in ERK1/2 phosphorylation induced by the latter reduces ischemic damage. The impact of additional factors, including cell surface receptor density and the extracellular matrix, may also result in distinct biological functions related to ERK1/2 activity [74, 75].
Activation and inhibition of the MEK/ERK1/2 signaling pathway in ischemic stroke. MEK: mitogen activated protein kinase; ERK1/2: extracellular signal- regulated kinase 1/2; CREB: cAMP -response element binding protein; BBB: blood-brain barrier. PI3K: phosphatidylinositol 3-kinase catalytic subunit type 3; AKT: protein kinase B. Lines with solid arrows represent stimulatory connections; lines with flattened ends represent inhibitory connections. Dashed lines represent pathways with reduced activity.
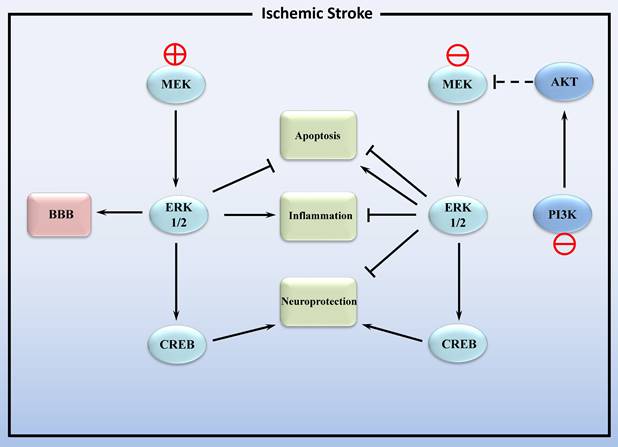
Intriguingly, a recent study demonstrated that lithium exerts its post-stroke neuroprotective activity through the PI3K/AKT pathway rather than the MEK/ERK pathway [77]. It has been reported that there is crosstalk between the PI3K/AKT and MAPK/ERK pathways in cerebral ischemia-reperfusion (Figure 2). Following cerebral ischemia, the PI3K/AKT signaling pathway is activated, leading to the inactivation of MAPK/ERK1/2. During the reperfusion process, the PI3K/AKT signaling pathway is inhibited, thereby restoring the activity of the MAPK/ERK1/2 signaling pathway [78].
Hence, it is essential to delve deeper into the role of the MAPK/ERK signaling pathway in ischemic stroke.
2.4 BDNF
Neurotrophic factors constitute a diverse family of soluble molecules implicated in various neurological functions, including cell growth, differentiation, and plasticity [78]. Among these factors, BDNF stands out as one of the extensively studied ones. BDNF, a factor with nerve growth-promoting activity, was first isolated and purified from the porcine brain in 1982 by Barde and others [79]. Neurotrophic factors exert their effects by binding to two types of transmembrane receptors: tropomyosin-related kinase (Trk) family and p75 neurotrophin receptor (p75NTR) [80]. Specifically, phosphorylated TrkB activates three major intracellular signaling pathways: the MAPK/ERK pathway, which influences cell growth and differentiation; the PI3K/AKT pathway, associated with cell survival; and the phospholipase C-γ (PLCγ)/Ca2+ pathway, which modulates synaptic plasticity [73, 81, 82]. Elevated BDNF levels regulate the PI3K/AKT pathway through TrkB, contributing to a neuroprotective role in cerebral ischemia-reperfusion injury [83].
HI significantly reduces BDNF levels in various brain regions [84]. HI induces BDNF/TrkB dysregulation, a phenomenon implicated in various neurological disorders, including ischemic stroke [85]. BDNF, through the PI3K/AKT/mTOR signaling pathway, induces autophagy, providing neuroprotection against hypoxic injury in vitro [86]. Experimental stroke models demonstrate that BDNF exerts neuroprotective effects, encompassing cognitive recovery promotion, neuroregeneration, anti-inflammatory and anti-neurotoxicity actions [85, 87]. The majority of evidence supporting the neuroprotective effects of BDNF comes from preclinical trials, but its precise role in stroke patients remains unclear [88]. Currently, there is insufficient evidence to consider BDNF as a biomarker for predicting functional outcomes in stroke [88]. Nevertheless, during the acute phase of stroke, an inverse correlation exists between stroke severity and BDNF levels [88]. Stroke patients prone to post-stroke depression (PSD) exhibit lower serum BDNF concentration levels in the early stroke phase compared to those without depression [89, 90]. The BDNF Val66Met, a common single nucleotide polymorphism (SNP) in the human BDNF gene, may impact post-stroke recovery [91].
Lithium treatment alleviates cognitive deficits associated with cerebral perfusion injury by upregulating BDNF expression [92] and activates the BDNF/TrkB pathway in cortical neurons to prevent glutamate excitotoxicity [93]. Both acute and chronic modes of lithium administration can elevate BDNF levels in the brain and reduce apoptotic levels, thereby achieving neuroprotective effects [94, 95]. The regulation of multiple genes within specific brain regions by CREB is associated with various phenomena in neuropsychiatric disorders, and BDNF serves as a major downstream regulator of CREB [96]. Lithium activates both the PI3K/AKT/CREB pathway and the cAMP/PKA/CREB pathway, leading to increased production of BDNF. This activation inhibits oxidative stress, inflammation, and apoptosis, achieving neuroprotective effects [97]. Additionally, the inhibition of GSK-3β/CREB/BDNF [98] and MAPK/ERK/CREB/BDNF [73] signaling pathways can upregulate BDNF expression, thereby alleviating cerebral ischemia-reperfusion injury.
The activation of metabotropic glutamate receptor (mGluR) 2/3 through the application of mGluR2/3 agonists increases BDNF expression in the brains of newborn mice. Therefore, considering the interaction between neurotrophic factors and the mGlu2/3 receptor could be viewed as a potential mechanism involved in neuroprotection [84].
2.5 mTOR
mTOR, a serine/threonine kinase, holds significance as a member of the phosphoinositide 3-kinase-related kinase (PIKK) family [99]. mTOR forms two distinct complexes, mTORC1, and mTORC2, through interactions with multiple chaperone proteins. The kinase activity of mTOR has been implicated in the pathogenesis of various diseases, including cancer and central nervous system disorders [100].
After the occurrence of cerebral ischemia, mTOR undergoes inhibition due to an insufficient energy supply, triggering autophagy [101]. mTOR exerts a neuroprotective effect by affecting autophagy [101-103]. Lithium chloride plays a crucial role in exerting its neuroprotective effects on ischemic stroke by activating the Wnt signaling pathways [54, 56] and inhibiting GSK-3 [31]. mTOR is regulated by both GSK-3 and the Wnt pathway [38, 39]. The activation of mTOR by lithium chloride through the Wnt/GSK-3β pathway inhibits autophagy, alleviating ischemic brain damage [63]. Furthermore, lithium chloride demonstrates the capacity to activate mTOR, restraining excessive autophagy and consequently ameliorating spatial cognitive deficits in murine models of cerebral ischemia-reperfusion [104]. In the case of neonatal rats following hypoxic-ischemic injury, lithium chloride exerts its influence through the GSK-3β/mTORC1 signaling pathway, inducing axon repair [49].
It is noteworthy to recognize that lithium's influence on mTOR extends beyond ischemic stroke. Lithium demonstrates the ability to suppress autophagy through the Wnt/GSK-3/mTOR pathway in diabetic rat myocardial cells [105]. Lithium facilitates physiological ventricular remodeling post-myocardial infarction by activating the PI3K/AKT/mTOR signaling pathway, suggesting potential implications for heart attack therapeutics. Additionally, lithium exhibits the capability to inhibit corticosteroid-induced chondrocyte autophagy [106, 107]. Methylamphetamine (MA) possesses neurotoxic properties, and lithium treatment enhances the phosphorylation of Akt/GSK-3β/mTOR pathways, thereby mitigating the detrimental effects of MA on neuronal cells [108]. Lithium treatment activates the AKT/mTOR pathway, leading to necrotic apoptosis and the demise of schwannoma cells, implying potential anti-tumor effects of lithium [109].
Ferroptosis, a form of cell death driven by iron-dependent lipid peroxidation, was identified as a distinct form of regulated cell death [110]. Ferroptosis was first proposed in 2012 by Dixon in cancer research [111], and recent evidence suggests that ferroptosis is also present in the post-ischemic brain [112, 113]. While it was once believed that ferroptosis remained impervious to other mechanisms, recent studies have contested this idea. The process of ferroptosis is not solely influenced by autophagy but is also affected by other factors, including inflammation [114, 115] and oxidative stress [116].
According to a recent report, ROS-induced autophagy promotes cellular ferroptosis [117]. Given that ROS-induced autophagy and subsequent ferroptosis may contribute to myocardial injury after a heart attack, idebenone attenuates ferroptosis by inhibiting excessive autophagy through the ROS/AMPK/mTOR pathway to preserve cardiac function post-infarction [118]. In trophoblast cells exposed to high concentrations of glucose and ferroptosis-inducing compounds, Sirtuin 3 (SIRT3) knockdown inhibits the AMPK/mTOR pathway and enhances glutathione peroxidase 4 (GPX4) levels to resist autophagy-dependent ferroptosis [119]. Ferroptosis induced by the type 3 ferroptosis inducer (Fin56) in bladder cancer cells is also autophagy-dependent, and inhibiting autophagy attenuates the degradation of ferritin and GPX4. Although Fin56 may induce autophagy independent of mTOR, the ferroptosis triggered by Fin56 can be facilitated through autophagy mediated by mTOR inhibition [120].
Notably, mTOR and GPX4 exhibit interactions: (a) mTORC1 inhibition sensitizes cancer cells or tumors to ferroptosis and acts synergistically with ferroptosis inducers to inhibit tumor growth [121], (b) mTORC1 inhibition reduces GPX4 levels [121, 122] and (c) RSL3 blocks mTOR activation [121, 123].
Based on the available studies, it can be inferred that lithium holds therapeutic potential for ferroptosis. It is worth exploring whether lithium can modulate autophagy through mTOR to inhibit ferroptosis in ischemic stroke models.
2.6 Glutamate receptors
Glutamate receptors play a crucial role in physiological processes such as memory, learning, and synaptic plasticity. These receptors can be broadly classified into two types: ionotropic and metabotropic. In the mammalian brain, three ionotropic glutamate receptors exist: the N-methyl-d-aspartate receptor (NMDAR), the α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor (AMPAR), and the kainate receptor (KAR) [124]. The conventional understanding of signal transduction by ionotropic glutamate receptors involves glutamate binding, which opens ion channels, allowing the passage of sodium, potassium, and calcium ions and generating excitatory signals [125]. Metabotropic glutamate receptors (mGluRs) are G protein-coupled receptors [126].
2.6.1 Ionic glutamate receptors
NMDAR are glutamate-gated ion channels widely expressed in the central nervous system and are crucial for neuronal communication [127]. NMDARs form tetrameric complexes composed of two obligatory GluN1 subunits along with two GluN2 or GluN3 subunits, of which there are four (GluN2A-GluN2D) and two subtypes (GluN3A and GluN3B) respectively [128]. GluN2A and GluN2B are the major subunits of functional NMDAR [129].
Recent studies have presented a paradox concerning the functional properties of NMDAR: only excessive activation of NMDAR leads to deleterious effects. In the context of ischemic brain injury, physiological activation of NMDAR is crucial for neuroplasticity and regeneration [127]. Two hypotheses, the "NMDAR subtype" and "NMDAR location," have been proposed to elucidate the dual role of NMDAR in neuronal survival and death [130].
The "NMDAR subtype" hypothesis suggests that GluN2AR promotes neuronal survival while GluN2BR induces neuronal death. The specifics of the "NMDAR location" hypothesis remain controversial: one is that activation of synaptic NMDARs facilitates survival, while activation of extra-synaptic NMDARs leads to neuronal death; the other is that activation of synaptic or extra-synaptic NMDARs alone promotes neuronal survival, while co-activation of synaptic and extra-synaptic NMDARs is responsible for stroke injury. Co-activation is necessary to induce stroke injury. Both hypotheses remain to be further confirmed [126, 130].
NMDAR-mediated excitotoxicity can also cause different forms of neuronal death during cerebral ischemia, such as autophagy, apoptosis, ferroptosis, and parthanatos [131]. The primary therapeutic approaches for managing excitotoxicity in ischemic stroke involve targeting glutamate, NMDAR, and downstream death signaling proteins [131]. However, the clinical utility of NMDAR antagonists is restricted by the considerable side effects and relatively low efficiency. In contrast, the modulation of glutamatergic transmission through mGluRs to mitigate neurological damage may represent a safer and more effective therapeutic strategy [132].
And the neuroprotective effect of lithium is related to the inhibition of NMDAR-mediated calcium inward flow and downstream signaling [31]. In addition, the synaptic protective effect of lithium is also mediated by activated NMDAR [133].
In the model of cerebral ischemia, lithium protects against excitotoxic damage by inhibiting the phosphorylation of NR2A and NR2B subunits and downregulating NMDAR [134, 135]. However, some investigators have demonstrated through ex vivo experiments that lithium chloride can increase NMDAR expression by inhibiting GSK-3β [136].
NMDAR is related to the ability of learning and memory functions [137, 138]. Prolonged lithium treatment impairs spatial memory function in rats [139], and NMDAR may contribute to the impact of lithium on inhibiting memory consolidation in rats [140]. Generally, lithium demonstrates neuroprotective effects in pathological conditions. In the process of cerebral ischemia, lithium chloride can mitigate cognitive impairment caused by cerebral ischemia by inhibiting excessive autophagy, suppressing apoptosis, and increasing BDNF expression [104, 141].
2.6.2 Metabotropic glutamate receptors
Based on sequence similarity, pharmacology, and intracellular signaling mechanisms, mGluRs are divided into group I, II, and III mGluRs. Group I mGluRs (mGluR1 and mGluR5) are associated with G proteins and coupled to phospholipase C (PLC). Group II (mGluR2 and mGluR3) and III mGluRs (mGluR4, mGluR6, mGluR7, and mGluR8) are associated with G proteins and negatively coupled to adenylate cyclase, and they are distributed in various brain regions [142, 143].
The regulation of mGluRs plays a role in various neuropsychiatric diseases, including Alzheimer's disease (AD) and Huntington's disease (HD) [144, 145]. The subsequent discussion delves into their roles in cerebral ischemia and investigates the potential involvement of lithium as a neuroprotective agent.
Group I mGluRs
mGluR1 antagonists exhibit neuroprotective effects in ischemic stroke, potentially linked to the phosphorylation of the NMDAR subunit NR2A [146, 147]. Notably, the neuroprotective efficacy of mGluR1 antagonists is comparatively weaker than that of mGluR5 antagonists [148].
The selective mGluR5 antagonist MPEP and agonist CHPG exhibit varying degrees of efficacy in alleviating ischemic brain injury in rat models [149, 150]. However, in the focal cerebral ischemia model induced by endothelin-1, the mGluR5 agonist CHPG does not enhance brain function [151]. The influence of mGluR5 on NMDAR function, including the inhibition of NR2A subunit phosphorylation, may be implicated in the neuronal cell death process in the ischemic stroke model [152, 153]. These findings underscore the controversy surrounding the role of mGluR5 in ischemic stroke, emphasizing the need for further research and clarification in this area.
Group II mGluRs
A study has demonstrated that pretreatment with agonists of mGluR2/3 reduces apoptosis levels and provides neuroprotection against brain injury induced by cerebral ischemia in neonatal rats [126]. However, another study adds evidence that the neuroprotection of mGluR2/3 agonists is mediated through the activation of mGluR3, not mGluR2 [154]. The selective mGluR2 negative allosteric modulators might alleviate ischemic neuronal death [155]. Hereditary deletion of mGluR2 in a model of transient ischemia exerts neuroprotective effects [156]. Notably, mGluRs and BDNF interact in the cerebral cortex. The gene expression of Group II mGluRs is negatively regulated by BDNF, whereas activation of group II mGluRs positively regulates BDNF expression levels [157]. In a neonatal rat model of cerebral ischemia, group II mGluR agonists also upregulate BDNF levels, raising the possibility that crosstalk between mGluR and BDNF could offer new therapeutic targets for ischemic stroke [84]. mGluR2/3 blockade induces behavioral deficits in mice, and clinically used antipsychotics such as lithium reverse the effects of mGluR2/3 antagonists [158]. Enhanced mGluR-dependent long-term depression (mGluR-LTD) at the Schaffer lateral branch of hippocampal CA1 pyramidal synapses is a feature of a mouse model of fragile X syndrome [159]. Both lithium and the group II mGluR antagonists treatment ameliorate enhanced mGluR-LTD in fragile X mice, a potential therapeutic agent for fragile X syndrome [159]. Whether acting in ischemic stroke models or not, studies in which lithium directly affects mGluR2/3 are limited and deserve further exploration.
Group III mGluRs
Earlier studies indicated increased mRNA levels of mGluR4 in the hippocampus and parietal cortex after transient ischemia [160, 161]. To investigate the role of mGluR4 in ischemic stroke, researchers utilized the middle cerebral artery occlusion (MCAO) model and the endothelin-1(Et-1) model of transient focal ischemia. They discovered that mGluR4 enhancer PHCCC administration reduced infarct size and improved sensorimotor function. In contrast, mice lacking mGluR4 exhibited greater brain damage than their wild counterparts from the same litter [162].
The group III mGluR agonist ACPT-I exerts neuroprotective effects in ischemic stroke and improves post-ischemic gait impairment [163]. ACPT-I exerts similar effects in spontaneously hypertensive rats (SHR) after MCAO/R [132, 164].
mGluR6 is not expressed in the brain but only in the retina [165]. mGluR7 activation may protect mouse neuronal cells from ischemic injury [166].
The role of lithium on metabotropic glutamate receptors is also worthy of consideration. Gq/PLC signaling is the target of lithium effects, and Gq/PLC-linked receptors include muscarinic receptors M1, 3, and 5, glutamatergic receptors mGluR1 and 5, adrenergic α1 receptors, and 5HT receptor subtype 2a-c [167].
Chronic lithium treatment of hippocampal neurons [168] and cortical neurons [169] resulted in a decrease in cell surface mGluR5 expression and a reduction in mGluR5-mediated intracellular calcium ion levels, while acute lithium treatment yielded null results. This observation may help explain the therapeutic effect of lithium in bipolar affective disorder. Altered excitatory/inhibitory synaptic transmission is also found in ischemic stroke [170]. Multiple in vitro and in vivo experiments have demonstrated that lithium can play a role in ischemic stroke models [25]. Lithium may benefit in improving clinical outcomes for stroke patients [25]. Therefore, it is worthwhile to investigate whether lithium can exert a neuroprotective role in ischemic stroke models through mGluR5-mediated effects.
3. Mitochondrial dysfunction
Mitochondrial dysfunction has been identified as one of the main mechanisms involved in cerebral ischemic injury. The relationship between mitochondrial dysfunction and ischemic stroke has been studied more in recent years [171]. Mitochondria are two-membrane-encapsulated organelles present in most eukaryotes [172]. The primary role of mitochondria is to provide energy to the cell in the form of ATP through oxidative phosphorylation via the mitochondrial electron transport chain (ETC). Physiologically, about 90% of cellular energy is provided by mitochondria through the ETC [173].
Cerebral ischemia disrupts the ultrastructure of mitochondria, leading to mitochondrial dysfunction. In this process, the impairment of the mitochondrial electron transport chain initiates, leading to a cessation of ATP production. The diminished ATP production further disrupts ion transport. As ischemia advances, ATP levels continue to decrease, culminating in the accumulation of mitochondrial ROS and calcium ions. This cascade of events ultimately results in the opening of the mitochondrial permeability transition pore (mPTP) [174]. mPTP opening has a significant effect on mitochondrial impairment [175], and it allows various solutes and water to enter the mitochondrial matrix, leading to the rupture of the outer membrane and swelling of the inner membrane, ultimately resulting in necrotic cell death [176]. Endogenous apoptosis also involves the opening of the mPTP and the release of cytochrome c (cyt c).
The NLRP3 inflammasome plays a role in the pathological process of ischemic stroke. Following mitochondrial dysfunction, the release of ROS, mitochondrial DNA (mtDNA) damage, increased cardiolipin levels, and calcium influx into the cytoplasm are also involved in activating the NLRP3 inflammasome [177].
The mitochondrial quality control system, which includes mitochondrial dynamics (fission and fusion) and mitochondrial autophagy, is responsible for maintaining mitochondrial homeostasis, improving cellular function, and mitigating post-stroke brain damage [178].
Mitochondrial fusion involves the merging of two mitochondria into a single entity, with Mitofusin1 (Mfn1) and Mfn2 mediating outer mitochondrial membrane (OMM) fusion, while optic atrophy protein 1 (Opa1) mediates inner mitochondrial membrane (IMM) fusion. Mitochondrial fission involves the division of a mitochondrion into two smaller mitochondria, with a crucial role played by dynamin-related GTPase 1 (Drp1) [179].
Mitochondrial autophagy, a form of macroautophagy, is a selective process for removing damaged or unwanted mitochondria through autophagy. Diverse molecular mechanisms of mitophagy have been identified, including the phosphatase and tensin homolog-induced kinase 1 (PINK1)-Parkin pathway, the BCL2 and adenovirus E1B19-kDa-interacting protein 3 (BNIP3)/NIX pathway, the FUN14 domain-containing 1 (FUNDC1) pathway, and the cardiolipin pathway [180]. Following a stroke, mitochondrial autophagy is inhibited [181]. Proper mitochondrial autophagy plays a neuroprotective role by eliminating damaged mitochondria. However, excessive mitochondrial autophagy can lead to the over-degradation of intracellular components. The dual role of mitochondrial autophagy is influenced by various factors, including cell type, stroke progression, and variations in upstream stimulants [171].
Interventions such as estrogen replacement therapy, fission inhibitors, fusion activators, and modulation of mitochondrial autophagy can ameliorate mitochondrial dysfunction induced by cerebral ischemia. Consequently, developing pharmaceutical agents targeting mitochondria holds promise for advancing stroke treatment [171, 182].
Lithium inhibits the release of mitochondrial cytochrome C and apoptotic factors, mitigating brain injury in the HI model [31]. Low concentrations of rotenone, a mitochondrial complex I inhibitor, induce mitochondrial dysfunction in cultured neurons in vitro. Lithium has shown potential in normalizing mitochondrial respiration and alleviating neuronal mitochondrial dysfunction by enhancing autophagy and modulating mitochondrial complex I activity [183]. The respiration of mitochondria isolated from the ischemic brain is inhibited, likely attributable to damage in the mitochondrial respiratory chain induced by ischemia. In contrast, respiratory inhibition is alleviated in mitochondria from ischemic brains treated with lithium chloride [184]. Further, in vivo experiments are needed to confirm the impact of lithium on mitochondrial dysfunction in ischemic stroke models.
4. Inflammatory markers
Inflammation plays a pivotal role in the pathogenesis of ischemic stroke. Ischemia triggers oxidative stress and excitotoxicity, leading to the activation of glial cells, which in turn secrete cytokines and matrix metalloproteinases (MMPs). Pro-inflammatory factors upregulate the expression of adhesion molecules on brain endothelial cells, facilitating the adhesion and infiltration of blood-borne leukocytes into the ischemic brain [185]. Furthermore, dying or dead cells activate the innate immune system by releasing various danger-associated molecular patterns (DAMP). Activated immune cells further stimulate glial cells by secreting pro-inflammatory factors. These pathological events lead to neuronal death and exacerbate damage in the ischemic brain [185].
Post-stroke inflammation plays a dual role in the pathophysiology of ischemic stroke. In addition to pro-inflammatory factors contributing to post-stroke brain damage, anti-inflammatory factors and cells play a role in tissue and functional recovery after cerebral ischemia [68, 186].
Potential inflammatory markers can be classified into brain-specific, non-specific, and vascular markers, holding promise for stroke diagnosis, treatment response, and patient outcomes [186]. Brain-specific inflammatory markers primarily encompass thiobarbituric acid reactive substances (TBARs), malondialdehyde (MDA), neuron-specific enolase (NSE), and heart-type fatty acid-binding protein (H-FABP). TBARs and MDA have shown predictive value for poor neurological outcomes, while NSE and H-FABP exhibit early elevation during cerebral ischemic events. Nevertheless, it is crucial to recognize that these markers possess limitations and are not currently employed in clinical practice [186]. Vascular markers predominantly encompass d-dimer, intracellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, and E-selectin. Existing evidence implies the potential of these markers to predict post-ischemic functional outcomes. However, further confirmation is required through additional studies in the field of research [186]. Non-specific inflammatory markers have been extensively investigated, mainly including C-reactive protein (CRP), interleukin (IL)-1β, IL-6, IL-10, and MMP-9 [186].
CRP, an acute-phase protein, plays a role in activating several inflammatory mechanisms [187]. Studies have identified inflammatory markers like CRP, IL-6, and IL-33 as predictors of initial stroke risk and post-stroke prognosis [188]. Low levels of the anti-inflammatory cytokine IL-10 may predict an increased risk of stroke. Pre-clinical research has suggested that IL-10-deficient mice exhibit larger lesion sizes following MCAO [68].
Some immune cells can also serve as inflammatory markers for predicting stroke outcomes. One study has elucidated the association between inflammatory markers and four outcomes, including mortality and three primary indicators defined by the National Institutes of Health Stroke Scale (NIHSS), Modified Rankin Scale (mRS), and Barthel Index (BI) scores for functional outcomes. In this context, inflammatory markers such as neutrophils, lymphocytes, CRP, and IL-6 play an important role in predicting stroke severity as per the NIHSS, disability as per the mRS, poor outcomes according to the BI, and overall mortality [189].
Cognitive impairment affects approximately half of stroke survivors. Higher concentrations of plasma inflammatory markers are associated with cognitive impairment up to 36 months after stroke [190]. In a study involving ischemic stroke patients, baseline neuropsychological assessments were conducted three months post-stroke, with annual psychological assessments over the next five years. The finding revealed that elevated IL-8 was independently associated with baseline cognitive impairment after ischemic stroke, while elevated IL-12 was linked to long-term cognitive decline [191]. Inflammatory markers (IL-6 and CRP) are associated with post-stroke cognitive impairment [192].
Post-stroke depression (PSD) emerges as the predominant psychological consequence among stroke patients [193]. Stroke-induced inflammatory responses are closely associated with the development of PSD [193]. Inflammatory markers can predict PSD, with elevated CRP levels during the acute phase indicating an increased risk [193]. IL-18 may also be involved in the development of PSD [194]. Plasma fibrinogen level upon admission can also serve as an inflammatory marker for predicting PSD [193].
Beyond their diagnostic and prognostic roles in stroke, some inflammatory markers show promise as targets for stroke treatment. Inhibiting tumor necrosis factor (TNF)-α, MMP-9, and ICAM-1 may alleviate ischemic brain damage [187].
Some inflammatory pathologies may influence inflammatory marker levels. Identifying inflammatory markers presents a formidable challenge. Definitive markers meeting clinical prediction and treatment needs for ischemic stroke have not yet been established [187, 195]. Further research is warranted to explore the potential utility of inflammatory markers for predicting functional outcomes and as targets for treating ischemic stroke.
Lithium treatment reduces microglia activation and the expression of pro-inflammatory factors following HI, promoting neuronal cell survival [196, 197]. Lithium activates the MAPK/ERK pathway, which inhibits MMP-9, leading to enhanced BBB stability and reduced leukocyte infiltration in the ischemic brain [197]. Co-administration of lithium and rutin reduces the expression of pro-inflammatory factors in a cerebral ischemia-reperfusion model [198]. Additionally, lithium inhibits the activation of NLRP3 inflammasome, mitigating cerebral ischemic injury [12].
5. Summary and outlook
The mechanism of action of lithium in ischemic stroke has been extensively studied, but its precise mechanism remains a subject of controversy. Existing studies have demonstrated lithium can affect the treatment of some psychiatric disorders. This review summarizes that lithium exerts neuroprotective effects on ischemic stroke models through multiple pathways. However, the potential role of lithium in post-stroke recovery has not been thoroughly tested in humans. The issues raised in the review may be worth further investigation:
5.1 The MAPK/ERK signaling pathway is activated after ischemia-reperfusion. However, current studies suggest that the effect of activating this pathway on stroke remains elusive.
5.2 The concept of ferroptosis is relatively recent, and exploring its association with ischemic stroke may emerge as a future research direction. Within this realm, the impact of lithium on ferroptosis warrants additional investigation.
5.3 Current research indicates that scholars globally have frequently explored ionotropic glutamate receptors in ischemic stroke. The impact of mGluRs on ischemic stroke primarily occurs through their metabotropic modulators. Further investigation is needed to determine whether drugs targeting ionotropic glutamate receptors can also influence mGluRs.
5.4 The clinical application of inflammatory markers as predictive factors for ischemic stroke requires further experimental evidence. Nonetheless, this also offers new possibilities in treating ischemic stroke, which merits further exploration.
Abbreviations
GSK-3: glycogen synthase kinase 3; MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; BDNF: brain-derived neurotrophic factor; mTOR: mammalian target of rapamycin; CNS: central nervous system; GBD: Global Burden of Diseases, Injuries, and Risk Factors Study; t-PA: tissue-type plasminogen activator; sICH: symptomatic intracranial hemorrhage; LVO: large vessel occlusion; OIRR: orthodontically induced root resorption; ATP: adenosine triphosphate; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B; PKC: protein kinase C; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; βArr2: βArr2β-arrestin-2; PP2A: protein phosphatase 2A; I-2: inhibitor-2; PP-1: protein phosphatase-1; mTORC: mammalian target of rapamycin complex; Foxk1: Foxk protein 1; OGD: oxygen-glucose deprivation; ROS: reactive oxygen species; HI: hypoxia-ischemia; rt-PA: recombinant tissue plasminogen activator; NLRP3: nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing protein 3; FoxO3a: forkhead box O3; PCP: planar cell polarity; BBB: blood-brain barrier; AMPK: AMP-activated protein kinase; OGD/R: oxygen-glucose deprivation/reoxygenation; JNK: c-Jun-N-terminal kinase; CREB: cAMP-response element binding protein; tMCAO: transient middle cerebral artery occlusion; MCAO/R: middle cerebral artery occlusion/reperfusion; Trk: tropomyosin-related kinase; PLCγ: phospholipase C-γ; p75NTR: p75 neurotrophin receptor; SNP: single nucleotide polymorphism; PSD-95: postsynaptic density protein 95; GAP-43: growth associated protein-43; mGluR: metabotropic glutamate receptor; PIKK: phosphoinositide 3-kinase-related kinase; SIRT3: Sirtuin 3; GPX4: glutathione peroxidase 4; Fin56: type 3 ferroptosis inducer; NMDAR: N-methyl-d-aspartate receptor; AMPA receptor: α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor; KA receptor: kainate receptor; mGluRs: metabotropic glutamate receptors; PLC: phospholipase C; AD: Alzheimer's disease; HD: Huntington's disease; mGluR-LTD: mGluR-dependent long-term depression; MCAO: middle cerebral artery occlusion; Et-1: endothelin-1; SHR: spontaneously hypertensive rats; ETC: electron transport chain; mPTP: mitochondrial permeability transition pore; cyt c: cytochrome c; mtDNA: mitochondrial DNA; Mfn1: mitofusin1; OMM: outer mitochondrial membrane; Opa1: optic atrophy protein 1; IMM: inner mitochondrial membrane; Drp1: dynamin-related GTPase 1; PINK1: phosphatase and tensin homolog-induced kinase 1; BNIP3: BCL2 and adenovirus E1B19-kDa-interacting protein 3; FUNDC1: FUN14 domain-containing 1; MMP: matrix metalloproteinase; DAMP: danger-associated molecular patterns; TBARs: thiobarbituric acid reactive substances; MDA: malondialdehyde; NSE: neuron-specific enolase; H-FABP: heart-type fatty acid-binding protein; ICAM: intracellular adhesion molecule; CRP: C-reactive protein; IL: interleukin; NIHSS: National Institutes of Health Stroke Scale; mRS: Modified Rankin Scale; BI: Barthel Index; PSD: post-stroke depression; TNF: tumour necrosis factor.
Acknowledgements
This work was supported by Yuan Du Scholars, Clinical Research Center of Affiliated Hospital of Weifang Medical University (No.2022WYFYLCYJ02), Weifang Key Laboratory, Yuan Du Scholars, Weifang Science and Technology Development Plan Project Medical category (No. 2022YX093).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Campbell BCV, Khatri P. Stroke. Lancet. 2020;396:129-142
2. GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021;20:795-820
3. Kleindorfer DO, Towfighi A, Chaturvedi S. et al. 2021 Guideline for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack: A Guideline From the American Heart Association/American Stroke Association. Stroke. 2021;52:e364-e467
4. Campbell B. Thrombolysis and Thrombectomy for Acute Ischemic Stroke: Strengths and Synergies. Seminars in Thrombosis and Hemostasis. 2016;43:185-190
5. Emberson J, Lees KR, Lyden P. et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet. 2014;384:1929-1935
6. Zhao Y, Zhang X, Chen X. et al. Neuronal injuries in cerebral infarction and ischemic stroke: From mechanisms to treatment (Review). Int J Mol Med. 2022;49:15
7. Kolahchi Z, Rahimian N, Momtazmanesh S. et al. Direct Mechanical Thrombectomy Versus Prior Bridging Intravenous Thrombolysis in Acute Ischemic Stroke: A Systematic Review and Meta-Analysis. Life (Basel). 2023;13:185
8. Yoshimura S, Sakai N, Uchida K. et al. Endovascular Therapy in Ischemic Stroke With Acute Large-Vessel Occlusion: Recovery by Endovascular Salvage for Cerebral Ultra-Acute Embolism Japan Registry 2. J Am Heart Assoc. 2018;7:e008796
9. Berkhemer OA, Fransen PS, Beumer D. et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372:11-20
10. Mosconi MG, Paciaroni M. Treatments in Ischemic Stroke: Current and Future. Eur Neurol. 2022;85:349-366
11. Shi H, Hu X, Leak RK. et al. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp Neurol. 2015;272:17-25
12. Chen B, Zhang M, Ji M. et al. The neuroprotective mechanism of lithium after ischaemic stroke. Commun Biol. 2022;5:105
13. Murru A, Manchia M, Hajek T. et al. Lithium's antiviral effects: a potential drug for CoViD-19 disease? Int J Bipolar Disord. 2020;8:21
14. Ferensztajn-Rochowiak E, Chłopocka-Woźniak M, Rybakowski JK. Ultra-long-term lithium therapy: all-important matters and a case of successful 50-year lithium treatment. Braz J Psychiatry. 2021;43:407-413
15. Gómez-Bernal G. Lithium for the 2019 novel coronavirus. Med Hypotheses. 2020;142:109822
16. Araldi E, Jutzeler CR, Ristow M. Lithium treatment extends human lifespan: findings from the UK Biobank. Aging (Albany NY). 2023;15:421-440
17. Fang Y, Chen B, Liu Z. et al. Age-related GSK3β overexpression drives podocyte senescence and glomerular aging. J Clin Invest. 2022;132:e141848
18. Ueda-Ichinose Y, Hotokezaka H, Miyazaki T. et al. Lithium reduces orthodontically induced root resorption by suppressing cell death, hyalinization, and odontoclast formation in rats. Angle Orthod. 2022;92:547-554
19. Li B, Wang W, Huang Y. et al. Lithium treatment promotes the activation of primordial follicles through PI3K/Akt signaling. Biol Reprod. 2022;107:1059-1071
20. Hamstra SI, Roy BD, Tiidus P. et al. Beyond its Psychiatric Use: The Benefits of Low-dose Lithium Supplementation. Curr Neuropharmacol. 2023;21:891-910
21. Delangre E, Pommier G, Tolu S. et al. Lithium treatment mitigates the diabetogenic effects of chronic cortico-therapy. Biomed Pharmacother. 2023;164:114895
22. Plotnikov E, Pukhnyarskaya D, Chernova A. Lithium and Microorganisms: Biological Effects and Mechanisms. Curr Pharm Biotechnol. 2023;24:1623-1629
23. Barbisan F, Azzolin VF, Monteiro GC. et al. Genetic or pharmacological superoxide-hydrogen peroxide imbalances modulate the in vitro effects of lithium on glycogen synthase kinase-3β. Gene. 2018;655:48-55
24. Barbisan F, Azzolin VF, Teixeira CF. et al. Xanthine-Catechin Mixture Enhances Lithium-Induced Anti-Inflammatory Response in Activated Macrophages In Vitro. Biomed Res Int. 2017;2017:4151594
25. Mastella MH, Roggia I, Turra BO. et al. The Protective Effect of Lithium Against Rotenone may be Evolutionarily Conserved: Evidence from Eisenia fetida, a Primitive Animal with a Ganglionic Brain. Neurochem Res. 2023;48:3538-3559
26. Selvarajan S, Srinivasan A, Sakkarabani P. et al. Genetic polymorphisms influencing response to lithium in early-onset Bipolar disorder from south India. Asian J Psychiatr. 2022;70:103018
27. Almeida OP, Singulani MP, Ford AH. et al. Lithium and Stroke Recovery: A Systematic Review and Meta-Analysis of Stroke Models in Rodents and Human Data. Stroke. 2022;53:2935-2944
28. Rippin I, Eldar-Finkelman H. Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration. Cells. 2021;10:262
29. Pandey MK, DeGrado TR. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics. 2016;6:571-593
30. Mancinelli R, Carpino G, Petrungaro S. et al. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid Med Cell Longev. 2017;2017:4629495
31. Chuang DM, Wang Z, Chiu CT. GSK-3 as a Target for Lithium-Induced Neuroprotection Against Excitotoxicity in Neuronal Cultures and Animal Models of Ischemic Stroke. Front Mol Neurosci. 2011;4:15
32. Chatterjee D, Beaulieu JM. Inhibition of glycogen synthase kinase 3 by lithium, a mechanism in search of specificity. Front Mol Neurosci. 2022;15:1028963
33. Yang C, Zhu B, Zhan M. et al. Lithium in Cancer Therapy: Friend or Foe? Cancers (Basel). 2023;15:1095
34. Puglisi-Allegra S, Ruggieri S, Fornai F. Translational evidence for lithium-induced brain plasticity and neuroprotection in the treatment of neuropsychiatric disorders. Transl Psychiatry. 2021;11:366
35. Damri O, Shemesh N, Agam G. Is There Justification to Treat Neurodegenerative Disorders by Repurposing Drugs?. The Case of Alzheimer's Disease, Lithium, and Autophagy. Int J Mol Sci. 2020;22:189
36. Pan HY, Valapala M. Regulation of Autophagy by the Glycogen Synthase Kinase-3 (GSK-3) Signaling Pathway. Int J Mol Sci. 2022;23:1709
37. Bowman CJ, Ayer DE, Dynlacht BD. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat Cell Biol. 2014;16:1202-1214
38. Azoulay-Alfaguter I, Elya R, Avrahami L. et al. Combined regulation of mTORC1 and lysosomal acidification by GSK-3 suppresses autophagy and contributes to cancer cell growth. Oncogene. 2015;34:4613-4623
39. Inoki K, Ouyang H, Zhu T. et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955-968
40. Bertrand FE. The cross-talk of NOTCH and GSK-3 signaling in colon and other cancers. Biochim Biophys Acta Mol Cell Res. 2020;1867:118738
41. Chen Y, Wu Z, Zhu X. et al. OCT4B-190 protects against ischemic stroke by modulating GSK-3β/HDAC6. Exp Neurol. 2019;316:52-62
42. Wang Y, Meng C, Zhang J. et al. Inhibition of GSK-3β alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int Immunopharmacol. 2019;68:234-241
43. Zhang Y, Wu Z, Huang Z. et al. GSK-3β inhibition elicits a neuroprotection by restoring lysosomal dysfunction in neurons via facilitation of TFEB nuclear translocation after ischemic stroke. Brain Res. 2022;1778:147768
44. Ali M, Tabassum H, Alam MM. et al. N-acetyl-L-cysteine ameliorates mitochondrial dysfunction in ischemia/reperfusion injury via attenuating Drp-1 mediated mitochondrial autophagy. Life Sci. 2022;293:120338
45. Li Y, Zhu J, Liu Y. et al. Glycogen Synthase Kinase 3β Influences Injury Following Cerebral Ischemia/Reperfusion in Rats. Int J Biol Sci. 2016;12:518-531
46. Diao MY, Zhu Y, Yang J. et al. Hypothermia protects neurons against ischemia/reperfusion-induced pyroptosis via m6A-mediated activation of PTEN and the PI3K/Akt/GSK-3β signaling pathway. Brain Res Bull. 2020;159:25-31
47. Venna VR, Benashski SE, Chauhan A. et al. Inhibition of glycogen synthase kinase-3β enhances cognitive recovery after stroke: the role of TAK1. Learn Mem. 2015;22:336-343
48. Wang W, Li M, Wang Y. et al. GSK-3beta inhibitor TWS119 attenuates rtPA-induced hemorrhagic transformation and activates the Wnt/beta-catenin signaling pathway after acute ischemic stroke in rats. Molecular neurobiology. 2016;53:7028-36
49. Xiong T, Qu Y, Wang H. et al. GSK-3β/mTORC1 Couples Synaptogenesis and Axonal Repair to Reduce Hypoxia Ischemia-Mediated Brain Injury in Neonatal Rats. J Neuropathol Exp Neurol. 2018;77:383-394
50. Rim EY, Clevers H, Nusse R. The Wnt Pathway: From Signaling Mechanisms to Synthetic Modulators. Annu Rev Biochem. 2022;91:571-598
51. Hayat R, Manzoor M, Hussain A. Wnt signaling pathway: A comprehensive review. Cell Biol Int. 2022;46:863-877
52. Nusse R. Wnt signaling. Cold Spring Harb Perspect Biol. 2012;4:a011163
53. Daneman R, Agalliu D, Zhou L. et al. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:641-646
54. Ji YB, Gao Q, Tan XX. et al. Lithium alleviates blood-brain barrier breakdown after cerebral ischemia and reperfusion by upregulating endothelial Wnt/β-catenin signaling in mice. Neuropharmacology. 2021;186:108474
55. Mo Z, Zeng Z, Liu Y. et al. Activation of Wnt/Beta-Catenin Signaling Pathway as a Promising Therapeutic Candidate for Cerebral Ischemia/Reperfusion Injury. Frontiers in Pharmacology. 2022;13:914537
56. Junde Z, Tingting L, Lu Z. et al. Lithium chloride promotes neural functional recovery after local cerebral ischaemia injury in rats through Wnt signalling pathway activation. Folia Morphol (Warsz). 2023;82:519-532
57. De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochimica et Biophysica Sinica. 2011;43:745-756
58. Kühl M, Geis K, Sheldahl L C. et al. Antagonistic regulation of convergent extension movements in Xenopus by Wnt/β-catenin and Wnt/Ca2+ signaling. Mechanisms of Development. 2001;106:61-76
59. Sato A, Yamamoto H, Sakane H. et al. Wnt5a regulates distinct signalling pathways by binding to Frizzled2. The EMBO Journal. 2010;29:41
60. Ji Y, Wang T, Gao Q. et al. Normalization of non-canonical Wnt signalings does not compromise blood-brain barrier protection conferred by upregulating endothelial Wnt/β-catenin signaling following ischemic stroke. CNS Neuroscience & Therapeutics. 2021;27:1085-1096
61. Libro R, Bramanti P, Mazzon E. The role of the Wnt canonical signaling in neurodegenerative diseases. Life Sciences. 2016;158:78-88
62. Ríos JA, Godoy JA, Inestrosa NC. Wnt3a ligand facilitates autophagy in hippocampal neurons by modulating a novel GSK-3β-AMPK axis. Cell Communication and Signaling : CCS. 2018;16:15
63. Chen C, Yu Q, Xu K. et al. Electroacupuncture pretreatment prevents ischemic stroke and inhibits Wnt signaling-mediated autophagy through the regulation of GSK-3β phosphorylation. Brain Research Bulletin. 2020;158:90-98
64. Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3-38
65. Zheng Y, Han Z, Zhao H. et al. MAPK: A Key Player in the Development and Progression of Stroke. CNS Neurol Disord Drug Targets. 2020;19:248-256
66. Slattery ML, Mullany LE, Sakoda LC. et al. The MAPK-Signaling Pathway in Colorectal Cancer: Dysregulated Genes and Their Association With MicroRNAs. Cancer Inform. 2018;17:1176935118766522
67. Barbosa R, Acevedo LA, Marmorstein R. The MEK/ERK Network as a Therapeutic Target in Human Cancer. Mol Cancer Res. 2021;19:361-374
68. Jin R, Liu L, Zhang S. et al. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res. 2013;6:834-85
69. Haupt M, Zechmeister B, Bosche B. et al. Lithium enhances post-stroke blood-brain barrier integrity, activates the MAPK/ERK1/2 pathway and alters immune cell migration in mice. Neuropharmacology. 2020;181:108357
70. Wang QC, Lu L, Zhou HJ. Relationship between the MAPK/ERK pathway and neurocyte apoptosis after cerebral infarction in rats. European Review for Medical and Pharmacological Sciences. 2019;23:5374-5381
71. Xu L, Ding L, Su Y. et al. Neuroprotective effects of curcumin against rats with focal cerebral ischemia-reperfusion injury. International Journal of Molecular Medicine. 2019;43:1879-1887
72. Schanbacher C, Bieber M, Reinders Y. et al. ERK1/2 Activity Is Critical for the Outcome of Ischemic Stroke. International Journal of Molecular Sciences. 2022;23:706
73. Ren Y, Ma X, Wang T. et al. The Cerebroprotein Hydrolysate-I Plays a Neuroprotective Effect on Cerebral Ischemic Stroke by Inhibiting MEK/ERK1/2 Signaling Pathway in Rats. Neuropsychiatric Disease and Treatment. 2021;17:2199-2208
74. Sawe N, Steinberg G, Zhao H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. Journal of Neuroscience Research. 2008;86:1659-1669
75. Kong T, Liu M, Ji B. et al. Role of the Extracellular Signal-Regulated Kinase 1/2 Signaling Pathway in Ischemia-Reperfusion Injury. Frontiers in Physiology. 2019;10:1038
76. Ates N, Caglayan A, Balcikanli Z. et al. Phosphorylation of PI3K/Akt at Thr308, but not phosphorylation of MAPK kinase, mediates lithium-induced neuroprotection against cerebral ischemia in mice. Experimental Neurology. 2022;351:113996
77. Zhou J, Du T, Li B. et al. Crosstalk Between MAPK/ERK and PI3K/AKT Signal Pathways During Brain Ischemia/Reperfusion. ASN NEURO. 2015;7:1759091415602463
78. Sato C. Releasing Mechanism of Neurotrophic Factors via Polysialic Acid. Vitamins and hormones. 2017;104:89-112
79. Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. The EMBO Journal. 1982;1:549-553
80. Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nature Reviews Neuroscience. 2005;6:603-614
81. von Bohlen und Halbach O, von Bohlen und Halbach V. BDNF effects on dendritic spine morphology and hippocampal function. Cell and Tissue Research. 2018;373:729-741
82. Numakawa T, Suzuki S, Kumamaru E. et al. BDNF function and intracellular signaling in neurons. Histology and Histopathology. 2010;25:237-258
83. Li Y, Xiang L, Wang C. et al. Protection against acute cerebral ischemia/reperfusion injury by Leonuri Herba Total Alkali via modulation of BDNF-TrKB-PI3K/Akt signaling pathway in rats. Biomedicine & Pharmacotherapy. 2021;133:111021
84. Bratek-Gerej E, Ziembowicz A, Salinska E. Group II Metabotropic Glutamate Receptors Reduce Apoptosis and Regulate BDNF and GDNF Levels in Hypoxic-Ischemic Injury in Neonatal Rats. International Journal of Molecular Sciences. 2022;23:7000
85. Chen A, Xiong LJ, Tong Y. et al. The neuroprotective roles of BDNF in hypoxic ischemic brain injury. Biomed Rep. 2013;1:167-176
86. Chen A, Xiong LJ, Tong Y. et al. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol Med Rep. 2013;8:1011-1016
87. Lau D, Bengtson CP, Buchthal B. et al. BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin A. Cell Rep. 2015;12:1353-1366
88. Karantali E, Kazis D, Papavasileiou V. et al. Serum BDNF Levels in Acute Stroke: A Systematic Review and Meta-Analysis. Medicina (Kaunas). 2021;57:297
89. Zhang E, Liao P. Brain-derived neurotrophic factor and post-stroke depression. J Neurosci Res. 2020;98:537-548
90. Xu HB, Xu YH, He Y. et al. Decreased Serum Brain-Derived Neurotrophic Factor May Indicate the Development of Poststroke Depression in Patients with Acute Ischemic Stroke: A Meta-Analysis. J Stroke Cerebrovasc Dis. 2018;27:709-715
91. Balkaya M, Cho S. Genetics of stroke recovery: BDNF val66met polymorphism in stroke recovery and its interaction with aging. Neurobiol Dis. 2019;126:36-46
92. Taliyan R, Ramagiri S. Delayed neuroprotection against cerebral ischemia reperfusion injury: putative role of BDNF and GSK-3β. J Recept Signal Transduct Res. 2016;36:402-410
93. Chiu CT, Chuang DM. Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol Ther. 2010;128:281-304
94. Bannova A V, Menshanov P N, Dygalo N N. The Effect of Lithium Chloride on the Levels of Brain-Derived Neurotrophic Factor in the Neonatal Brain. Neurochemical Journal. 2019;13:344-348
95. Tai S H, Huang S Y, Chao L C. et al. Lithium upregulates growth-associated protein-43 (GAP-43) and postsynaptic density-95 (PSD-95) in cultured neurons exposed to oxygen-glucose deprivation and improves electrophysiological outcomes in rats subjected to transient focal cerebral ischemia following a long-term recovery period. Neurological Research. 2022;44:870-878
96. Carlezonjr W, Duman R, Nestler E. The many faces of CREB. Trends in Neurosciences. 2005;28:436-445
97. Ghanaatfar F, Ghanaatfar A, Isapour P. et al. Is lithium neuroprotective?. An updated mechanistic illustrated review. Fundam Clin Pharmacol. 2023;37:4-30
98. Ramagiri S, Taliyan R. Remote limb ischemic post conditioning during early reperfusion alleviates cerebral ischemic reperfusion injury via GSK-3β/CREB/ BDNF pathway. European Journal of Pharmacology. 2017;803:84-93
99. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25-32
100. Mao B, Zhang Q, Ma L. et al. Overview of Research into mTOR Inhibitors. Molecules. 2022;27:5295
101. Hou W, Hao Y, Sun L. et al. The dual roles of autophagy and the GPCRs-mediating autophagy signaling pathway after cerebral ischemic stroke. Molecular Brain. 2022;15:14
102. Motoi Y, Shimada K, Ishiguro K. et al. Lithium and Autophagy. ACS Chemical Neuroscience. 2014;5:434-442
103. Sarkar S, Krishna G, Imarisio S. et al. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Human Molecular Genetics. 2008;17:170-178
104. Xiao Y, Fan M, Jin W. et al. Lithium chloride ameliorated spatial cognitive impairment through activating mTOR phosphorylation and inhibiting excessive autophagy in the repeated cerebral ischemia-reperfusion mouse model. Experimental and Therapeutic Medicine. 2020;20:109
105. Wei H, Qu H, Wang H. et al. 1,25-Dihydroxyvitamin-D3 prevents the development of diabetic cardiomyopathy in type 1 diabetic rats by enhancing autophagy via inhibiting the β-catenin/TCF4/GSK-3β/mTOR pathway. The Journal of Steroid Biochemistry and Molecular Biology. 2017;168:71-90
106. Lee TM, Lin SZ, Chang NC. Effect of lithium on ventricular remodelling in infarcted rats via the Akt/mTOR signalling pathways. Biosci Rep. 2017;37:BSR20160257
107. Wang Q, Zhang W, Hu J. et al. Lithium prevents glucocorticoid-induced chondrocyte autophagy: An in vitro study. Mol Med Rep. 2023;28:183
108. Wu J, Zhu D, Zhang J. et al. Lithium protects against methamphetamine-induced neurotoxicity in PC12 cells via Akt/GSK3β/mTOR pathway. Biochem Biophys Res Commun. 2015;465:368-373
109. Wang Y, Zhang Q, Wang B. et al. LiCl Treatment Induces Programmed Cell Death of Schwannoma Cells through AKT- and MTOR-Mediated Necroptosis. Neurochem Res. 2017;42:2363-2371
110. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401-2421
111. Dixon S J, Lemberg K M, Lamprecht M R. et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell. 2012;149:1060-1072
112. Hanke N, Rami A. Cerebral Ischemia Induces Iron Deposit, Ferritin Accumulation, NuclearReceptor Coactivator 4-depletion, and Ferroptosis. Current Neurovascular Research. 2022;19:47-60
113. Tuo Q, Zhang S, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Medicinal Research Reviews. 2022;42:259-305
114. Wang F, He J, Xing R. et al. Molecular mechanisms of ferroptosis and their role in inflammation. International Reviews of Immunology. 2023;42:71-81
115. Wang C, Chen S, Guo H. et al. Forsythoside A Mitigates Alzheimer's-like Pathology by Inhibiting Ferroptosis-mediated Neuroinflammation via Nrf2/GPX4 Axis Activation. International Journal of Biological Sciences. 2022;18:2075-2090
116. Ren JX, Li C, Yan XL. et al. Crosstalk between Oxidative Stress and Ferroptosis/Oxytosis in Ischemic Stroke: Possible Targets and Molecular Mechanisms. Oxidative Medicine and Cellular Longevity. 2021;2021:6643382
117. Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death & Disease. 2019;10:822
118. Li D, Zhang G, Wang Z. et al. Idebenone attenuates ferroptosis by inhibiting excessive autophagy via the ROS-AMPK-mTOR pathway to preserve cardiac function after myocardial infarction. European Journal of Pharmacology. 2023;943:175569
119. Han D, Jiang L, Gu X. et al. SIRT3 deficiency is resistant to autophagy-dependent ferroptosis by inhibiting the AMPK/mTOR pathway and promoting GPX4 levels. Journal of Cellular Physiology. 2020;235:8839-8851
120. Sun Y, Berleth N, Wu W. et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death & Disease. 2021;12:1028
121. Zhang Y, Swanda RV, Nie L. et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nature Communications. 2021;12:1589
122. Liu Y, Wang Y, Liu J. et al. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Therapy. 2021;28:55-63
123. Sekhar KR, Hanna DN, Cyr S. et al. Glutathione peroxidase 4 inhibition induces ferroptosis and mTOR pathway suppression in thyroid cancer. Scientific Reports. 2022;12:19396
124. Lai MC, Yang SN. Perinatal Hypoxic-Ischemic Encephalopathy. Journal of Biomedicine and Biotechnology. 2011;2011:609813
125. Rajani V, Sengar AS, Salter M W. Tripartite signalling by NMDA receptors. Molecular Brain. 2020;13:23
126. Bratek - Gerej E, Bronisz A, Ziembowicz A. et al. Pretreatment with mGluR2 or mGluR3 Agonists Reduces Apoptosis Induced by Hypoxia-Ischemia in Neonatal Rat Brains. Oxidative Medicine and Cellular Longevity. 2021;2021:8848015
127. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383-400
128. Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327-335
129. Zhou X, Ding Q, Chen Z. et al. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J Biol Chem. 2013;288:24151-24159
130. Ge Y, Chen W, Axerio-Cilies P. et al. NMDARs in Cell Survival and Death: Implications in Stroke Pathogenesis and Treatment. Trends in Molecular Medicine. 2020;26:533-551
131. Mao R, Zong N, Hu Y. et al. Neuronal Death Mechanisms and Therapeutic Strategy in Ischemic Stroke. Neuroscience Bulletin. 2022;38:1229-1247
132. Domin H, Przykaza Ł, Kozniewska E. et al. Neuroprotective effect of the group III mGlu receptor agonist ACPT-I after ischemic stroke in rats with essential hypertension. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2018;84:93-101
133. Hong N, Park JS, Kim HJ. Synapto-protective effect of lithium on HIV-1 Tat-induced synapse loss in rat hippocampal cultures. Animal Cells and Systems. 2022;26:1-9
134. Hashimoto R, Hough C, Nakazawa T. et al. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. Journal of Neurochemistry. 2002;80:589-597
135. Ma J, Zhang GY. Lithium reduced N-methyl-d-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neuroscience Letters. 2003;348:185-189
136. Monaco SA, Ferguson BR, Gao WJ. Lithium Inhibits GSK3β and Augments GluN2A Receptor Expression in the Prefrontal Cortex. Frontiers in Cellular Neuroscience. 2018;12:16
137. Wang M, Yang Y, Wang CJ. et al. NMDA Receptors Subserve Persistent Neuronal Firing During Working Memory In Dorsolateral Prefrontal Cortex. Neuron. 2013;77:736-749
138. Cui Y, Jin J, Zhang X. et al. Forebrain NR2B Overexpression Facilitating the Prefrontal Cortex Long-Term Potentiation and Enhancing Working Memory Function in Mice. PLoS ONE. 2011;6:e20312
139. Yousef M, Kavraal Ş, Artış AS. et al. Effects of Chronic and Acute Lithium Treatment on the Long-term Potentiation and Spatial Memory in Adult Rats. Clinical Psychopharmacology and Neuroscience. 2019;17:233-243
140. Amiri S, Jafari-Sabet M, Keyhanfar F. et al. Hippocampal and prefrontal cortical NMDA receptors mediate the interactive effects of olanzapine and lithium in memory retention in rats: the involvement of CAMKII-CREB signaling pathways. Psychopharmacology. 2020;237:1383-1396
141. Fan M, Jin W, Zhao H. et al. Lithium chloride administration prevents spatial learning and memory impairment in repeated cerebral ischemia-reperfusion mice by depressing apoptosis and increasing BDNF expression in hippocampus. Behavioural Brain Research. 2015;291:399-406
142. Levite M. Glutamate receptor antibodies in neurological diseases. Journal of Neural Transmission. 2014;121:1029-1075
143. Vahidinia Z, Joghataei MT, Beyer C. et al. G-Protein-Coupled Receptors and Ischemic Stroke: a Focus on Molecular Function and Therapeutic Potential. Molecular Neurobiology. 2021;58:4588-4614
144. Crupi R, Impellizzeri D, Cuzzocrea S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Frontiers in Molecular Neuroscience. 2019;12:20
145. Ribeiro F M, Vieira LB, Pires RGW. et al. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacological Research. 2017;115:179-191
146. Kohara A, Takahashi M, Yatsugi S ichi. et al. Neuroprotective effects of the selective type 1 metabotropic glutamate receptor antagonist YM-202074 in rat stroke models. Brain Research. 2008;1191:168-179
147. Murotomi K, Takagi N, Takayanagi G. et al. mGluR1 antagonist decreases tyrosine phosphorylation of NMDA receptor and attenuates infarct size after transient focal cerebral ischemia. Journal of Neurochemistry. 2008;105:1625-1634
148. Szydlowska K, Kaminska B, Baude A. et al. Neuroprotective activity of selective mGlu1 and mGlu5 antagonists in vitro and in vivo. European Journal of Pharmacology. 2007;554:18-29
149. Bao WL, Williams AJ, Faden AI. et al. Selective mGluR5 receptor antagonist or agonist provides neuroprotection in a rat model of focal cerebral ischemia. Brain Research. 2001;922:173-179
150. Cavallo D, Landucci E, Gerace E. et al. Neuroprotective effects of mGluR5 activation through the PI3K/Akt pathway and the molecular switch of AMPA receptors. Neuropharmacology. 2020;162:107810
151. Riek-Burchardt M, Henrich-Noack P, Reymann KG. No improvement of functional and histological outcome after application of the metabotropic glutamate receptor 5 agonist CHPG in a model of endothelin-1-induced focal ischemia in rats. Neuroscience Research. 2007;57:499-503
152. Takagi N, Besshoh S, Morita H. et al. Metabotropic glutamate mGlu5 receptor-mediated serine phosphorylation of NMDA receptor subunit NR1 in hippocampal CA1 region after transient global ischemia in rats. European Journal of Pharmacology. 2010;644:96-100
153. Takagi N, Besshoh S, Marunouchi T. et al. Effects of metabotropic glutamate mGlu5 receptor antagonist on tyrosine phosphorylation of NMDA receptor subunits and cell death in the hippocampus after brain ischemia in rats. Neuroscience Letters. 2012;530:91-96
154. Corti C, Battaglia G, Molinaro G. et al. The Use of Knock-Out Mice Unravels Distinct Roles for mGlu2 and mGlu3 Metabotropic Glutamate Receptors in Mechanisms of Neurodegeneration/Neuroprotection. The Journal of Neuroscience. 2007;27:8297-8308
155. Motolese M, Mastroiacovo F, Cannella M. et al. Targeting type-2 metabotropic glutamate receptors to protect vulnerable hippocampal neurons against ischemic damage. Molecular Brain. 2015;8:66
156. Mastroiacovo F, Moyanova S, Cannella M. et al. Genetic deletion of mGlu2 metabotropic glutamate receptors improves the short-term outcome of cerebral transient focal ischemia. Molecular Brain. 2017;10:39
157. Suzuki S, Koshimizu H, Adachi N. et al. Functional interaction between BDNF and mGluR II in vitro: BDNF down-regulated mGluR II gene expression and an mGluR II agonist enhanced BDNF-induced BDNF gene expression in rat cerebral cortical neurons. Peptides. 2017;89:42-49
158. Bespalov A, Jongen-Rêlo AL, van Gaalen M. et al. Habituation deficits induced by metabotropic glutamate receptors 2/3 receptor blockade in mice: reversal by antipsychotic drugs. J Pharmacol Exp Ther. 2007;320:944-950
159. Choi CH, Schoenfeld BP, Bell AJ. et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106-119
160. Rosdahl D, Seitzberg DA, Christensen T. et al. Changes in mRNA for metabotropic glutamate receptors after transient cerebral ischaemia. NeuroReport. 1994;5:593
161. Iversen L, Mulvihill E, Haldeman B. et al. Changes in Metabotropic Glutamate Receptor mRNA Levels Following Global Ischemia: Increase of a Putative Presynaptic Subtype (mGluR4) in Highly Vulnerable Rat Brain Areas. Journal of Neurochemistry. 1994;63:625-633
162. Moyanova SG, Mastroiacovo F, Kortenska LV. et al. Protective role for type 4 metabotropic glutamate receptors against ischemic brain damage. Journal of Cerebral Blood Flow & Metabolism. 2011;31:1107-1118
163. Domin H, Przykaza Ł, Jantas D. et al. Neuroprotective potential of the group III mGlu receptor agonist ACPT-I in animal models of ischemic stroke: In vitro and in vivo studies. Neuropharmacology. 2016;102:276-294
164. Domin H. Group III metabotropic glutamate receptors as promising targets for neuroprotective therapy: Particular emphasis on the role of mGlu4 and mGlu7 receptors. Pharmacology Biochemistry and Behavior. 2022;219:173452
165. Nakajima Y, Iwakabe H, Akazawa C. et al. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L-2-amino-4-phosphonobutyrate. Journal of Biological Chemistry. 1993;268:11868-11873
166. Domin H, Jantas D, Śmiałowska M. Neuroprotective effects of the allosteric agonist of metabotropic glutamate receptor 7 AMN082 on oxygen-glucose deprivation- and kainate-induced neuronal cell death. Neurochemistry International. 2015;88:110-123
167. Sánchez Triviño CA, Landinez MP, Duran S. et al. Modulation of Gq/PLC-Mediated Signaling by Acute Lithium Exposure. Frontiers in Cellular Neuroscience. 2022;16:838939
168. Sourial-Bassillious N, Rydelius PA, Aperia A. et al. Glutamate-mediated calcium signaling: a potential target for lithium action. Neuroscience. 2009;161:1126-1134
169. Khayachi A, Ase A, Liao C. et al. Chronic lithium treatment alters the excitatory/inhibitory balance of synaptic networks and reduces mGluR5-PKC signalling in mouse cortical neurons. Journal of Psychiatry & Neuroscience : JPN. 2021;46:E402-E414
170. Mayor D, Tymianski M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology. 2018;134:178-188
171. Yang M, He Y, Deng S. et al. Mitochondrial Quality Control: A Pathophysiological Mechanism and Therapeutic Target for Stroke. Front Mol Neurosci. 2022;14:786099
172. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85-100
173. Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J. 2015;29:4766-71
174. Kaur MM, Sharma DS. Mitochondrial repair as potential pharmacological target in cerebral ischemia. Mitochondrion. 2022;63:23-31
175. Liu Y, Liu XJ, Sun D. Ion transporters and ischemic mitochondrial dysfunction. Cell Adh Migr. 2009;3:94-98
176. Wu MY, Yiang GT, Liao WT. et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46:1650-1667
177. He Z, Ning N, Zhou Q. et al. Mitochondria as a therapeutic target for ischemic stroke. Free Radic Biol Med. 2020;146:45-58
178. Liu F, Lu J, Manaenko A. et al. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018;9:924-937
179. Zhou X, Chen H, Wang L. et al. Mitochondrial Dynamics: A Potential Therapeutic Target for Ischemic Stroke. Front Aging Neurosci. 2021;13:721428
180. Wu M, Gu X, Ma Z. Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury. Mol Neurobiol. 2021;58:5253-5271
181. Wang H, Chen S, Zhang Y. et al. Electroacupuncture ameliorates neuronal injury by Pink1/Parkin-mediated mitophagy clearance in cerebral ischemia-reperfusion. Nitric Oxide. 2019;91:23-34
182. Jia J, Jin H, Nan D. et al. New insights into targeting mitochondria in ischemic injury. Apoptosis. 2021;26:163-183
183. Damri O, Natour S, Agam G. Do Autophagy Enhancers/ROS Scavengers Alleviate Consequences of Mild Mitochondrial Dysfunction Induced in Neuronal-Derived Cells? Int J Mol Sci. 2021;22:5753
184. Silachev DN, Gulyaev MV, Zorova LD. et al. Magnetic resonance spectroscopy of the ischemic brain under lithium treatment. Link to mitochondrial disorders under stroke. Chem Biol Interact. 2015;237:175-182
185. Jayaraj RL, Azimullah S, Beiram R. et al. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16:142
186. Bonaventura A, Liberale L, Vecchié A. et al. Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. Int J Mol Sci. 2016;17:1967
187. Simats A, García-Berrocoso T, Montaner J. Neuroinflammatory biomarkers: From stroke diagnosis and prognosis to therapy. Biochim Biophys Acta. 2016;1862:411-424
188. Winbeck K, Poppert H, Etgen T. et al. Prognostic relevance of early serial C-reactive protein measurements after first ischemic stroke. Stroke. 2002;33:2459-2464
189. Zhang XG, Xue J, Yang WH. et al. Inflammatory markers as independent predictors for stroke outcomes. Brain Behav. 2021;11:e01922
190. Sandvig HV, Aam S, Alme KN. et al. Plasma Inflammatory Biomarkers Are Associated With Poststroke Cognitive Impairment: The Nor-COAST Study. Stroke. 2023;54:1303-1311
191. Narasimhalu K, Lee J, Leong YL. et al. Inflammatory markers and their association with post stroke cognitive decline. Int J Stroke. 2015;10:513-518
192. Couch C, Mallah K, Borucki DM. et al. State of the science in inflammation and stroke recovery: A systematic review. Ann Phys Rehabil Med. 2022;65:101546
193. Zhu J, Wang L, Shao H. et al. Higher Plasma Fibrinogen Level at Admission Is Associated with Post-Stroke Depression at Discharge. Brain Sci. 2022;12:1032
194. Wu D, Zhang G, Zhao C. et al. Interleukin-18 from neurons and microglia mediates depressive behaviors in mice with post-stroke depression. Brain Behav Immun. 2020;88:411-420
195. Xue Y, Zeng X, Tu WJ. et al. Tumor Necrosis Factor-α: The Next Marker of Stroke. Dis Markers. 2022;2022:2395269
196. Li H, Li Q, Du X. et al. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J Cereb Blood Flow Metab. 2011;31:2106-2115
197. Haupt M, Bähr M, Doeppner TR. Lithium beyond psychiatric indications: the reincarnation of a new old drug. Neural Regen Res. 2021;16:2383-2387
198. Rana AK, Kumar R, Shukla DN. et al. Lithium co-administration with rutin improves post-stroke neurological outcomes via suppressing Gsk-3β activity in a rat model. Free Radic Biol Med. 2023;207:107-119
Author contact
![]() Corresponding authors: Youkui Shi; E-mail: syk2101603com. Yanqiang Wang; E-mail: wangyq6sysu.edu.cn.
Corresponding authors: Youkui Shi; E-mail: syk2101603com. Yanqiang Wang; E-mail: wangyq6sysu.edu.cn.