Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2023; 20(12):1616-1630. doi:10.7150/ijms.85114 This issue Cite
Research Paper
GAS1 Promotes Ferroptosis of Liver Cells in Acetaminophen-Induced Acute Liver Failure
Jinqiu Tao1#, Cailin Xue2#, Xiaodong Wang3#, Huihui Chen4, Qiaoyu Liu4, Chunping Jiang1 ![]() , Wenjie Zhang1,4
, Wenjie Zhang1,4 ![]()
1. Department of General Surgery, Nanjing Drum Tower Hospital Clinical College of Nanjing Medical University, Nanjing, 210008 Jiangsu Province, China.
2. Department of Hepatobilliary Surgery, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, 210008 Jiangsu Province, China.
3. Department of General Surgery, Northern Jiangsu People's Hospital, Yangzhou, 225001 Jiangsu Province, China.
4. Department of Hepatobilliary Surgery, The first affiliated hospital of Anhui medical university, Hefei, 230022 Anhui Province, China.
#These authors contributed equally to this work.
Received 2023-4-9; Accepted 2023-9-15; Published 2023-9-25
Abstract

Purpose: Acute liver failure (ALF) is a clinically fatal disease that leads to the rapid loss of normal liver function. Acetaminophen (APAP) is a leading cause of drug-induced ALF. Ferroptosis, defined as iron-dependent cell death associated with lipid peroxide accumulation, has been shown to be strongly associated with APAP-induced liver injury. Growth arrest-specific 1 (GAS1) is a growth arrest-specific gene, which is closely related to the inhibition of cell growth and promotion of apoptosis. However, the functional role and underlying mechanism of GAS1 in APAP-induced ferroptosis remain unknown.
Methods: We established liver-specific overexpression of GAS1 (GAS1AAV8-OE) mice and the control (GAS1AAV8-vector) mice by tail vein injection of male mice with adeno-associated virus. APAP at 500 mg/kg was intraperitoneally injected into these two groups of mice to induce acute liver failure. The shRNA packaged by the lentivirus inhibits GAS1 gene expression in human hepatoma cell line HepaRG (HepaRG-shNC and HepaRG-shGAS1-2) and primary hepatocytes of mice with liver-specific overexpression of GAS1 were isolated and induced by APAP in vitro to further investigate the regulatory role of GAS1 in APAP-induced acute liver failure.
Results: APAP-induced upregulation of ferroptosis, levels of lipid peroxides and reactive oxygen species, and depletion of glutathione were effectively alleviated by the ferroptosis inhibitor, ferrostatin-1, and downregulation of GAS1 expression. GAS1 overexpression promoted ferroptosis-induced lipid peroxide accumulation via p53, inhibiting its downstream target, solute carrier family 7 member 11.
Conclusion: Collectively, our findings suggest that GAS1 overexpression plays a key role in aggravating APAP-induced acute liver injury by promoting ferroptosis-induced accumulation of lipid peroxides.
Keywords: growth arrest-specific gene 1, acute liver failure, acetaminophen, ferroptosis
Introduction
Acute liver failure (ALF) is characterized by the rapid loss of liver function. In >50% of cases, ALF is characterized by liver dysfunction, coagulopathy, encephalopathy, multiorgan failure and death1,2. ALF is characterized by severe hepatic inflammation that results in apoptosis and fulminant necrosis of the liver. ALF has several causes including, but not limited to, drug toxicity, viruses, toxins, and ischemia3. The antipyretic and analgesic drug, paracetamol (APAP, also known as acetaminophen), one of the most commonly used drugs in clinical practice, is also a major cause of acute liver injury and even death1,4. N-acetylcysteine is widely recognized by the Food and Drug Administration of the United States as a clinical therapeutic drug. It is one of the few drugs that can effectively treat APAP-induced acute liver injury5,6. Although it is effective in treating APAP-induced acute liver injury in the early stages, it also results in a few undesirable side effects.
APAP overdose-induced acute liver injury occurs via a complex mechanism that has not yet been fully elucidated. Accumulation of APAP leads to the production of toxic metabolites, including N-acetyl-p-benzoquinoneimine (NAPQI)7. Excess NAPQI in the body consumes glutathione (GSH) in the cytoplasm and mitochondria, while simultaneously harming the liver cells8. In addition, abnormal production of reactive oxygen species (ROS) leads to primary hepatotoxicity, GSH, which is a key antioxidant in liver cells that plays a crucial role in the antioxidant defense system of the body9,10. Increase in ROS levels results in a reduction in the activity of antioxidant enzymes, such as glutathione peroxidase (GPX), via lipid peroxidation, thereby aggravating acute liver injury caused by APAP accumulation11,12.
In recent years, ferroptosis, another form of non-apoptotic cell death, has been shown to exhibit distinct morphological and biochemical characteristics13. ROS accumulation and iron-driven lipid peroxidation characterize ferroptosis, and cancer, neurological diseases, acute renal failure, and liver injury are associated with dysfunctional ferroptosis14,15. Recent evidence indicates that ferroptosis may cause various liver diseases, including drug-induced liver damage, viral hepatitis, and ALF16,17. Ferroptosis can also be caused by the depletion of GSH or inactivation of GPX4 and the accumulation of iron-dependent lipid peroxides18-20. In ferroptosis, GPX4 is the only metabolic enzyme in the GPX subtype that is capable of regulating the lipid peroxide and ROS levels21,22. According to current research on ferroptosis, lipid peroxidation resulting from iron overload is the primary cause of ferroptosis13,23. Activation of intracellular toxic ROS can be enhanced by iron, which acts as a catalyst to promote oxidative stress reactions and aggravate liver injury caused by oxidative stress. According to previous studies, cytosolic iron overload can lead to toxic ROS production via the Fenton reaction, further aggravating APAP-induced oxidative stress24. A recent study has identified various regulatory pathways, such as the p53 pathway and factors, such as the cystine/glutamate antiporter (SystemXc-) and inhibitory compounds, involved in ferroptosis25. These studies suggest that GSH depletion causes APAP-induced liver injury, which may contribute to ALF.
Growth arrest-specific1 (GAS1) has been identified as a growth arrest gene that inhibits cell growth and cell transition from G0 to S phase. Evidence suggests that this region of the chromosome is frequently lost in tumors, particularly in human leukemia, bone marrow malignancies, colorectal carcinomas and bladder cancer26-29. GAS1 has been reported to selectively express and inhibit DNA replication in quiescent NIH3T3 fibroblasts, suggesting its potential role as a tumor suppressor gene30. In B16-F10 melanoma cells with high metastasis, GAS1 shows significantly low expression, which inhibits its metastasis, partly by promoting apoptosis31; GAS1 expression has been reported to promote cell differentiation in differentiated hind limb muscle cells26,32. Overexpression of GAS1 reduces the size, proliferation, and malignancy of liver tumors33. Studies have shown that overexpression of GAS1 blocks cell proliferation in a p53-dependent manner and that the transactivation function of p53 is optional for GAS1-induced growth arrest, consequently, other intrinsic non-activated functions of p53 (possibly related to apoptosis regulation) may play a role in GAS1-induced growth arrest34,35. However, whether GAS1 plays a role in drug-induced liver injury remains unclear.
In this study, we found that ferroptosis is significantly increased by the exposure of liver-specific overexpression of GAS1 (GAS1AAV8-OE) mice to APAP, suggesting GAS1 as a critical component of acute liver damage. Therefore, targeting GAS1 may be a novel approach to treat APAP-induced hepatotoxicity.
Materials and Methods
Vector preparation
AAV8 vectors were designed to express mouse GAS1 with a thyroxine-binding globulin (TBG) promoter produced by WZ Biosciences, Inc (Jinan, Shandong, China). The carrier was stored between -60 and -80°C. After dilution in sterile phosphate-buffered saline (PBS), the final carrier product was stored at 4 °C or on wet ice.
Mice
Male C57BL/6J mice were obtained from Gempharmatech Co., Ltd (Nanjing, Jiangsu, China) at 6 weeks-of-age. All mice were raised in specific-pathogen free animal rooms, where constant temperature and humidity were maintained in the animal room and in the standard feed; light was providedin12:12h light/day cycle. The design and operation of all animal experiments were approved by the Laboratory Animal Ethics Committee of Nanjing Drum Tower Hospital Clinical College of Nanjing Medical University. In all studies, the Council for the International Organizations of Medical Sciences' International Guiding Principles for Biomedical Research Involving Animals was followed.
Clinical samples and cell culture
Tissues used in this study were obtained from the Department of Hepatobiliary Surgery at the drum tower hospital affiliated with the Nanjing University Medical College. During hepatectomy for hepatic hemangiomas, normal liver tissues were obtained, while ALF liver tissues were obtained from patients with ALF receiving liver transplants. The written consent was obtained from all patients and the Ethics Committee of Drum Tower Hospital affiliated with Nanjing University Medical College before the use of patient tissue samples in this study. HepaRG cell line was purchased from the American Type Culture Collection (ATCC). In the culture of the HepaRG cell line, Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) (Vazyme Company, Nanjing, Jiangsu, China) was used. It was maintained at 37°C and 5% CO2.
Reagents and antibodies
APAP (HY-66005), ferrostatin-1 (HY-100579), RSL (HY-100218A) and erastin (HY-15763) were purchased from MedChemExpress (New Jersey, USA). Anti-α-tubulin (ab7291) antibody was purchased from Abcam (Cambridge, UK), anti-GPX4 (381958) antibody was purchased from Zenbio (California USA). Anti-P53 (2527S) antibody was purchased from Cell Signaling Technology (Massachusetts, USA). Anti-GAS1 (17903-1-AP), anti-solute carrier family 7 member 11 (SLC7A11) (26864-1-AP), anti-mouse IgG-horseradish peroxidase (HRP) (SA00001-1), and anti-rabbit IgG-HRP (SA00001-2) antibodies were purchased from Proteintech (Wuhan, Hubei, China).
Establishment of an APAP-induced liver injury model
We selected nine-weeks-old male mice with weights similar to those of the model mice. We starved the mice for 16 h before administration of APAP (dissolved in PBS at 60°C and cooled to 37°C before injection). APAP (500 mg/kg) was administered to the mice at four time points (0, 6, 12 and 24 h) after stimulation. After administering anesthesia with isoflurane, eyeball blood was collected in a 1.5-mL centrifuge tube and centrifuged at 3000 rpm for 15 min at 4°C. Cervical vertebrae of the mice were dissected. After disinfection, the abdominal cavity was opened to dissect and separate the livers. Liver tissues (50 mg) were taken and placed in a refrigerator at -80°C for subsequent protein and RNA detection. The remaining liver tissue was fixed with paraformaldehyde and subjected to hematoxylin and eosin (H&E) and immunohistochemical (IHC) staining.
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
Total RNA was extracted and RT-qPCR was performed as previously described36. The following primers were used: GAS1 (Homo), forward 5′-TGACCTACTGCGGCAAAGTC-3′ and reverse 5′-CTTGACCGACTCGCAGATGG-3′; GAS1 (Mus), forward 5′-CGGCAAGCTTTTCAACGGG-3′ and reverse 5′-TCTCTTTGACCGATTCGCAG-3′; glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Homo), forward 5′-GATATTGTTGCCATCAATGAC-3′ and reverse 5′-TTGATTTTGGAGGGATCTCG-3′; GAPDH (Mus), forward 5′-CCCAGCTTAGGTTCATCAGGT-3′ and reverse 5′-GGTCAATGAAGGGGTCGTTG-3′. GAPDH was used as the endogenous control for mRNA expression.
Lentivirus transfection and stable establishment of interfering GAS1 cell line
Several short hairpin RNA (shRNA) sequences, including a nonsense sequence shNC (5′-GTTCTGGATCGTCAGGT-3′) packaged in lentivirus vectors, shGAS1-1 (5′-GGGCTGTCTATTAGCATATTT-3′) and shGAS1-2 (5′-GGGCTGTCTATTAGCATATTT-3′), were designed and synthesized by GenePharma (Shanghai, China) using Lipofectamine 3000 (Invitrogen, California, USA), according to the manufacturer's instructions.
H&E staining
For H&E staining, tissues were fixed in paraformaldehyde for 24 h and subjected to gradient dehydration and paraffin embedding. H&E staining was performed on 4 mm-thick paraffin sections prior to sealing.
IHC and immunofluorescence (IF) staining
After paraffinizing the liver tissues, they were incubated at 37 °C overnight. The sections were then deparaffinized and treated with 3% hydrogen peroxide for 15 min, microwaved and naturally cooled for 40 min. Finally, the samples were incubated with anti-GAS1 antibodies, and fluorescent or biotin-labeled secondary antibodies were added to the liver tissues to observe the protein expression.
Measurement of mitochondrial membrane potential (MMP) and ROS levels
For ROS quantification, Gas1AAV8-OE and Gas1AAV8-vector primary hepatocytes and HepaRG-shNC and HepaRG-shGAS1-2 cells were treated with 10 mM APAP for 24 h and loaded with MitoTracker Red CMXRos (TRC) and ROS fluorescent probe DHE obtained from KeyGEN BioTECH (Nanjing, Jiangsu, China) and Beyotime Biotech (Shanghai, China), respectively. TRC and ROS levels were determined by measuring the fluorescence excitation/emission spectra at 518/605 and 579/599 nm, respectively, using a laser confocal microscope (Leica, Heidelberg, Germany).
Liver biochemistry and measurement of MDA and GSH levels
Plasma samples were analyzed using Beckman AU680 (Bria, California, USA) to determine the alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine, and lactate dehydrogenase (LDH) levels. We also determined the malondialdehyde (MDA) and GSH levels in HepaRG cells and primary hepatocyte lysates using the MDA assay kit (Beyotime, Shanghai, China) and GSH assay kit (Nanjing Jiancheng, Nanjing, Jiangsu, China), respectively, according to the manufacturer's instructions.
Primary hepatocyte culture
Primary hepatocytes were isolated from anesthetized mice after perfusion with collagenase solution37-39. To expose the abdominal vasculature, the intestine was gently displaced using a blunt instrument. The inferior abdominal vena cava (IVC) was located and a suture was placed beneath it, distal to the renal vein. The catheter and needle were placed beyond the suture level of the IVC, distal to the suture. To hold the catheter in place, a suture was tied around it. The needle was then removed carefully. If performed correctly, blood flows through the catheter. The tubing was carefully attached to the catheter. The lung cavity was dissected open to gently reflect the superior liver lobes to expose the diaphragm. Using sharp tip scissors, the diaphragm was punctured and a lateral incision was made to expose the pleural cavity, taking care to avoid the gall bladder and pleural vessels. A bulldog clamp was placed around the inferior thoracic vena cava, proximal to the hepatic vein. Liver was perfused for 5 min with the Liver Perfusion Medium by cutting the portal vein and turning on the pump at a rate of 3-4 mL/min. After 5 min of incubation, the pump was stopped, and the tubing was transferred to the Liver Digest Medium. The pump was later restarted, and the liver was perfused for another 10-15 min. After carefully dissecting the liver, the gallbladder was removed, and the liver was placed in a 10-cm tissue culture dish. Then, 10 mL of the medium was added to the liver in a biosafety cabinet. The liver was gently scraped to remove the hepatocytes. In a 50-mL centrifug tube, the suspension was filtered through a 100-μm cell strainer. The plate was washed with 10mL of cell medium, and the cells were pooled in a conical tube. Cells were centrifuged for 5 min at 350×g and 4 °C. The pellet was resuspendedin 10 mL of cell medium, 10% colloidal silica coated with polyvinylpyrrolidone was added, mixed gently, and centrifuged for 5 min at 4 °C. Dead cells appeared at the top of the mixture after centrifugation, while live cells settled at the bottom. The dead cells and medium were aspirated. Cells were washed twice with 20 mL of cell medium, centrifuged for 5 min at 350×g and 4 °C, and resuspendedin 10 mL of medium.
Transmission electron microscopy (TEM)
Left liver lobe was resected, fixed in glutaraldehyde, dehydrated with gradientethanol, and cut into ultrathin slices. The slices were then stained with lead citrate and uranyl acetate and observed under a Hitachi HT-7700 transmission electron microscope (Hitachi. Co. Ltd., Tokyo, Japan).
Western blot
Liver tissues or cells were extracted using protein extraction kit (Beyotime, Shanghai, China) and protein content was quantified using BCA protein assay kits (Beyotime, Shanghai, China). Protein samples from each group were electrophoresed on a 10-15% sodium dodecyl sulfate-polyacrylamide gel and transferred to polyvinylidene difluoridemembranes. The blots were hatched for 24 h at 4°C with primary antibodies (GAS1, GPX4, SLC7A11, P53 and α-tubulin) after blocking with non-fat dry milk (5%). The blots were washed with tris-buffered saline with tween 20 and incubated with secondary antibodies at 37 °C for 120 min after blocking with non-fat dry milk (5%). An ECL kit was used to detect the bands.
Statistical analysis
All data are represented as the mean ± standard deviation (mean ± SD). Kaplan-Meier analysis was performed to determine the rate of survival. Student's t-test was used to compare the data of two groups. Statistical software (SPSS 23.0) was used to analyze the data and GraphPad Prism version 9.0.0 was used to draw a chart. P-values < 0.05 was considered as statistically significant.
Results
GAS1 expression is upregulated in ALF
Using real-time PCR, we analyzed GAS1 expression levels in the liver tissues of 18 patients with ALF and 30 controls. The control group consisted of normal liver tissues from patients with benign liver tumors who underwent partial hepatectomy. We found that GAS1 mRNA expression levels were significantly higher in the liver tissues of patients with ALF than in those of the control. We also evaluated the protein levels of GAS1 in seven randomly selected ALF and normal liver tissues using western blot. IHC and IF were also used to determine the expression levels of GAS1 in ALF liver tissues. Expression levels of GAS1 protein were high in ALF liver tissues, but low in normal liver tissues (data not shown).
Establishment of a mice model with liver-specific ectopic expression of GAS1 using AAV8 with the liver-specific promoter TBG
GAS1 was ectopically expressed in the liver tissue using AAV8, which is highly efficient in liver transfection, and the liver-specific promoter, TBG40. Structural design of theAAV8-TBG-GAS1 carrier and experimental flow chart are shown in Figure 1A. After three weeks, we divided 9-week-old male C57BL/6J mice into three groups, namely the wild-type (WT), vector and GAS1 overexpression groups, to study the effects of GAS1 on APAP-induced liver injury in mice. After APAP treatment, RT-qPCR analysis of revealed an increase in GAS1 mRNA levels in the livers of mice overexpressing GAS1 compared to those in WT and vector mice (Figure 1B). We then conducted western blot to determine the protein expression levels of GAS1 in the three groups and found that the results were consistent with the RT-qPCR results (Figure 1C).
Construction and in vivo liver specific transfection of AAV8-TBG-GAS1. (A) Schematic showing the experimental strategy used for GAS1 to aggravate APAP-induced ALF in mice. (B) RT-qPCR confirmed the overexpression of GAS1 in the mouse liver. (C) Western blot confirmed the overexpression of GAS1 in the mouse liver. Unpaired Student's t-test; *P ≤ 0.05, ***P ≤ 0.001.
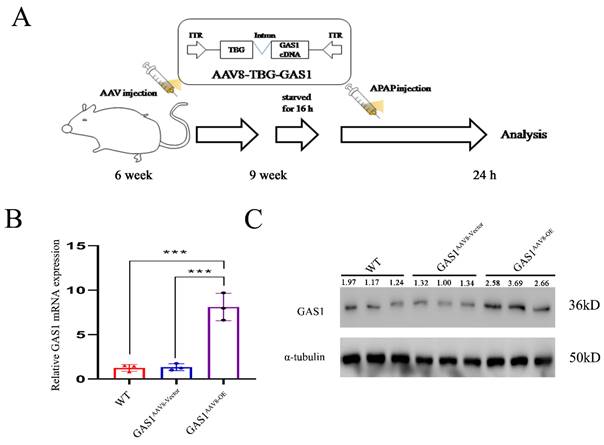
Liver-specific ectopic expression of GAS1 aggravates tissue damage after ALF in mice
After 24 h of APAP treatment, H&E staining revealed that the degree of damage in the GAS1 overexpression group was significantly higher than that in the carrier group, with edema and necrosis of hepatocytes observed around the central vein of the hepatic lobule (Figure 2A). Meanwhile, GAS1 immunohistochemical expression levels in the livers of the overexpression group were significantly higher than those in the overexpression group without APAP treatment (Figure 2B). Moreover, IHC and IF results in the liver tissues also demonstrated that mice overexpressing GAS1 in hepatocytes had significantly higher levels of GAS1 staining in their livers compared with 0h, indicating that the expression of GAS1 protein is significantly higher after ALF than that without ALF (Figure 2B and 2C). Levels of serum biochemical indicators were also determined (Figures 2D, 2E and 2F). Levels of ALT, AST and LDH increased significantly after APAP treatment. Levels of the biochemical indicators, ALT, AST and LDH were significantly different between the liver-specific overexpression GAS1 and control groups. Additionally, we determined the 48h survival rate of mice in the control and GAS1 overexpression groups following liver injury induced by 500 mg/kg APAP. We found that only 35.71% of mice in the GAS1 overexpression group survived compared to 63.64% of mice that survived in the control group (Figure 2G). GSH of GAS1AAV8-OE and GAS1AAV8-vector mice liver after 1 hour of APAP administration were assessed (Figure S1).
Overexpression of GAS1 aggravated the hepatotoxicity induced by APAP (500mg/kg, 24h) in mice. (A) Liver tissues were harvested 24 h after application of APAP and processed for hematoxylin and eosin (H&E) staining; (B-C) Immunohistochemical and immunofluorescence staining of GAS1 in the liver tissues of mice treated with APAP for 24 h; (D-F) Levels of the serum markers, AST, ALT and LDH were measured; (G) Survival curve of GAS1 vector and OE mice treated with a dose of APAP. All mice were observed for survival 48 h after APAP treatment. Unpaired Student's t-test; *P ≤ 0.05, ***P ≤ 0.001.
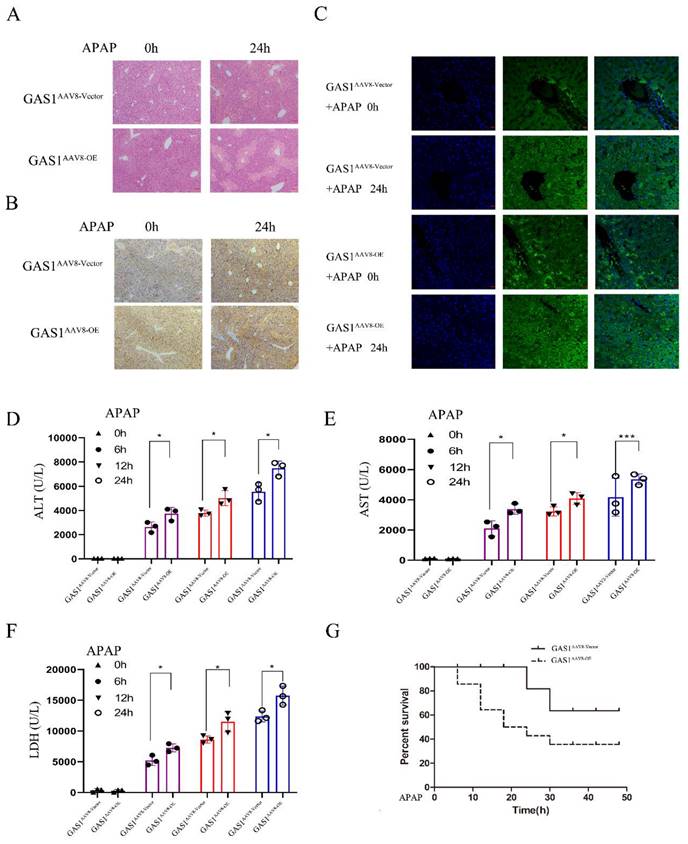
Knockdown of GAS1 inhibits APAP-induced ferroptosis in HepaRG
In vitro studies of APAP hepatotoxicity commonly use 10 mM APAP as the median lethal dose for hepatocytes41,42. We established an in vitro APAP model using HepaRG cells. RT-qPCR and western blot analysis were conducted using the constructed stably transfected cell line knocked down by GAS1 (Figure 3A). For subsequent experiments, we selected the stable cell line with the highest interference efficiency. After APAP treatment, GAS1 expression was significantly increased in HepaRG-shGAS1-2 and HepaRG-shNC cells in a time-dependent manner, and indicated that the expression of GAS1 protein is significantly higher after ALF than that without ALF (Figure 3B). Additionally, to verify whether interference with GAS1 expression can inhibit APAP-induced oxidative stress and MMP changes in hepatocytes, we tested the levels of corresponding indicators. TRC and DHE staining results showed that after 24h of APAP treatment, the decrease in MMP in the GAS1 knockdown group was significantly lower than that in the control group (Figure 3C), while ROS levels were reduced in the HepaRG-shGAS1-2 group compared to those the control group (Figure 3D). Ferroptosis is a type of regulated cell death caused by the accumulation of redox-active iron, depletion of GSH and lipid oxidation43,44. To determine whether GAS1 was involved in the regulation of APAP caused ferroptosis in HepaRG cells, we evaluated the effects of ferroptosis inducers (erastin 10μM) and inhibitors (ferrostatin-1 1μM) on these cells. We found that APAP decreased GSH and increased MDA levels in HepaRG cells, whereas treatment with ferrostatin-1 had the opposite effect and no significant difference between MDA and GSH levels in HepaRG-shGAS1-2 and HepaRG-shNC cells. These results indicate that APAP stimulates HepaRG cells to activate ferroptosis (Figure 3E and 3F). These results indicate that in vitro inhibition of GAS1 inhibits ROS accumulation, alleviates oxidative damage and inhibits ferroptosis caused by APAP.
Knockdown of GAS1 alleviates APAP-induced ALF by reducing ferroptosis in HepaRG cells. (A) HepaRG cells stably inhibited by shGAS1-2 were examined via western blotting and RT-PCR. (B) Expression levels of GAS1 in liver tissues at a specific time point after APAP treatment of HepaRG cells determined via western blotting. (C-D) For HepaRG-shNC and HepaRG-shGAS1-2, mitochondrial membrane potential (MMP) and ROS levels were measured 24h after ALF was induced by APAP. (E-F) After treatment with ferrostatin-1(1μM) and/or erastin (10μM) for 24 h, malondialdehyde (MDA) and glutathione (GSH) levels in HepaRG-shNC and HepaRG-shGAS1-2 cells were determined in the presence or absence of APAP. Unpaired Student's t-test; *P ≤ 0.05, ***P ≤ 0.001.
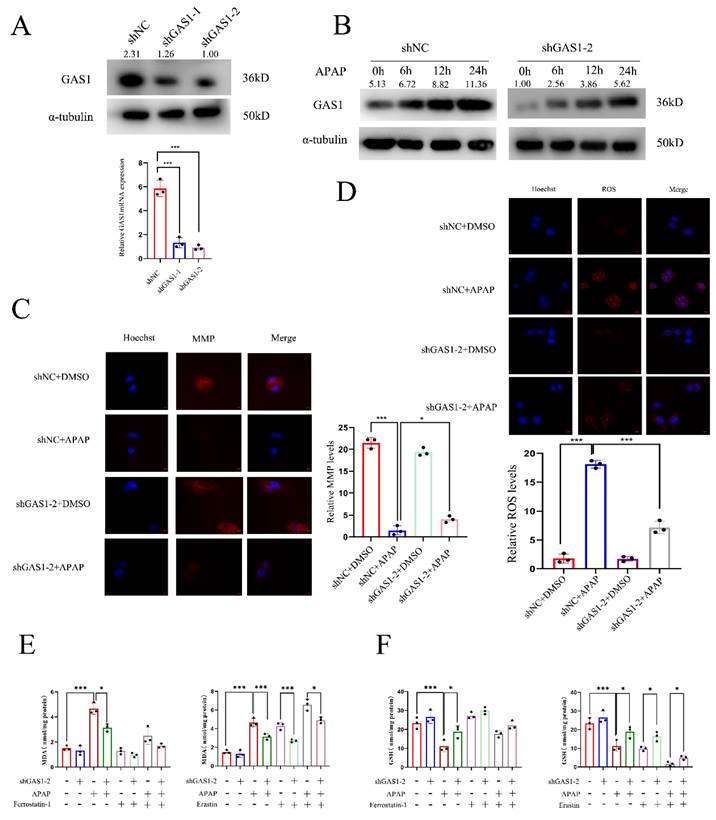
Overexpression of GAS1 aggravates APAP-induced ferroptosis of primary mice hepatocytes
To further assess the effects of GAS1 on APAP-induced liver injury, we induced ferroptosis in primary mouse hepatocytes. We isolated primary mouse hepatocytes from mice with liver-specific overexpression of GAS1 as well as from those in the vector group. After APAP treatment, GAS1 expression was significantly increased in the GAS1AAV8-Vector and GAS1AAV-OE groups in a time-dependent manner, and indicated that the expression of GAS1 protein is significantly higher after ALF than that without ALF (Figure 4A). APAP treatment of GAS1AAV8-Vector and GAS1AAV-OE groups worked as expected, with GAS1AAV-OE producing more MDA and consuming more GSH than GAS1AAV8-Vector. Erastin (10μM) and ferrostatin-1 (1μM) were evaluated for the participation of GAS1 in primary hepatocyte ferroptosis. Erastin decreased the levels of GSH and increased the levels of MDA induced by APAP in GAS1AAV8-OE mice, whereas ferrostatin-1 increased the levels of GSH and MDA (Figure 4B and 4C). Similarly, we treated GAS1AAAV8-Vector and GAS1AAAV-OE primary mouse hepatocytes with APAP. MMP and ROS levels were measured after 24h. MMP was decreased in GAS1AAV8-OE cells compared to that in GAS1 AAV8-Vector cells, while ROS levels were increased in GAS1AAAV-OE cells compared to that in GAS1AAV8-Vector (Figure 4D and 4E). Ferroptosis is distinguishable by morphological changes in mitochondria. The morphology of mitochondria was captured using TEM to determine the root cause of aggravated liver injury. We found that APAP caused significant mitochondrial abnormalities, including the reduction or even disappearance of mitochondrial cristae or rupture of the mitochondrial outer membranes. Overexpression of GAS1 further aggravated mitochondrial damage induced by APAP via ferroptosis (Figure 4F).
Overexpression of GAS1 in mice primary hepatocytes aggravates APAP-induced ALF by promoting ferroptosis. (A) Expression levels of GAS1 in the liver tissues at a specific time point after APAP treatment of mice primary hepatocytes determined via western blotting. (B-C) After treatment with ferrostatin-1(1μM) and/or erastin (10 μM) for 24 h, malondialdehyde (MDA) and glutathione (GSH) levels in GAS1AAV8-Vector and GAS1AAV8-OE mice primary hepatocytes were determined in the presence or absence of APAP. (D-E) In GAS1AAV8-Vector and GAS1AAV8-OE mice primary hepatocytes, MMP and ROS levels were measured 24h after ALF was induced by APAP. (F) Representative images showed by TEM. The red arrow indicates representative mitochondria in GAS1AAV8-Vector and GAS1AAV8-OE mice primary hepatocytes treated by APAP or not. Unpaired Student's t-test; *P ≤ 0.05, ***P ≤ 0.001.
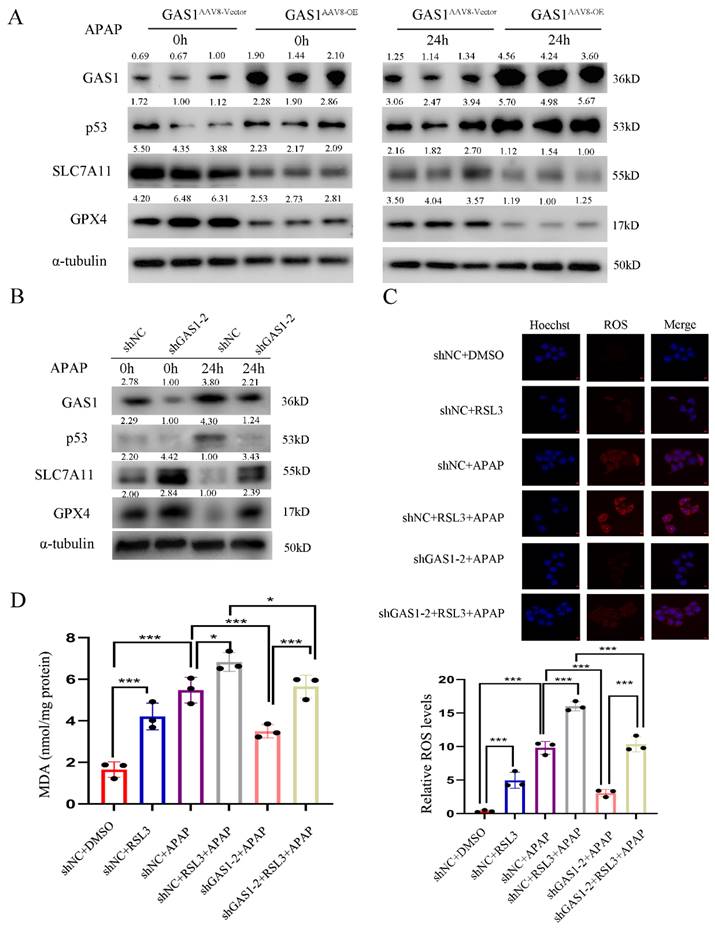
GAS1 promotes APAP-induced hepatotoxicity via the p53/SLC7A11 signaling pathway
Ferroptosis is a new pathway of cell death that is characterized by an increase in Fe2+ levels and MDA, accumulation of ROS and reduction in the synthesis of GSH, which results in the damage of the mitochondrial cristae or rupture of the outer membrane of mitochondria, ultimately leading to cell death13. SLC7A11, a part of System Xc- is involved in the production of GSH; therefore, inhibiting its expression can reduce GSH activity. p53 has been reported to inhibit SLC7A11 expression to promote ferroptosis45,46. Following our demonstration of the role of GAS1 in APAP-induced liver injury, we isolated primary mouse hepatocytes and explored the molecular signaling mechanisms of GAS1 involved in ferroptosis promotion in primary mouse hepatocytes and HepaRG cells. As a result of exposure to APAP, p53 expression levels were significantly higher, but SLC7A11 and GPX4 expression levels were significantly lower in GAS1AAV8-OE primary mouse hepatocytes than in GAS1AAV8-Vector primary mouse hepatocytes (Figure 5A). Inhibition of GAS1 expression in HepaRG cells further confirmed this result (Figure 5B). RSL3 (1μM) significantly increased ROS and MDA production in HepaRG-shNC and HepaRG-shGAS1-2 cells, suggesting that GAS1 may regulate ferroptosis through GPX4 (Figure 5C and 5D). These results suggest that GAS1 aggravates hepatocyte injury caused by APAP by promoting ferroptosis.
Activation of the p53/SLC7A11/GPX4 pathway is critical for GAS1 to affect ferroptosis in APAP-induced ALF. (A) Expression levels of p53, SLC7A11 and GPX4 in GAS1AAV8-Vector and GAS1AAV8-OE mice primary hepatocytes induced by APAP were determined using western blot. (B) Western blot analysis of ferroptosis signaling pathway-related proteins, p53, SLC7A11 and GPX4 after transfection of HepaRG cells with shGAS1-2 induced by APAP. (C-D) After treatment with RSL3 (1μM) for 24 h, ROS and MDA levels in HepaRG-shNC and HepaRG-shGAS1-2 cells were determined in the presence or absence of APAP. Unpaired Student's t-test; *P ≤ 0.05, ***P ≤ 0.001.
Discussion
ALF is a severe condition. Globally, APAP overdose is the leading cause of acute liver damage and even liver failure, with increasing prevalence in China2,4. However, relevant treatment methods for it are still limited and safer and more effective treatment strategies urgently needed. Researchers have found that APAP-induced liver oxidative stress and mitochondrial dysfunction are responsible for APAP-induced liver injury caused by oxidative stress and mitochondrial dysfunction47,48. APAP is believed to cause liver injury as follows: excessive intake of APAP is saturated by liver glucuronidation and sulphuration, and APAP is metabolized to form toxic NAPQI primarily by CYP4502E1, resulting in its degradation by GSH in the liver49, eventually causing various forms of liver cell death. Therefore, it is necessary to determine the underlying mechanism of cell death to aid in the clinical treatment of acute liver injury and liver failure caused by excessive APAP.
Recently, ferroptosis has been identified as a new form of non-apoptotic cell death induced by iron-dependent lipid peroxide accumulation, and it is distinct from other forms of cell death based on its morphological and biochemical characteristics13. In addition to causing oxidative stress, fibrosis can also result in liver injury as a result of acute liver inflammation and injury 18,50. An experimental study found that ferroptosis can be induced by depletion of GSH, inactivation of GPX4, and accumulation of iron-dependent lipid peroxide19,51. Ferroptosis is mediated by the GPX4 metabolizing enzyme, which is the only GPX subtype enzyme capable of regulating the lipid peroxide and ROS levels12,52. Lipid peroxidation caused by iron overload is the primary cause of fibrosis. Iron is a catalyst for intracellular toxic ROS, which promote oxidative stress reactions and enhance liver injury caused by oxidative stress. Through the Fenton reaction, cytosolic iron overload can result in toxic ROS production, which may exacerbate the oxidative stress-induced damage caused by APAP12,53. The metallothionein, SLC7A11, is part of System Xc-; therefore, inhibiting its expression can reduce the GSH activity. p53 has been reported to inhibit SLC7A11 expression to promote ferroptosis25,45,54,55.
Studies have shown that GAS1 expression is generally downregulated in many tumor tissues, suggesting its potential function as a tumor suppressor26,27,30. GAS1 can inhibit the proliferation of malignant tumor cells and promote apoptosis in cancers of the rectal wall and melanoma29,31. In this study, we confirmed that GAS1 expression levels were significantly higher in ALF liver tissues than in normal liver tissues. We established a mouse liver failure model based on APAP-induced liver injury. The results showed that GAS1AAV8-OE mice showed significantly worse in liver injury and inflammation than the control GAS1AAV8-vector mice. Several indices of mouse hepatocytes and HepaRG cells were analyzed before and after exposure of GAS1AAV8-OE and GAS1AAV8-Vector mice and HepaRG-shNC and HepaRG-shGAS1-2 cells to APAP. Use of ferroptosis inhibitor, ferrostatin-1, and downregulation of GAS1 effectively alleviated the APAP-induced upregulation of lipid peroxide and ROS levels and depletion of GSH. Therefore, inhibition of GAS1 may reduce the accumulation of ROS in the liver, thereby reducing liver lipid peroxide levels and further inhibiting ferroptosis and liver injury. The objective of this study was to determine the pathway through which GAS1 plays a role in ferroptosis and which pathway GAS1 acts as the main regulatory pathway of ferroptosis, including antioxidant, iron metabolism, and lipid metabolism pathways. Studies have shown that the overexpression of GAS1 blocks cell proliferation in a p53-dependent manner and that the trans-activation function of p53 is optional for GAS1-induced growth arrest. Therefore, other intrinsic non-activated functions of p53 (possibly related to apoptosis regulation) may play a role in GAS1-induced growth arrest34. p53 functions as a tumor suppressor gene. It regulates the cell growth cycle, affects the metabolism of tumor cells, induces apoptosis, and promotes DNA damage56. p53 inhibits the activity of System Xc- and the entry of cystine into cells, reduces the synthesis of GSH and decreases the activity of GPX4 after acetylation, thereby promoting ferroptosis57,58. Recent studies have shown that p53 increases glutaminase 2 activity to catalyze the production of glutamic acid. Because cystine and glutamic acid are transported in a 1:1 ratio, the high concentration of glutamic acid in cells inevitably reduces the entry of cystine into the cells, reducing ferroptosis resulting from GSH synthesis57. These studies outline the significance of the relationship between GAS1 and ferroptosis in APAP-induced liver injury mediated by p53. Therefore, we hypothesized that the knockdown or overexpression of GAS1 could attenuate or aggravate APAP-induced ALF via the p53/SLC7A11 signaling pathway. Western blot was used to detect the expression levels of p53, SLC7A11, and GPX4 in GAS1AAV8-OE and GAS1AAV8-Vector primary mouse hepatocytes and HepaRG-shNC and HepaRG-shGAS1-2 cells. Indeed, p53 expression was positively correlated with GAS1 expression, whereas SLC7A11 and GPX4 expression levels were negatively correlated with GAS1 expression.
As many others, this study has some shortcomings. More than one type of hepatocyte death is involved in acute and chronic liver injury, including apoptosis, necroptosis, ferroptosis, pyroptosis and autophagy59,60. The tandem relationship between hepatocyte death and many other types of cell death in ALF shows that not only ferroptosis plays an important role in acute liver injury, but also other types of hepatocyte death, which provides a direction for our follow-up research. Ferroptosis, a new type of cell death caused by the accumulation of iron-dependent lipid peroxides, has been associated with ALF61. Studies have shown that GPX4 regulates the regulatory cell death, including apoptosis, necrosis and autophagy. Previous studies have indicated that the knockout of receptor-interacting protein kinase 1 (RIPK1) or RIPK3 increases the susceptibility of mouse cells to ferroptosis, while inhibition of GPX4 facilitates the induction of apoptosis by apoptosis activators. Recent studies have also demonstrated the relationship between ferroptosis and apoptosis, although further in-depth studies are required to confirm this relationship62. GPX4 is an important component of ferroptosis. Overexpression of GPX4 has been reported to inhibit ROS-mediated autophagy and death of immune cells owing to its enhanced antioxidant capacity63. Therefore, ALF is characterized not only by single hepatocyte death but also ferroptosis, a relatively new mode of cell death identified in recent years. Although only a few studies have focused on ALF, the tandem relationship between ALF and many other types of cell death suggests that it may play an important role in acute liver injury.
The present study has several limitations. Firstly, the clinical samples we selected included ALF caused by prescription drugs, Chinese herbs, viruses, etc., and APAP-induced liver failure was only an experimental model we used. APAP hepatotoxicity mouse model can well reproduce human pathophysiology, so this clinically relevant model is often used in experimental studies. However, no model can fully restore all the characteristics of human acute liver injury. Different modeling methods involve different experimental procedures, and each animal model has its own advantages, but at the same time there are certain shortcomings. In spite of these limitations, we also found a high expression of GAS1 in the APAP-induced liver failure mouse model, suggesting a need to add additional liver failure models for more detailed study. Based on the clinical data, we presented subsequent hypotheses, which represent a direction for follow-up research. Secondly, our study contains a small sample size and there may be type I or type II statistical errors. The article does not mention the effects of ferroptosis inducers and inhibitors at different doses. Our previous pre-experiments evaluated the effects of different doses of ferroptosis inhibitors and inducers, but were not reported in the article. We selected a dose with the highest effect as the follow-up experiment64. We will, if necessary, increase the number of samples and concentration gradient for future validation. At last, the mechanism of APAP-induced ALF is still unclear, and there are different voices in the academic community. The role of lipid peroxidation in APAP-induced ALF has been debated for decades. Some scholars believe that APAP-induced acute liver failure is due to the nitrification of proteins65, this view has been supported by evidence showing that, while tocopherol supplementation can reduce APAP-induced lipid peroxidation (LPO), it does not attenuate APAP-induced liver injury, possibly due to its inability to affect peroxynitrite formation66. Another study found that a diet deficient in vitamin E and high in soybean oil increased hepatic polyunsaturated fatty acid levels, and this could be the primary pathway of APAP hepatotoxicity. However, in mice, the increase of LPO was very limited in the APAP-induced liver injury, and the amount was insufficient to cause cell death65,67. In addition, MnSOD catalyzes the distortion of superoxide in mitochondria and prevents the formation of peroxynitrite, MnSOD also produces hydrogen peroxide. Depending on the redox balance, it can be detoxified by glutathione peroxidase or catalase68. It has been found that MnSOD deficiency substantially exacerbates APAP-induced protein nitrification and liver injury69. On the contrary, some scholars believe that there is ferroptosis in the process of APAP-induced ALF12,17,70,71. In previous studies, ferroptosis has been demonstrated to contribute to APAP-induced ALF in mouse models, is prevented by the ferroptosis-specific inhibitor ferrostatin-1, the iron chelator deferoxamine (DFO) and α-tocopherol and APAP-induced liver injury, LPO and GSH depletion have been reduced by ferroptosis inhibitors. These results suggest that GSH depletion-driven iron depletion may be a major mechanism of APAP-induced toxicity. Mass spectrometry and genetic inhibition of ACSL4 demonstrate that free radical oxidation of n-6 polyunsaturated fatty acids causes APAP-driven hepatocyte ferroptosis17. There have been numerous studies examining the role of ferrostatin-1, a specific ferroptosis inhibitor, in APAP-induced cell death in vitro. Ferrostatin-1 is a free radical scavenger and has been demonstrated to protect liver cells from the cellular damage caused by APAP72. As demonstrated by Schnellmann et al., DFO inhibits APAP-induced liver injury in a dose-dependent manner. Ferrostatin-1 prevented APAP-induced cell death both in vitro and in vivo. Although ferrostatin-1 had no effect on CYP2E1 expression, pretreatment with this inhibitor resulted in increased GSH levels three hours following APAP treatment.17,72. According to RNA-seq data and qPCR validation in another group of mice, MnSOD mRNA levels in mouse livers were not upregulated after APAP challenge. However, whether SOD-mediated endogenous defenses are toxic is unknown73. In our study, a strong occurrence of both cell death and lipid peroxidation was observed in the livers of APAP-treated mice, accompanied by decreased GPX4 expression and decreased GSH content. The ferroptosis inhibitor ferrostatin-1 greatly attenuated the above changes caused by APAP. This evidence may support that ferroptosis is involved in the pathogenesis of ALF. In addition, our findings show that inhibiting the expression of GAS1 had significant protective and therapeutic effects on APAP-induced ALF. However, the mechanism of APAP-induced ALF is unclear. More research is needed to clarify this in the future.
Conclusion
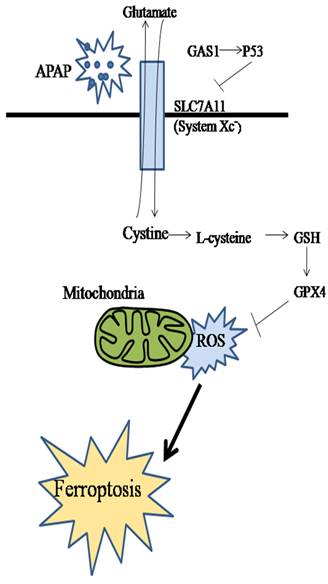
In conclusion, we studied the effect of GAS1 on ferroptosis in APAP-induced ALF and found that lipid peroxidation and redox imbalance play key roles in this process. Inhibiting the expression of GAS1 had significant protective and therapeutic effects on APAP-induced ALF (Figure 6). Therefore, ferroptosis may be an effective target for the treatment of APAP-induced ALF. Moreover, GAS1 may be a potential target for ferroptosis that can be used in the clinical treatment of ALF in the future.
Schematic diagram of the effect and underlying mechanism of GAS1 on ferroptosis in APAP-induced ALF.
Abbreviations
ALF, acute liver failure; APAP, acetaminophen; GAS1, growth arrest-specific1; GPX4, glutathione peroxidase 4; NAPQI, N-acetyl-p-benzoquinone imineh; ROS, reactive oxygen species; MDA, malondialdehyde; GSH, glutathione; ALT, alanine transaminase; AST, aspartate transaminase; LDH, lactic dehydrogenase; RT-qPCR, reverse transcription quantitative polymerase chain reaction; shRNA, short hairpin RNA; AAV, adeno-associated virus; DMSO, dimethyl sulfoxide; MMP, mitochondrial membrane potential.
Supplementary Material
Supplementary figure.
Acknowledgements
The manuscript was copy edited for English language by wiley editing services, with regard to grammar, punctuation, spelling, and clarity.
Funding
This study was supported by the Key Project supported by Medical Science and Technology Development Foundation of Nanjing Department of Health (YKK16108 and YKK19071) and National Natural Science Foundation of China (82371850 and 82171737).
Author Contributions
WJZ and CPJ designed the study and wrote the protocol. JQT and CLX performed the experiments and analyzed the data. XDW, HHC and QYL managed the figure drawing. JQT wrote the first draft of the manuscript. All authors contributed to improve the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rajaram P, Subramanian R. Acute Liver Failure. Semin Respir Crit Care Med. 2018;39(5):513-522
2. Squires JE, McKiernan P, Squires RH. Acute Liver Failure: An Update. Clin Liver Dis. 2018;22(4):773-805
3. Bantel H, Schulze-Osthoff K. Mechanisms of cell death in acute liver failure. Front Physiol. 2012;3:79
4. Rumack BH, Bateman DN. Acetaminophen and acetylcysteine dose and duration: past, present and future. Clin Toxicol (Phila). 2012;50(2):91-98
5. Craig DG, Bates CM, Davidson JS, Martin KG, Hayes PC, Simpson KJ. Staggered overdose pattern and delay to hospital presentation are associated with adverse outcomes following paracetamol-induced hepatotoxicity. Br J Clin Pharmacol. 2012;73(2):285-294
6. Pakravan N, Waring WS, Sharma S, Ludlam C, Megson I, Bateman DN. Risk factors and mechanisms of anaphylactoid reactions to acetylcysteine in acetaminophen overdose. Clin Toxicol (Phila). 2008;46(8):697-702
7. Krenkel O, Mossanen JC, Tacke F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg Nutr. 2014;3(6):331-343
8. Yuan L, Kaplowitz N. Mechanisms of drug-induced liver injury. Clin Liver Dis. 2013;17(4):507-518 vii
9. Noh JR, Kim YH, Hwang JH. et al. Sulforaphane protects against acetaminophen-induced hepatotoxicity. Food Chem Toxicol. 2015;80:193-200
10. Ding Y, Li Q, Xu Y. et al. Attenuating Oxidative Stress by Paeonol Protected against Acetaminophen-Induced Hepatotoxicity in Mice. PLoS One. 2016;11(5):e0154375
11. Zhang J, Guo J, Yang N, Huang Y, Hu T, Rao C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022;13(12):1051
12. Li H, Weng Q, Gong S. et al. Kaempferol prevents acetaminophen-induced liver injury by suppressing hepatocyte ferroptosis via Nrf2 pathway activation. Food Funct. 2023;14(4):1884-1896
13. Dixon SJ, Lemberg KM, Lamprecht MR. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072
14. Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26(3):165-176
15. Wang Y, Chen Q, Shi C, Jiao F, Gong Z. Mechanism of glycyrrhizin on ferroptosis during acute liver failure by inhibiting oxidative stress. Mol Med Rep. 2019;20(5):4081-4090
16. Macias-Rodriguez RU, Inzaugarat ME, Ruiz-Margain A, Nelson LJ, Trautwein C, Cubero FJ. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis-A Novel Form of Non-Apoptotic Cell Death? Int J Mol Sci. 2020 21(5)
17. Yamada N, Karasawa T, Kimura H. et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020;11(2):144
18. Stockwell BR, Friedmann Angeli JP, Bayir H. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273-285
19. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73(11-12):2195-2209
20. Uchiyama A, Kim JS, Kon K. et al. Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology. 2008;48(5):1644-1654
21. Brigelius-Flohe R, Flohe L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid Redox Signal. 2020;33(7):498-516
22. Yang WS, SriRamaratnam R, Welsch ME. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1-2):317-331
23. Kagan VE, Mao G, Qu F. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81-90
24. Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2-3):65-87
25. Huang C, Yang M, Deng J, Li P, Su W, Jiang R. Upregulation and activation of p53 by erastin-induced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol Rep. 2018;40(4):2363-2370
26. Mellstrom B, Cena V, Lamas M, Perales C, Gonzalez C, Naranjo JR. Gas1 is induced during and participates in excitotoxic neuronal death. Mol Cell Neurosci. 2002;19(3):417-429
27. Segovia J, Zarco N. Gas1 is a pleiotropic regulator of cellular functions: from embryonic development to molecular actions in cancer gene therapy. Mini Rev Med Chem. 2014;14(14):1139-1147
28. Simoneau AR, Spruck CH 3rd, Gonzalez-Zulueta M. et al. Evidence for two tumor suppressor loci associated with proximal chromosome 9p to q and distal chromosome 9q in bladder cancer and the initial screening for GAS1 and PTC mutations. Cancer Res. 1996;56(21):5039-5043
29. Konstantinova LN, Fleischman EW, Knisch VI, Perevozchikov AG, Kopnin BP. Karyotype peculiarities of human colorectal adenocarcinomas. Hum Genet. 1991;86(5):491-496
30. Del Sal G, Ruaro ME, Philipson L, Schneider C. The growth arrest-specific gene, gas1, is involved in growth suppression. Cell. 1992;70(4):595-607
31. Gobeil S, Zhu X, Doillon CJ, Green MR. A genome-wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes Dev. 2008;22(21):2932-2940
32. Leem YE, Han JW, Lee HJ. et al. Gas1 cooperates with Cdo and promotes myogenic differentiation via activation of p38MAPK. Cell Signal. 2011;23(12):2021-2029
33. Sacilotto N, Castillo J, Riffo-Campos AL. et al. Growth Arrest Specific 1 (Gas1) Gene Overexpression in Liver Reduces the In Vivo Progression of Murine Hepatocellular Carcinoma and Partially Restores Gene Expression Levels. PLoS One. 2015;10(7):e0132477
34. Del Sal G, Ruaro EM, Utrera R, Cole CN, Levine AJ, Schneider C. Gas1-induced growth suppression requires a transactivation-independent p53 function. Mol Cell Biol. 1995;15(12):7152-7160
35. Evdokiou A, Cowled PA. Tumor-suppressive activity of the growth arrest-specific gene GAS1 in human tumor cell lines. Int J Cancer. 1998;75(4):568-577
36. Li Z, Wang JW, Wang WZ. et al. Natriuretic peptide receptor A inhibition suppresses gastric cancer development through reactive oxygen species-mediated G2/M cell cycle arrest and cell death. Free Radic Biol Med. 2016;99:593-607
37. Akie TE, Cooper MP. Determination of Fatty Acid Oxidation and Lipogenesis in Mouse Primary Hepatocytes. J Vis Exp. 2015(102):e52982.
38. Zhang W, Zhangyuan G, Wang F. et al. The zinc finger protein Miz1 suppresses liver tumorigenesis by restricting hepatocyte-driven macrophage activation and inflammation. Immunity. 2021;54(6):1168-1185 e1168
39. Jin K, Shi Y, Zhang H. et al. A TNFalpha/Miz1-positive feedback loop inhibits mitophagy in hepatocytes and propagates nonalcoholic steatohepatitis. J Hepatol. 2023
40. Wang Z, Chen J, Zeng Z. et al. The LOX-1 receptor ectopically expressed in the liver alleviates atherosclerosis by clearing Ox-LDL from the circulation. Mol Med. 2022;28(1):26
41. Liu K, Chen X, Ren Y. et al. Identification of a novel farnesoid X receptor agonist, kaempferol-7-O-rhamnoside, a compound ameliorating drug-induced liver injury based on virtual screening and in vitro validation. Toxicol Appl Pharmacol. 2022;454:116251
42. Zhao L, Zhang J, Hu C. et al. Apigenin Prevents Acetaminophen-Induced Liver Injury by Activating the SIRT1 Pathway. Front Pharmacol. 2020;11:514
43. Wang J, Zhu Q, Li R, Zhang J, Ye X, Li X. YAP1 protects against septic liver injury via ferroptosis resistance. Cell Biosci. 2022;12(1):163
44. Bertrand RL. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med Hypotheses. 2017;101:69-74
45. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124-137
46. Dixon SJ, Patel DN, Welsch M. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523
47. Shan S, Shen Z, Song F. Autophagy and acetaminophen-induced hepatotoxicity. Arch Toxicol. 2018;92(7):2153-2161
48. Cai X, Cai H, Wang J. et al. Molecular pathogenesis of acetaminophen-induced liver injury and its treatment options. J Zhejiang Univ Sci B. 2022;23(4):265-285
49. Sun YK, Zhang YF, Xie L. et al. Progress in the treatment of drug-induced liver injury with natural products. Pharmacol Res. 2022;183:106361
50. Xie Y, Hou W, Song X. et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369-379
51. Bloomer SA, Kregel KC, Brown KE. Heat stress stimulates hepcidin mRNA expression and C/EBPalpha protein expression in aged rodent liver. Arch Gerontol Geriatr. 2014;58(1):145-152
52. Wang Y, Zheng L, Shang W. et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. 2022;29(11):2190-2202
53. Li J, Lu Q, Peng M. et al. Water extract from Herpetospermum pedunculosum attenuates oxidative stress and ferroptosis induced by acetaminophen via regulating Nrf2 and NF-kappaB pathways. J Ethnopharmacol. 2023;305:116069
54. Lewerenz J, Hewett SJ, Huang Y. et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18(5):522-555
55. Capelletti MM, Manceau H, Puy H, Peoc'h K. Ferroptosis in Liver Diseases: An Overview. Int J Mol Sci. 2020 21(14)
56. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170(6):1062-1078
57. Jiang L, Kon N, Li T. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57-62
58. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162-168
59. Eguchi A, Wree A, Feldstein AE. Biomarkers of liver cell death. J Hepatol. 2014;60(5):1063-1074
60. Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90(3):1165-1194
61. Mishima E, Sato E, Ito J. et al. Drugs Repurposed as Antiferroptosis Agents Suppress Organ Damage, Including AKI, by Functioning as Lipid Peroxyl Radical Scavengers. J Am Soc Nephrol. 2020;31(2):280-296
62. Schoeneberger H, Belz K, Schenk B, Fulda S. Impairment of antioxidant defense via glutathione depletion sensitizes acute lymphoblastic leukemia cells for Smac mimetic-induced cell death. Oncogene. 2015;34(31):4032-4043
63. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021-1032
64. Liu J, Song X, Kuang F. et al. NUPR1 is a critical repressor of ferroptosis. Nat Commun. 2021;12(1):647
65. Jaeschke H, Adelusi OB, Ramachandran A. Ferroptosis and Acetaminophen Hepatotoxicity: Are We Going Down Another Rabbit Hole? Gene Expr. 2021;20(3):169-178
66. Knight TR, Fariss MW, Farhood A, Jaeschke H. Role of lipid peroxidation as a mechanism of liver injury after acetaminophen overdose in mice. Toxicol Sci. 2003;76(1):229-236
67. Jaeschke H, Ramachandran A. Oxidant Stress and Lipid Peroxidation in Acetaminophen Hepatotoxicity. React Oxyg Species (Apex). 2018;5(15):145-158
68. Ramachandran A, Jaeschke H. Oxidant Stress and Acetaminophen Hepatotoxicity: Mechanism-Based Drug Development. Antioxid Redox Signal. 2021;35(9):718-733
69. Ramachandran A, Lebofsky M, Weinman SA, Jaeschke H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2011;251(3):226-233
70. An Y, Luo Q, Han D, Guan L. Abietic acid inhibits acetaminophen-induced liver injury by alleviating inflammation and ferroptosis through regulating Nrf2/HO-1 axis. Int Immunopharmacol. 2023;118:110029
71. Zeng Y, Wu R, Wang F. et al. Liberation of daidzein by gut microbial beta-galactosidase suppresses acetaminophen-induced hepatotoxicity in mice. Cell Host Microbe. 2023;31(5):766-780 e767
72. Lorincz T, Jemnitz K, Kardon T, Mandl J, Szarka A. Ferroptosis is Involved in Acetaminophen Induced Cell Death. Pathol Oncol Res. 2015;21(4):1115-1121
73. Niu B, Lei X, Xu Q. et al. Protecting mitochondria via inhibiting VDAC1 oligomerization alleviates ferroptosis in acetaminophen-induced acute liver injury. Cell Biol Toxicol. 2022;38(3):505-530
Author contact
![]() Corresponding author: drzhangwjcom, chunpingjiangcom.
Corresponding author: drzhangwjcom, chunpingjiangcom.