Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Overview of the molecular...
Critical genetic markers and...
The progression of glioma models
Gene editing techniques in...
Conclusions and Perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2022; 19(14):2071-2079. doi:10.7150/ijms.77287 This issue Cite
Review
Current understanding of gliomagenesis: from model to mechanism
Juanjuan Luo1,2#, Muhammad Junaid1,3#, Naima Hamid1,3#, Jin-Jing Duan1, Xiaojun Yang2 ![]() , De-Sheng Pei1
, De-Sheng Pei1 ![]()
1. School of Public Health and Management, Chongqing Medical University, Chongqing 400016, China.
2. Guangdong Provincial Key Laboratory of Infectious Disease and Molecular Immunopathology, Shantou University Medical College, Shantou 515041, China.
3. University of Chinese Academy of Sciences, Beijing 100049, China.
#These authors contributed equally to this work.
Received 2022-7-20; Accepted 2022-11-3; Published 2022-11-14
Abstract

Glioma, a kind of central nervous system (CNS) tumor, is hard to cure and accounts for 32% of all CNS tumors. Establishing a stable glioma model is critically important to investigate the underlying molecular mechanisms involved in tumorigenesis and tumor progression. Various core signaling pathways have been identified in gliomagenesis, such as RTK/RAS/PI3K, TP53, and RB1. Traditional methods of establishing glioma animal models have included chemical induction, xenotransplantation, and genetic modifications (RCAS/t-va system, Cre-loxP, and TALENs). Recently, CRISPR/Cas9 has emerged as an efficient gene editing tool with high germline transmission and has extended the scope of stable and efficient glioma models that can be generated. Therefore, this review will highlight the documented evidence about the molecular characteristics, critical genetic markers, and signaling pathways responsible for gliomagenesis and progression. Moreover, methods of establishing glioma models using gene editing techniques and therapeutic aspects will be discussed. Finally, the prospect of applying gene editing in glioma by using CRISPR/Cas9 strategy and future research directions to establish a stable glioma model are also included in this review. In-depth knowledge of glioma signaling pathways and use of CRISPR/Cas9 can greatly assist in the development of a stable, efficient, and spontaneous glioma model, which can ultimately improve the effectiveness of therapeutic responses and cure glioma patients.
Keywords: Glioma, Tumorigenesis, Signaling pathways, CRISPR/Cas9, Model
Introduction
Glioma is a common human brain tumor that accounts for approximately 50% of intracranial tumors. Although the current incidence of center nervous system (CNS) tumors is 14.3% of all tumors, 49.1% of nervous system tumors are malignant gliomas [1]. Advanced gliomas, which are also named primary glioblastoma (GBM), are considered one of the deadliest malignant tumors in the world [2]. Glioma incidence varies by age; for example, common central nervous system (CNS) tumors in children include pilocytic astrocytomas and embryonal tumors, whereas common CNS tumors in adults include meningiomas, pituitary tumors, and malignant gliomas [3]. In addition to age differences, glioma incidence varies by sex, ethnicity, tumor histology, and tumor subtype. Besides, the CNS microenvironment is naturally equipped to control proliferative cells and lead to cancer development [4-6]. Recently, a new classification system for glioma tumors by integrating both tumor morphology and biomolecular features [7]. The classification evaluated the correlation between microarray expression profiling of gliomas and tumor grades, progression, malignancy, and prognosis to identify the molecular subtypes [8]. A recent study indicated that the characteristics of gene expression in the gliomas derived from transgenic zebrafish supported the notion that different molecular mutation-selective pressures drive different progression of gliomagenesis [9].
In recent decades, the survival rate of glioma patients has remained low due to poor prognosis, with less than 5% five-year survival rate, and the prognosis is even worse in elderly patients with glioma [10]. Drug-based curative measures are not ideal for treating glioma because of insufficient understanding of the molecular mechanisms associated with gliomagenesis [11, 12]. In addition, the limited and vague identification of early-stage gliomas also leads to delays in glioma treatment. Recently, genetic markers were proven to be effective clinical signs that can be measured with precision to predict glioma outcome [13]. To date, three core signaling pathways have been identified for high-grade glioma, namely, the receptor tyrosine kinase (RTK)/phosphatidylinositol 3'-kinase (PI3K)/alpha serine-threonine protein kinase (AKT), phosphoprotein53 (TP53), and retinoblastoma (RB1) signaling pathways [14-16]. Therefore, it is necessary to modify the current techniques and establish new methods to improve the prognosis of glioma patients.
Cancer cells have the fundamental features of growth and survival beyond a nominal homeostatic or favorable environment, and their growth, survival, oncogenic proliferation, and apoptosis are governed by signaling pathways [12]. Several methods of genetic modification, including RCAS/t-va, Cre-loxP, zinc finger nucleases (ZFNs), and Tal-effector nuclease (TALENs), have introduced a new dimension in the development of genome manipulation at the molecular level [17-20]. However, these gene editing techniques have various limitations, such as high cost, difficult design, labor intensiveness, low efficiency, and time consumption [21]. Specifically, CRISPR/Cas9 system, a powerful gene-editing tool, includes a guide RNA (gRNA) and programmable nuclease (Cas9) that can process double-strand breaks (DSBs) at specific target sites efficiently and accurately via homology-directed repair (HDR) or nonhomologous end joining (NHEJ) [22, 23]. Therefore, it is essential to highlight the benefits of developing a new glioma model using CRISPR/Cas9 to better understand the molecular mechanisms of gliomagenesis and establish better therapeutic methods.
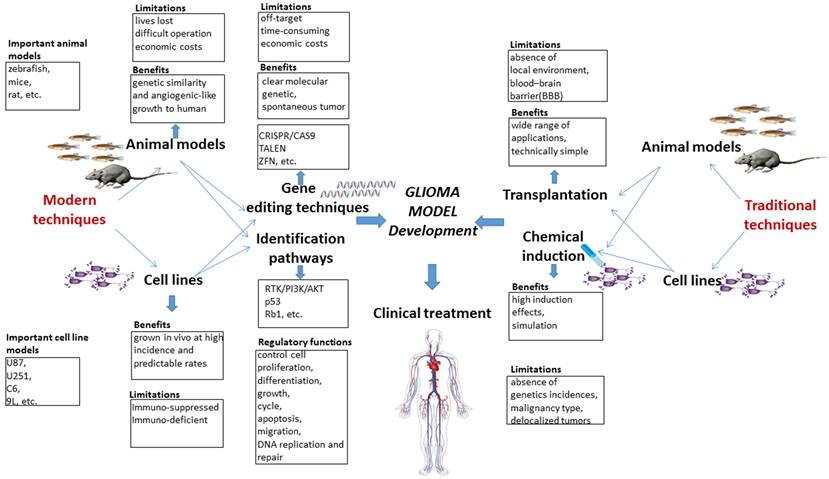
Here, we presented a systematic view of the molecular characteristics of gliomagenesis in the context of genetic markers and core signaling pathways. Furthermore, comparisons are drawn among traditional and modern methods for establishing glioma models. Moreover, the advantages and limitations of traditional and modern gene editing techniques are critically discussed. The potential applications of CRISPR/Cas9 system for generating glioma models are specifically highlighted, and the clinical trials associated with gene therapy are briefly discussed. Finally, clinical trials and available glioma therapies are also briefly discussed in the current study. A summary highlighting the pros and cons of traditional and modern glioma models, gene editing techniques, key signaling pathways and their regulatory function is presented in the form of a schematic diagram (Fig. 1).
Schematic diagram highlighting the pros and cons of traditional and modern glioma models, gene editing techniques and key pathways and their regulatory functions.
Overview of the molecular classification of gliomas
Previous studies indicated that the new molecular classification system for gliomas might be valuable for predicting the prognostic of the glioma patients [24, 25]. We therefore summarized the molecular signatures associated with tumor aggressiveness and progression, as well as the correlation between these signatures and the signaling pathways implicated in gliomagenesis [8, 17]. Several subtypes, including classical, neural, proneural and mesenchymal subtypes, were named based on the expression of signature genes (Fig. 2).
Characteristics of the molecular classification of gliomas.
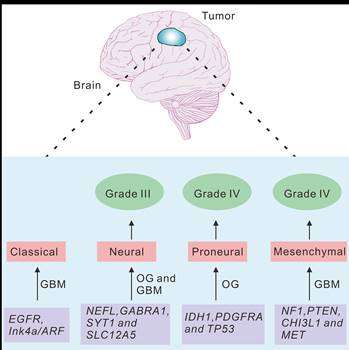
Classical subtype
Chromosome 10 (Chr 10) loss combined with chromosome 7 (Chr 7) amplification was detected in all classical subtype, and frequently occurred in GBM patients. Although the Chr 7 amplification was also observed in other subtypes of glioma, the highly expression of EGFR was infrequently determined in other subtypes. It is known that TP53 mutation is one of the most frequently event in GBM patients. However, compared with the highly frequency of EGFR mutation, TP53 mutation is rarely observed in classical subtype [14]. In addition, homozygous deletion of 9p21.3, which targets CDKN2A, was a frequent and significantly associated event in classical subtype, and accompanied with EGFR amplification in most of classical subtype samples. Furthermore, previous report showed that the neural precursor and stem cell marker NES, and the regulators of Notch and Sonic hedgehog signaling pathways, including NOTCH3, JAG1, LFNG, SMO, GAS1 and GLI2, were frequently amplified in classical subtype [16].
Neural subtype
The neural subtype was identified by the amplification of several neuron markers, including SYT1, GABRA1, NEFL and SLC12A5, which were involved in the processes of neuron projection and axon and synaptic transmission [16]. In addition, extensive pathological analysis confirmed the diagnosis of GBM events in samples of this subtype [16]. This classification offers a markedly better prognosis and includes the expression of certain genes, which were involved in the process of neurogenesis in the normal brain [8].
Proneural subtype
The mutations of PDGFRA and IDH1 were two major features in proneural subtype. Although PDGFRA was amplified in almost all subtypes of GBM, it was amplified with a much higher rate in proneural subtype [8]. However, the characteristic signature of PDGFRA in proneural samples is accompanied by focal amplification and highly expression of PDGFRA gene [16, 26]. In addition, ten out of sixteen PIK3CA/PIK3R1 mutations were observed in proneural subtype, most of which did not have PDGFRA abnormalities, whereas eleven out of twelve mutations of IDH1 gene were determined in this subtype with no PDGFRA abnormality [16]. It is noted that the mutations of TP53 were also frequently detected in this subtype. In contrast, the classical GBM event, the amplification of Chr 7 paired with the loss of Chr 10, were distinctly less prevalent and only occurred in 54% proneural subtype samples. The highly expressed oligodendrocyte development genes, including PDGFRA, NKX2-2 and OLIG2, were detected in proneural subtype [27]. Notably, the amplification of OLIG2 could promote proliferation and induce tumorigenesis through downregulating CDKN1A expression [28].
Mesenchymal subtype
The mesenchymal subtype mainly exhibited the amplification of the mesenchymal markers, including CHI3L1 and MET [8]. Focal hemizygous deletions of NF1 predominantly occurred in the mesenchymal subtype [16]. Verhaak et al. reported that, 70% samples were defined as mesenchymal subtype in NF1 mutated samples. In addition, most of mesenchymal subtype samples were detected as NF1 and PTEN dual mutations, which play an important role in the RTK/PI3K pathway [16].
Critical genetic markers and signaling pathways in gliomagenesis
With the continuous development of sequencing technologies and high-throughput gene editing, it is possible to analyze the genetic and epigenetic changes in tumors [29]. In 2009, The Cancer Genome Atlas Network (TCGA) analyzed the variations in 601 types of tumor-associated genes by utilizing gene sequencing in more than 200 glioma samples [14]. TCGA analysis of DNA methylation, DNA copy number and other genetic mutations in glioma patients revealed the core glioma pathways, and their regulatory functions were summarized in the RTK/PI3K/AKT, TP53 and RB1 signaling pathways. Chow et al. found gene mutations (in Pten, Tp53 and Rb1) in mice that induced high-degree malignant tumors in astrocytes [26]. They showed that the cooperation within the aberrant Pten, Tp53, and Rb1 pathways can induce high-grade astrocytoma in the mouse brain. In this context, NF1 and PTEN, which are both involved in PI3K/AKT pathway, abrogated the major negative regulator restraining PI3K activation. RB1 regulated cell cycle, and TP53 is a tumor suppressor that regulates cell death. In addition, in glioma patients, several studies have indicated that at least one aberrant pathway among the RTK/PI3K/AKT, TP53 and RB1 signaling pathways was identified in 80-90% of glioblastomas [30, 31], which highlights their potential as therapeutic targets.
RTK/PI3K/AKT pathway
In RTK/Ras/PI3K pathway, receptor tyrosine kinases (RTKs) can mediate many growth signals through diffusive growth factors (Fig. 3A). Several ligands of RTK, including PDGFR, EGFR and VEGFR [32], are frequently overexpressed in GBM specimens [33]. In this context, the amplification of EGFR, which is the most common RTK target mutation, often couples with intragenic deletion to result in a constitutively activated form, and ultimately induces primary GBMs [34]. It is known that VEGF and its receptors are the critical regulators in glioma angiogenesis [35]. In addition, the local degradation of the vascular basement membrane and extracellular matrix play important roles in glioma angiogenesis through promoting he phosphorylation of focal adhesion kinase (FAK) [35]. Moreover, EGFR amplification is often associated with Ink4a mutation and TP53 mutation in the EGFR signaling pathway in primary GBMs [36], and the overexpression of the dual mutations of EGFR Ink4a-Arf led to the development of glioma-like lesions [37].
Critical genetic markers and signaling pathways in gliomagenesis. (A) The growth-factor-RTK signaling pathway. The ligands of RTKs, including PDGFR and EGFR, activate three downstream cascades in glioma. RAF/MEK/MAPK, PI3K/AKT, and CDC42 pathways are responsible for cell survival, metabolism, proliferation, and cytoskeletal organization, respectively. (B) TP53 and RB1 pathways. MDM2 gene contains a TP53 site, which can bind to TP53 and abolish its transcriptional activity. P14ARF could interact directly with MDM2, thereby resulting in the stabilization of both TP53 and MDM2. TP53 can promote the transcription of p21, which is capable of silencing CDKs. CDK4/6 and CDK2 can bind to D-type cyclins and E-type cyclins, respectively, and induce the release of E2F transcription factor, which activates the expression of a set of genes critical for regulating G1/S transition. CDKN2A and CDKN2B bind to CDK4/6, respectively, and inhibit CDK4/cyclin D1 complex, resulting in the inactivation of RB1-mediated G1/S transition.
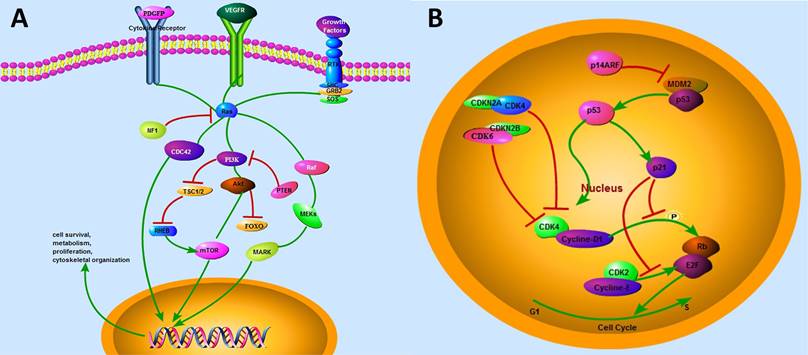
As a major effector of RTK/PI3K pathway, RAS is responsible for the activation of downstream cascades and regulates cellular proliferation and differentiation in various types of cells [38]. The RTK/RAS/PI3K pathways promote cell survival. PI3Ks, a family of heterodimeric kinases, have been reported to contain genetic alterations in 88% of GBM cases [39]. This pathway is also activated via the loss of negative regulators, such as NF1 and PTEN. As a tumor suppressor gene, NF1 and its inactivity can lead to the formation of several malignant tumors [40]. NF1 gene inactivation in glial cells and gliomas enhances the activation potential of RAS and its downstream effectors [41]. In addition, NF1 mutant-associated and sporadic astrocytoma, as well as the activations of RAF/MEK/MAPK and PI3K/AKT signaling pathways were determined in glioma patients [16]. Moreover, another regulator, PTEN acts as a dual-specificity protein phosphatase and may also impart glioma pathogenesis. PTEN mutations are frequently observed in many malignant cancers, including breast cancer and glioma [42, 43]. Moreover, mutated PTEN can cause the activation of the AKT signaling pathway, and subsequently regulate the multiple downstream AKT substrates, including FOXO and mTOR [44-46], which are usually downregulated in different types of cancers, including GBMs [47].
TP53 Pathway
In gliomas, another commonly identified pathway is TP53 signaling pathway (Fig. 3B). As the most common tumor suppressor gene, TP53 mutation is critical for the progression of glioma [48]. TP53 regulates multiple genes that control DNA repair, cell cycle, apoptosis, and progression [49]. TP53 pathway also regulates p21 (Waf1/Cip1), which is responsible for silencing cyclin-dependent protein kinases (CDKs) that are essential for the G1 to S transition. Li-Fraumeni syndrome, an inherited disorder caused by the presence of a germline mutation in TP53 gene, predisposes patients to the development of various brain tumors, including astrocytomas [50]. In addition, primary Tp53-/- astrocytes have an increased susceptibility to tumor transformation and growth [51]. Interestingly, either Tp53 homozygous (Tp53-/-) or heterozygous (Tp53+/-) mice and zebrafish fail to induce gliomagenesis [9, 52], indicating that the single Tp53 mutation might be insufficient to initiate gliomagenesis.
RB1 Pathway
RB1 pathway and its downstream effectors (Fig. 3B), were also genetically and epigenetically regulates in various malignant tumors [53, 54]. Specifically, frequent alterations in Rb1 pathway have been observed in 40-70% GBM patients [14, 54]. This pathway plays an important role in DNA repair and replication, cellular development, differentiation, migration, mitosis, and apoptosis [55-57]. RB1 pathway comprises five protein families: INK4 (p16INK4a, p15INK4b, p18INK4c and p19INK4d), cyclin-dependent protein kinases (CDK6 and CDK4), D-type cyclins (cyclins D3, D2, and D1), RB family proteins (p107, p130 and RB1), and transcription factors (E2F1-8) [58]. In this context, p16INK4a and p15INK4b are encoded by the CDKN2A and CDKN2B genes and bind to CDK4 and CDK6, respectively, to inhibit CDK4/cyclin D1 complex, which results in the inactivation of RB1-mediated G1 to S transition [59]. Therefore, the inhibition of the components of RB1 pathway might be a promising strategy for the treatment of various malignant cancer types, including astrocytoma, adenocarcinoma, basal cell carcinoma, and gastrointestinal tract endocrine tumor [60-64].
The progression of glioma models
A suitable animal model can be a useful tool for studying the mechanisms and potential treatments of glioma. Considerable variations exist in brain tumors on the basis of genetic, histological and physiological characteristics, which ultimately contributes to their different malignancies, prognoses, and invasive phenotypes [12, 65, 66]. The utility of animal models enables the systematic identification of molecular features, which contribute to the initiation and progression pathways in glioma. The identification of these molecular features could improve the therapeutic strategies of the treatment of glioma. Although the molecular mechanisms of gliomagenesis were widely investigated in cultured cells, the limitations for modeling invasion, angiogenesis and metastasis still exist. An ideal glioma animal model should have several features, including genetic similarity to human glioma, the invasive and angiogenic-like growth, the imitation of the therapeutic response, to allow for more accurately predict the clinical outcome [67]. The reviewed literature showed that mice, rats and zebrafish are the most widely used glioma animal models with methods such as chemical induction, xenografting, and bioengineering. To date, several models for brain tumors have been established, as shown in Table 1.
Traditional and modern techniques for establishing glioma animal models
| Tumor | Method | Genotype/cell line | Animal model | Remarks |
|---|---|---|---|---|
| GBM | Xenograft | U87MG and U251MG cell lines | zebrafish | A cost-effective approach to investigate glioma invasion, and high-throughput screen or evaluate anti-glioma invasion/metastasis compounds [68, 69]. |
| GBM | Chemical-induced | 9L, C6, CNS-1, F98, and RG2 | rats | Allows the immune system to interact with the developing tumor, but the genetic mechanism is not clear and the biological process is not stable [70]. |
| GBM | Xenograft | U251MG and U87MG cell lines | mice | Applied widely but is immunosuppressant [71]. |
| GBMOA | Transgenic/GFAP promoter | V12Ha-ras and EgfrvIII | mice | The EgfrvIII mutation leads to a constitutively active receptor with impaired internalization, resulting in the activation of pro-invasive signaling pathways [72]. |
| GBM | GFAP-T121 | Pten+/- | mice | Astrocytoma development is accelerated in a Pten+/- but not a Tp53+/- background [73]. |
| GBM | Gene knockout | Flox Nf1 + Tp53 | mice | A mouse model of astrocytoma with Tp53 and Nf1 mutations [74]. |
| GBMA, AA, OA | RCAS/tv-a;cre-lox system - deletion of Pten | Pdgfb and Ink4a-Arf-/- | mice | The mutations of Tp53, Arf, or Ink4a-Arf can induce higher-grade gliomas [17]. |
| GBMOA | Transgenic mice/GFAP | Pdgfpb and Tp53-/- | mice | Tp53 pathway mutations can mediate the transition from low- to high-grade glioma [29]. |
| HGA | GFAP-CreER; PtenloxP/loxP with Tp53; Rb1 double-floxed | Pten-/-, Tp53-/- and Rb-/- | mice | The combination of the mutations in Pten, Tp53, and Rb1 signaling pathways leads to GBMs in the adult brain of transgenic mice [26]. |
| GBMOA | TALEN-mediated somatic inactivation | rb-/- | zebrafish | TALEN-mediated the somatic inactivation of Rb1 induces tumorigenesis in genetic mosaic adult zebrafish [20]. |
| GBMA, AA, OA | CRISPR/Cas9 system | Pten-/-, Tp53-/- and Rb-/- | mice | CRISPR/Cas9-based strategies for establishing brain tumor model and investigating pathogenesis of brain tumors [75]. |
| GBM | U251 MG and SNB cell lines | VEGPF was correlative with vascularity and peritumoral edema in CNS tumor [76]. | ||
| GBM | Gene overexpression | EGFR and PDGFR | human tumor tissue | Overexpression of EGFR and PDGFR in glioblastomas [77]. |
| GBMA | Overexpression and deletion | p16, Rb1, CDK4 | The expression of cell cycle regulatory genes is rare in secondary glioblastomas [78]. | |
| GBM | Overexpression EGFR and deletion p16, p53 | EGFR and p16, TP53 | human tumor and blood samples | The genetic subtypes were divided into EGFR amplification, TP53 mutation, and CDKN2 deletion in GBM patients [79, 80]. |
| GBM | GFAP-Cre; Nf1flox/mut | Nf1+/- | mice | mTOR inhibition can suppress Nf1 optic glioma growth [41]. |
| glioma | Transplantation U87 cells | EGFR | zebrafish | The xenografted glioma cells can induce angiogenesis through regulating VEGF expression in zebrafish [81, 82]. |
| GBMAA | RCAS/t-va | Tp53-/-, Ink4a-Arf-/- | mice | The application of RCAS/t-va technology for establishing glioma mouse model [17]. |
| glioma | CRISPR/Cas9 system | nf1-/-, tp53-/- and rb1-/- | zebrafish | The crosstalk of three core signaling pathways, including RTK/Ras/PI3K, RB, and TP53 pathways, in gliomagenesis [9]. |
A, astrocytoma; AA, anaplastic astrocytoma; GS, gliosarcoma; OA, oligoastrocytoma; OG, oligodendroglioma.
Chemical Induction
Chemical induction was the first method attempted to induce brain tumors in animal models [67]. These induced tumor models did not have a defined genetic background because induction occurred through random chemical substances of bases that gave rise to mispairing or point mutations. Therefore, the location of tumorigenesis, malignancy type, and the incidence conditions were greatly various in each chemical induction study [70, 83, 84]. In addition, the tumors induced by the exposure to chemicals is hard to reflect a true representation of the tumorigenesis and tumor progression involved in the patients. Although the application of chemical substances in the generation of glioma models can produce substantial effects and simulate the natural conditions of human glioma, the genetic mechanisms were unclear, and the biological pathways were unspecified [85]. Therefore, these critical flaws in traditional glioma models further limit the applied perspective.
Xenograft Tumor Models
Xenograft tumor models are helpful for studying tumor developmental stages, angiogenesis, invasion, metastasis and the relationship to the host [86, 87]. By transplanting CD133-positive and CD133-negative U87 glioma cells in zebrafish, our previous study indicated that glioma stem cells have higher invasion ability than differentiated glioma cells through regulating MMP9 expression [68]. A previous report also indicated that the transplantation of U251 cells into zebrafish larvae could evaluate the efficacy and toxicity for anti-cancer molecular compounds [69]. Oka et al. showed an adenovirus-mediated REIC/Dkk-3 gene therapy in GL261 glioma cells xenografted mouse model [88]. However, although in the ideal state, these xenograft tumor models are technically simple and have lower morbidity rates, these models still do not perfectly represent the actual circumstances of human glioma because of the lack of immunoreactivity in the xenotransplantation model of human glioma cells in immunosuppressed or immunodeficient hosts [89].
Transgenic Tumor Models
Transgenic models involve molecular-level manipulation of the animal genome in the form of gene editing strategy to induce tumorigenesis [90]. Brinster et al. first developed characteristic brain tumors within the choroid plexus via microinjection with the plasmid contains the SV40 early region genes and a metallothionein fusion gene in transgenic mice [91]. In addition, previous studies indicated several altered signaling pathways, including ErbB family [92], EGFR and EGFRvIII signaling [93], were detected in most of human gliomas. Moreover, TP53 and PTEN were mutated in 30-40% glioma patients, suggesting these two tumor suppressor genes are the most frequently altered genes in gliomagenesis. Notably, although these two mutations are commonly found in human gliomas from low-grade to GBMs, previous study showed that the tumors generated in transgenic mice with double mutation of Pten and TP53 were high-grade gliomas (grades III and IV) [36]. Further investigation indicated that Tp53, Pten or Rb1 signaling pathway plays different role during gliomagenesis in transgenic mice and zebrafish, and their cooperation could result in high-grade astrocytomas (grades III and IV) in astrocytes and neural precursors in transgenic mice and zebrafish, suggesting that they have different functions for the initiation and progression of gliomagenesis [9, 26].
Gene editing techniques in studying gliomagenesis
Genome editing nucleases, including ZFNs, TALENS and CRISPR/Cas9, are also emerging tools to detect abnormal gene function in cancer. Gene editing technology has been successfully utilized in mice to study abnormal gene expression during tumorigenesis in liver and lung cancers [94, 95]. A previous study established a glioblastoma zebrafish model by TALEN-mediated somatic inactivation of Rb1 using two independent TALEN pairs in zebrafish embryos, which resulted in high-frequency tumor development, mainly in the brain [20]. In addition, RCAS/tv-a technology is also commonly used to generate brain tumor models. RCAS-PDGFB-injected SVZ can potentially cause higher grade glioma with a 100% incidence rate and shorter latency. It is noted that the mutations of several tumor suppressor genes, including Arf, Ink4a-Arf and Tp53, has also been determined during tumorigenesis [17]. However, although these gene editing technologies could effectively and precisely perform genome editing, their application has been restricted due to factors such as high cost and the difficulty of designing these endonucleases.
In contrast to ZFN and TALEN systems, CRISPR/Cas9 system could efficiently identify any target sequences with convenient design [75, 96]. This system based on gRNA, which enables the recognition of the targeted DNA sequences by Cas9 endonuclease, which can subsequently cleave both strands of interest [97-100]. In addition, CRISPR/Cas9 is a better technique because of the ease of plasmid construction. The annealing oligonucleotides specify the target site and link into the Cas9 gene-contained vector, which is much easier than the construction of the vector that contains new DNA-binding domain in TALENs or ZFNs system [22]. Moreover, the simultaneous generation of multiple mutations is another benefit of CRISPR/Cas9 system, which is very important for establishing disease models because many diseases develop as a result of the abnormal expression of multiple genes rather than dysfunction in a single gene. Thus, CRISPR/Cas9-based glioma models are more suitable for studying the molecular mechanisms of gliomagenesis [21].
Tumorigenesis is often caused by factors such as chromosome aberrations, base deletions or other mutations that ultimately lead to tumor-related genetic modifications. With this novel genomic technology, interactions among molecular mechanisms can be identified in different stages of glioma development. Coupled with the exploitation of relevant signaling pathways, various glioma subtypes can be classified via specific gene expression profiles. CRISPR/Cas9 system is therefore specific, fast, and easy, can lead to stable glioma tumor outcomes and can overcome the limitations of traditional gene editing methods. The CRISPR system can also provide a method to establish tumor models that will be used to study various novel drug target genes and resistance genes for drug treatments [21, 101]. Several efforts to establish brain tumor models via CRISPR/Cas9 technology have been successful (Table 1), suggesting that the CRISPR/Cas9-based gene editing technique might open the door to modeling pathological conditions to disclose the mysteries of diverse neurological diseases, including glioma. A previous study indicated the CRISPR/Cas9-mediated inhibition of miR-10b could abolished the neoplastic transformation of normal astrocytes in human glioma cells [102]. Luo et al also established a series of transgenic fish lines to tissue-specifically knockout several tumor suppressor genes, including nf1, tp53, or rb1, in gliocytes of brain tissue [9]. Therefore, this gene editing technology might provide a promising platform for genetically studying tumorigenesis and make it convenient to explore treatment methods for cancer drug targets inspired by CRISPR/Cas9-based genetic screens [103].
Conclusions and Perspectives
In conclusion, this review highlights the molecular aspects of glioma induction and provides a theoretical basis for establishing a glioma model. Furthermore, the expression and regulation of relevant genes in terms of core pathways have been discussed. Moreover, the method of establishing glioma models using the novel gene editing technology CRISPR/Cas9 and the significance of targeted gene therapy were explained. We believe that the CRISPR/Cas9 technology has a significant potential to enhance the understanding of the mechanisms of gliomagenesis, and enable the development of glioma models and novel and efficient treatment methods.
Acknowledgements
This work was supports by grants from the High-level Talents Project of Chongqing Medical University (R4014), CAS Team Project of the Belt and Road, Research Program of Chongqing Science and Technology Commission (cstc2019jcyj-zdxmX0035, CSTCCXLJRC201714).
Author Contributions
JL, XY and DSP discussed and designed the study. JL, MJ, NH and JD drafted the manuscript. XY and DSP revised the article. All authors contributed to the article and approved the submitted version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ostrom QT, Cioffi G, Waite K. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol. 2021;23:iii1-iii105
2. Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol. 2014;16:334-51
3. McNeill KA. Epidemiology of brain tumors. Neurol Clin. 2016;34:981-98
4. Altieri R, Barbagallo D, Certo F. et al. Peritumoral microenvironment in high-grade gliomas: from FLAIRectomy to microglia-glioma cross-talk. Brain Sci. 2021;11:200
5. De Luca C, Virtuoso A, Papa M. et al. Regional development of glioblastoma: the anatomical conundrum of cancer biology and its surgical implication. Cells. 2022;11:1349
6. Longhitano L, Vicario N, Forte S. et al. Lactate modulates microglia polarization via Igfbp6 expression and remodels tumor microenvironment in glioblastoma. Cancer Immunol Immunother. 2022 [Epub ahead of print]
7. Louis DN, Perry A, Wesseling P. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231-51
8. Phillips HS, Kharbanda S, Chen RH. et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157-73
9. Luo J, Liu P, Lu C. et al. Stepwise crosstalk between aberrant Nf1, Tp53 and Rb signalling pathways induces gliomagenesis in zebrafish. Brain. 2021;144:615-35
10. Van Meir EG, Hadjipanayis CG, Norden AD. et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA-Cancer J Clin. 2010;60:166-93
11. Kane JR, Miska J, Young JS. et al. Sui generis: gene therapy and delivery systems for the treatment of glioblastoma. Neuro Oncol. 2015;17:Ii24-Ii36
12. Wong MLH, Kaye AH, Hovens CM. Targeting malignant glioma survival signalling to improve clinical outcomes. J Clin Neurosci. 2007;14:301-8
13. Law M, Young RJ, Babb JS. et al. Gliomas: predicting time to progression or survival with cerebral blood volume measurements at dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging. Radiology. 2008;247:490-8
14. Chin L, Meyerson M, Aldape K. et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8
15. Johnson BE, Mazor T, Hong C. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189-93
16. Verhaak RGW, Hoadley KA, Purdom E. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98-110
17. Hambardzumyan D, Amankulor NM, Helmy KY. et al. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol. 2009;2:89-95
18. Marumoto T, Tashiro A, Friedmann-Morvinski D. et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110-6
19. Hilton IB, Gersbach CA. Enabling functional genomics with genome engineering. Genome Res. 2015;25:1442-55
20. Solin SL, Shive HR, Woolard KD. et al. Rapid tumor induction in zebrafish by TALEN-mediated somatic inactivation of the retinoblastoma1 tumor suppressor rb1. Sci Rep. 2015;5:13745
21. Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15:387-95
22. Jiang WY, Marraffini LA. CRISPR-Cas: new tools for genetic manipulations from bacterial immunity systems. Annu Rev Microbiol. 2015;69:209-28
23. Tu ZC, Yang WL, Yan S. et al. CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol Neurodegener. 2015;10:35
24. Freije WA, Castro-Vargas FE, Fang ZX. et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503-10
25. Nutt CL, Mani DR, Betensky RA. et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003;63:1602-7
26. Chow LML, Endersby R, Zhu XY. et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19:305-16
27. Noble M, Proschel C, Mayer-Proschel M. Getting a GR(i)P on oligodendrocyte development. Dev Biol. 2004;265:33-52
28. Ligon KL, Huillard E, Mehta S. et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503-17
29. Weiss WA, Burns MJ, Hackett C. et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. 2003;63:1589-95
30. Liu KW, Feng HZ, Bachoo R. et al. SHP-2/PTPN11 mediates gliomagenesis driven by PDGFRA and INK4A/ARF aberrations in mice and humans. J Clin Invest. 2011;121:905-17
31. Zhu H, Acquaviva J, Ramachandran P. et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. P Natl Acad Sci USA. 2009;106:2712-6
32. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669-76
33. Liu TR, Ma WJ, Xu HN. et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat Commun. 2018;9:3439
34. Eskilsson E, Rosland GV, Solecki G. et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2018;20:743-52
35. Ahir BK, Engelhard HH, Lakka SS. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol Neurobiol. 2020;57:2461-78
36. Zheng HW, Ying HQ, Yan HY. et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129-33
37. Saadeh FS, Mahfouz R, Assi HI. EGFR as a clinical marker in glioblastomas and other gliomas. Int J Biol Marker. 2018;33:22-32
38. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287-92
39. Samuels Y, Wang ZH, Bardelli A. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554
40. Rosenbaum T, Wimmer K. Neurofibromatosis type 1 (NF1) and associated tumors. Klin Padiatr. 2014;226:309-15
41. Kaul A, Toonen JA, Cimino PJ. et al. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol. 2015;17:843-53
42. Abounader R. Interactions between PTEN and receptor tyrosine kinase pathways and their implications for glioma therapy. Expert Rev Anticanc. 2009;9:235-45
43. Ngeow J, Sesock K, Eng C. Breast cancer risk and clinical implications for germline PTEN mutation carriers. Breast Cancer Res Treat. 2017;165:1-8
44. Vogt PK, Hart JR, Gymnopoulos M. et al. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol. 2011;347:79-104
45. Perez-Ramirez C, Canadas-Garre M, Molina MA. et al. PTEN and PI3K/AKT in non-small-cell lung cancer. Pharmacogenomics. 2015;16:1843-62
46. Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Bio. 2018;19:547-62
47. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729-34
48. Chiocca EA, Abbed KM, Tatter S. et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958-66
49. Madhumalar A, Smith DJ, Verma C. Stability of the core domain of p53: insights from computer simulations. BMC Bioinformatics. 2008;9:S17
50. Amadou A, Achatz MIW, Hainaut P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li-Fraumeni syndrome. Curr Opin Oncol. 2018;30:23-9
51. Hrstka R, Coates PJ, Vojtesek B. Polymorphisms in p53 and the p53 pathway: roles in cancer susceptibility and response to treatment. J Cell Mol Med. 2009;13:440-53
52. Donehower LA, Harvey M, Slagle BL. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215-21
53. Collins VP. Brain tumours: Classification and genes. J Neurol Neurosurg Psychiatry. 2004;75:2-11
54. Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009;100:2235-41
55. Cam H, Dynlacht BD. Emerging roles for E2F: Beyond the G1/S transition and DNA replication. Cancer Cell. 2003;3:311-6
56. Cao LH, Peng B, Yao L. et al. The ancient function of RB-E2F pathway: insights from its evolutionary history. Biol Direct. 2010;5:55
57. Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785-97
58. Pinheiro DD, Ferreira WAS, Borges BD. INK4/ARF and gastric carcinogenesis. Transl Gastrointest Cancer. 2015;4:265-71
59. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353-67
60. Li AFY, Li ACH, Tsay SH. et al. Alterations in the p16(INK4a)/Cyclin D1/RB pathway in gastrointestinal tract endocrine tumors. Am J Clin Pathol. 2008;130:535-42
61. Borges BD, Burbano RMR, Harada ML. Analysis of the methylation patterns of the p16(INK4A), p15(INK4B), and APC genes in gastric adenocarcinoma patients from a Brazilian population. Tumor Biol. 2013;34:2127-33
62. Kanellou P, Zaravinos A, Zioga M. et al. Deregulation of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in basal cell carcinoma. Brit J Dermatol. 2009;160:1215-21
63. Ushio Y, Tada K, Shiraishi S. et al. Correlation of molecular genetic analysis of p53, MDM2, P16, PTEN, and EGFR and survival of patients with anaplastic astrocytoma and glioblastoma. Front Biosci. 2003;8:E281-8
64. Ferreira WAS, Araujo MD, Anselmo NP. et al. Expression analysis of genes involved in the RB/E2F pathway in astrocytic tumors. PLoS One. 2015;10:e0137259
65. Tomaszewski W, Sanchez-Perez L, Gajewski TF. et al. Brain tumor microenvironment and host state: implications for immunotherapy. Clin Cancer Res. 2019;25:4202-10
66. Lah TT, Novak M, Breznik B. Brain malignancies: Glioblastoma and brain metastases. Semin Cancer Biol. 2020;60:262-73
67. Stylli SS, Luwor RB, Ware TMB. et al. Mouse models of glioma. J Clin Neurosci. 2015;22:619-26
68. Yang XJ, Cui W, Gu A. et al. A novel zebrafish xenotransplantation model for study of glioma stem cell invasion. PLoS One. 2013;8:e61801
69. Geiger GA, Fu WL, Kao GD. Temozolomide-mediated radiosensitization of human glioma cells in a zebrafish embryonic system. Cancer Res. 2008;68:3396-404
70. Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J Neuro-Oncol. 2009;94:299-312
71. Pontén J. Neoplastic human glia cells in culture. Boston, USA: Springer. 1975
72. Ding H, Shannon P, Lau N. et al. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res. 2003;63:1106-13
73. Xiao A, Wu H, Pandolfi PP. et al. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell. 2002;1:157-68
74. Reilly K, Loisel DA, Bronson RT. et al. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nature Genet. 2000;26:109-13
75. Mao XY, Dai JX, Zhou HH. et al. Brain tumor modeling using the CRISPR/Cas9 system: state of the art and view to the future. Oncotarget. 2016;7:33461-71
76. Berkman R, Merrill M, Reinhold WC. et al. Expression of the vascular permeability factor/vascular endothelial growth factor gene in central nervous system neoplasms. J Clin Invest. 1993;91:153-9
77. Fleming T, Saxena A, Clark WC. et al. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992;52:4550-3
78. Biernat W, Tohma Y, Yonekawa Y. et al. Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 1997;94:303-9
79. Hayashi Y, Ueki K, Waha A. et al. Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol. 1997;7:871-5
80. Simmons M, Lamborn KR, Takahashi M. et al. Analysis of complex relationships between age, p53, epidermal growth factor receptor, and survival in glioblastoma patients. Cancer Res. 2001;61:1122-8
81. Li D, Li XP, Wang HX. et al. VEGF induces angiogenesis in a zebrafish embryo glioma model established by transplantation of human glioma cells. Oncol Rep. 2012;28:937-42
82. Yang XJ, Chen GL, Yu SC. et al. TGF-beta 1 enhances tumor-induced angiogenesis via JNK pathway and macrophage infiltration in an improved zebrafish embryo/xenograft glioma model. Int Immunopharmacol. 2013;15:191-8
83. Schmidek H, Nielsen SL, Schiller AL. et al. Morphological studies of rat brain tumors induced by N-nitrosomethylurea. J Neurosurg. 1971;34:335-40
84. Cinatl J, Cinatl J, Mainke M. et al. In vitro differentiation of human neuroblastoma cells induced by sodium phenylacetate. Cancer Lett. 1993;70:15-24
85. Koestner A, Swenberg JA, Wechsler W. Transplacental production with ethylnitrosourea of neoplasms of the nervous system in Sprague-Dawley rats. Am J Pathol. 1971;63:37-56
86. Etchin J, Kanki JP, Look AT. Zebrafish as a model for the study of human cancer. Method Cell Biol. 2011;105:309-37
87. Feitsma H, Cuppen E. Zebrafish as a cancer model. Mol Cancer Res. 2008;6:685-94
88. Oka T, Kurozumi K, Shimazu Y. et al. A super gene expression system enhances the anti-glioma effects of adenovirus-mediated REIC/Dkk-3 gene therapy. Sci Rep. 2016;6:33319
89. Houchens D, Ovejera AA, Riblet SM. et al. Human brain tumor xenografts in nude mice as a chemotherapy model. Eur J Cancer Clin Oncol. 1983;19:799-805
90. Toledo C, Yu D, Hoellerbauer P. et al. Genome-wide CRISPR-Cas9 screens reveal loss of redundancy between PKMYT1 and WEE1 in patient-derived glioblastoma stem-like cells. Cell Rep. 2015;13:2425-39
91. Brinster R, Chen HY, Messing A. et al. Transgenic mice harboring SV40 T-antigen genes develop characteristic brain tumors. Cell. 1984;37:367-79
92. Burgess AW. EGFR family: Structure physiology signalling and therapeutic targets. Growth Factors. 2008;26:263-74
93. Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009;16:748-54
94. Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R. et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428-431
95. Xue W, Chen SD, Yin H. et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514:380-4
96. Mussolino C, Cathomen T. RNA guides genome engineering. Nat Biotechnol. 2013;31:208-9
97. Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347-55
98. Hammond A, Galizi R, Kyrou K. et al. A CRISPR-Cas9 gene drive system-targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol. 2016;34:78-83
99. Malina A, Cameron CJF, Robert F. et al. PAM multiplicity marks genomic target sites as inhibitory to CRISPR-Cas9 editing. Nat Commun. 2015;6:10124
100. Cong L, Ran FA, Cox D. et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819-23
101. Mou HW, Kennedy Z, Anderson DG. et al. Precision cancer mouse models through genome editing with CRISPR-Cas9. Genome Med. 2015;7:53
102. El Fatimy R, Subramanian S, Uhlmann EJ. et al. Genome editing reveals glioblastoma addiction to microRNA-10b. Mol Ther. 2017;25:368-78
103. Shi JW, Wang E, Milazzo JP. et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661-7
Author contact
![]() Corresponding authors: De-Sheng Pei, E-mail: peidsac.cn; Xiaojun Yang, E-mail: yangxedu.cn.
Corresponding authors: De-Sheng Pei, E-mail: peidsac.cn; Xiaojun Yang, E-mail: yangxedu.cn.