Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Background
Neuronal cell development
Neurodegenerative diseases
Oxidative stress
Tau protein phosphorylation
Neuroinflammation
Roles of c-Abl blockers
Conclusions
Abbreviations
Facts
Open questions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2022; 19(12):1753-1761. doi:10.7150/ijms.73740 This issue Cite
Review
Roles for c-Abl in postoperative neurodegeneration
Long Feng1*, Shihui Fu2,3 ![]() *, Yao Yao4,5*, Yulong Li3*, Longhe Xu6
*, Yao Yao4,5*, Yulong Li3*, Longhe Xu6 ![]() , Yali Zhao7
, Yali Zhao7 ![]() , Leiming Luo3
, Leiming Luo3 ![]()
1. Department of Anesthesiology, Hainan Hospital of Chinese People's Liberation Army General Hospital, Sanya, China.
2. Department of Cardiology, Hainan Hospital of Chinese People's Liberation Army General Hospital, Sanya, China.
3. Department of Geriatric Cardiology, Chinese People's Liberation Army General Hospital, Beijing, China.
4. Center for the Study of Aging and Human Development and Geriatrics Division, Medical School of Duke University, North Carolina, USA.
5. Center for Healthy Aging and Development Studies, National School of Development, Peking University, Beijing, China.
6. Department of Anesthesiology, The Third Medical Center of Chinese People's Liberation Army General Hospital, Beijing, China.
7. Central Laboratory, Hainan Hospital of Chinese People's Liberation Army General Hospital, Sanya, China.
*Co-first authors.
Received 2022-4-15; Accepted 2022-9-1; Published 2022-9-28
Abstract

The nonreceptor tyrosine kinase c-Abl is inactive under normal conditions. Upon activation, c-Abl regulates signaling pathways related to cytoskeletal reorganization. It plays a vital role in modulating cell protrusion, cell migration, morphogenesis, adhesion, endocytosis and phagocytosis. A large number of studies have also found that abnormally activated c-Abl plays an important role in a variety of pathologies, including various inflammatory diseases and neurodegenerative diseases. c-Abl also plays a crucial role in neurodevelopment and neurodegenerative diseases, mainly through mechanisms such as neuroinflammation, oxidative stress (OS), and Tau protein phosphorylation. Inhibiting expression or activity of this kinase has certain neuroprotective and anti-inflammatory effects and can also improve cognition and behavior. Blockers of this kinase may have good preventive and treatment effects on neurodegenerative diseases. Cognitive dysfunction after anesthesia is also closely related to the abovementioned mechanisms. We infer that alterations in the expression and activity of c-Abl may underlie postoperative cognitive dysfunction (POCD). This article summarizes the current understanding and research progress on the mechanisms by which c-Abl may be related to postoperative neurodegeneration.
Keywords: Alzheimer's disease, Nonreceptor tyrosine kinase, Oxidative stress, Parkinson's disease, Postoperative cognitive dysfunction, Postoperative neurodegeneration
Background
Neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS) and frontotemporal dementia, are common central nervous system (CNS) diseases [1-5]. These diseases lead to the gradual impairment of CNS functions, such as activity, memory, learning, judgement and coordination. Besides, it often leads to postoperative cognitive dysfunction (POCD) after surgery with anesthesia in the elderly, which is more common after cardiac surgery than other types of surgery [6]. Developmental neurotoxicity induced by general anesthetics can cause acute widespread neuronal cell death affecting long-term memory and learning defects of the infants and young children [7-8]. The incidence of POCD decreases over time, with the rate being the highest (30% to 70%) at hospital discharge, 12% to 21% 3 months after anesthesia and noncardiac surgery, 20% to 30% 6 months after surgery, and 15% to 25% after 12 months of follow-up [9-13]. POCD can prolongs hospitalization and rehabilitation time, increases the incidence of disability, and decreases life quality and survival, thus resulting in a heavy economic burden on the health care system [5,14-15]. Compared with common neurodegenerative diseases and POCD, not only do they share the same characteristic symptoms, at the same time brain structural changes are similar, including cerebral white matter changes, central neuroinflammation, neuronal apoptosis, and so on [16-20].
Brain aging is a complex process that affects everything from the subcellular level to the organ level, starting early in life and accelerating with aging process [21]. Morphologically, brain aging is mainly characterized by brain volume loss, ventricular enlargement, cortical thinning, vitrification loss and white matter degradation. Pathophysiologically, brain aging is associated with neuron atrophy, dendritic degeneration, metabolic slowing, microglial activation, demyelinating disease, small vessel disease, and formation of white matter lesions (WML) [21]. WML is caused by cerebral hypoperfusion, observed in the elderly, related to cognitive decline, and prevalent in AD patients [22]. Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of CNS that causes focal VML and diffuse neurodegeneration throughout the brain. Range of MS lesions is in relation to inflammatory processes [23]. Furthermore, apoptosis is a programmed cell death that plays a key role in nervous system development and chronic neurodegenerative diseases, including AD and MS [24-26]. Neuroinflammation and neurodegeneration were caused by surgery and general anesthesia. Patients with POCD had significantly more white matter lesions and greater gray matter loss (medial temporal lobe) [27]. Different anesthetics and neuroinflammation will cause the apoptosis of neurons and increase the incidence of POCD [28-30].
c-Abl was first discovered in Aberson murine leukemia virus by Ozanne et al. in 1982 [31]. c-Abl (ABL, Abl1) and Abelson (ABL)-related genes (Arg, Abl2) have been identified as members of the c-Abl family of tyrosine kinases. c-Abl family members are highly conserved among different species and are involved in many cell regulatory processes, including regulation of actin cytoskeleton, cell cycle, stress-induced apoptosis and cycle arrest [32]. Under normal circumstances, apoptosis is an important physiological process that maintains the stability of internal environment and ensures normal tissue development and growth. Previous research has found that activated c-Abl can participate in apoptosis by interacting with multiple factors [33]. The gene is inactive under normal conditions. Upon activated and overexpression, this kinase can cause cell apoptosis, cycle alterations and pathological changes that may be related to neurodegenerative diseases [32-40]. Other studies have shown that Abl family kinases, especially c-Abl, play a critical role in neurodevelopmental and neurodegenerative diseases. Selective inhibition of c-Abl expression and activity has neuroprotective effects [32-39]. In addition, abnormally activated c-Abl is associated with a variety of neurodegenerative diseases. In studies using AD and PD models, it was found that c-Abl inhibitors can promote amyloid clearance and reduce the neural inflammation, which are two key drivers of nerve cell death [41]. Therefore, a large amount of literature has proposed that this kinase is crucial in regulating neurodegenerative diseases. The main mechanism of anesthesia-induced POCD is very similar to the mechanism by which c-Abl is involved in the pathogenesis of neurodegenerative diseases.
c-Abl activation regulates neuronal death response to Aβ fibrils. Intraperitoneal administration of imatinib rescued cognitive decline, Tau phosphorylation, and caspase-3 activation in neurons surrounding Aβ deposits [42]. Imatinib reduces plasma β-Oligomers and brain features, such as Oligomer accumulation, neural inflammation and cognitive deficits. The results support the role of c-Abl in Aβ accumulation of neurodegenerative diseases, and the efficacy of imatinib in the treatment of these diseases [43]. Besides, the c-Abl-drp1 signaling pathway regulates oxidative stress-induced mitochondrial fragmentation and cell death, which may be a potential target for the treatment of neurodegenerative diseases [44]. Furthermore, previous studies have found that intravenous anesthetic propofol significantly reduces c-Abl expression, but reducing c-Abl expression by propofol did not impair learning or memory function [45]. This study illustrates the involvement of c-Abl in the POCD process.
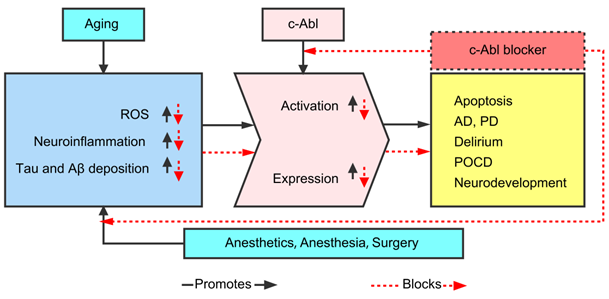
Therefore, this paper hypothesized that the expression and activation of c-Abl are abnormally elevated in various neurodegenerative diseases (AD, PD, ALS, MS, POCD, etc.), mainly as a result of abnormality in the Tau and Aβ proteins caused by neuroinflammation and oxidative stress (OS). In addition, various c-Abl blockers (imatinib, nilotinib, dasatinib, ladotinib and other drugs) can effectively reduce abnormal proteins and ameliorate cognitive dysfunction. As shown in Figure 1, this article summarizes the current understanding and research progress on the mechanisms by which c-Abl may be related to neurodegenerative diseases.
The current understanding and research progress on the mechanisms by which c-Abl may be related to neurodegenerative diseases.
Neuronal cell development
ABL and Arg tyrosine kinases play an important role in the development and function of neuronal systems. Abl1 and Abl2 are downstream targets of ABL family to regulate cell growth and transformation. They may have unique and common functions in the development of CNS. Changed phosphorylation and molecular weight of ABL protein that occur during the maturation of CNS suggest that Abl1 and Abl2 may be involved in the signaling events that are responsible for regulating neural cell development [46]. Koleske and others found that ABL and Arg kinases are expressed at the highest levels in the neuroepithelium at Embryos day9 (E9) [47]. Arg is more abundant in the adult mouse brain, especially in the synaptic-rich regions. Arg deficient mice develop normally but show several behavioral abnormalities, indicating the existence of brain defects in these mice [47]. Embryos lacking both ABL and Arg develop neurodevelopmental defects [47]. ABL kinase-mediated signal transduction from different cell surface receptors also regulates cell proliferation and survival during cell development and homeostasis [48]. In addition, during normal axonal development, axonal growth is promoted by the binding of kinesin-1 to c-Abl and their interaction. The c-Jun interacting protein-1 (JIP1) is an important regulator of axonal development and a key target of c-Abl-dependent pathway in controlling axonal growth [49]. In addition, during neurite growth, c-Abl binds to and activates cyclin-dependent kinase 5 (CDK5), affecting neuronal migration and growth [35]. In addition to its typical functions in the pathogenesis of leukemia, c-Abl is also considered to play an important role in neuronal development, neuronal migration, axon extension and synaptic plasticity [37-39]. Miller et al found that c-Abl and ataxia-telangiectasia mutation (ATM) are very important for development and survival, especially after genotoxic stress, and that they have obvious selectivity for the developing nervous system [50]. In addition, c-Abl functionally interacts with p53 during cell development, and mice lacking them are unlikely to be viable [51]. Therefore, c-Abl is associated with a variety of cellular processes, including the regulation of cell growth and survival, as c-Abl deficient mice are embryonic or neonatal lethal [52,53].
Neurodegenerative diseases
A large number of studies have found that c-Abl is activated in human neurodegenerative diseases, especially AD [32-41]. Bowser's team found that the phosphorylation of c-Abl at Y412 and granulovacuolar degeneration (GVD) in the brain are common in patients with AD [54]. Some studies have also found in PD that c-Abl expression is increased in the striatum and that the tyrosine phosphorylation is also increased [55]. However, the pathogenesis of AD and POCD is still unclear. The possible mechanisms underlying the diseases may involve the following processes. First, some scholars believe that amyloid cascade leads to the formation of Aβ fibrils. Increased aggregation of these fibrils leads to neuroinflammation, which in turn alters the physiology of neurons and induces oxidative stress in these cells, ultimately leading to kinase activation, the formation of tangles and reduced nerve cells. In addition, a study suggested that the mechanism of AD may be related to an abnormal neuroinflammatory response caused by initial injury [56]. The signaling pathway activated by c-Abl may be related to growth factors, cell adhesion and OS [46-47].
Oxidative stress
Upon aging and disease development, the body's ability to deal with OS and deoxyribonucleic acid (DNA) damage during normal cell processes is weakened, resulting in the accumulation of oxygen free radicals and DNA damage. OS can regulate neurodegeneration through various mechanisms, including protein and lipid-related processes, Aβ deposition, cytokine production, mitochondrial dysfunction, proteasome dysfunction, and the formation of advanced glycation products, oxidation of nucleic acids and activation of glial cells [47]. c-Abl generally exists in cells in inactive and activated states, with the activation of c-Abl being tightly regulated by intramolecular bonds. c-Abl protein complexes can also be linked to the membrane through an amino-terminal myristoyl group. Reactive oxygen species (ROS) may activate c-Abl by triggering ATM kinase activation through OS. In addition, a subtype of protein kinase C can activate and phosphorylate c-Abl under hydrogen peroxide (H2O2) stimulation [41,46,49]. Some authors even believe that ROS can directly activate c-Abl [40]. c-Abl can eventually cause cell dysfunction through DNA damage and increase the level of free radicals under stress conditions [32]. Alvarez et al. found that c-Abl expression is upregulated in response to OS and the presence of Aβ fibrils in cultured neurons [57]. In addition, c-Abl can be activated under OS, dopaminergic stress, and genotoxic stress, and exposure to these stresses leads to reduced and destructed neuronal cells [58-59]. Therefore, OS is considered a potential therapeutic target for the prevention and treatment of neurodegenerative diseases through regulation of oxygen free radicals or alleviation of their harmful effects [60].
Tau protein phosphorylation
Tau is a microtubule-associated protein and a major component of neurofibrillary tangles (NFTs). It is normally involved in the formation of microtubules and cytoskeletal dynamics, and abnormal phosphorylation of Tau protein can cause microtubule entanglement and instability. Hyperphosphorylation of wild-type Tau protein results in the formation of NFTs, which is one of the hallmark pathologies of individuals with AD [61]. Excessive accumulation of toxic Tau protein can cause death and dysfunction of nerve cells and glial cells, thereby causing disease symptoms. Studies have shown that the exposure of cultured neuronal cells to various Aβ peptides can activate tyrosine kinases, in turn causing tyrosine phosphorylation of Tau protein. The E3 ubiquitin ligase is a common player to play a neuroprotective role in AD and PD through scavenging misfolded proteins, such as Aβ peptides and phosphorylated Tau protein [41]. However, during OS response in neurodegenerative diseases, ABL translocates to mitochondria, where it phosphorylates Parkin, causing activity loss of E3 ubiquitin ligase. This causes abnormal accumulation of Aβ peptides and Tau protein and is responsible for neuronal apoptosis and cognitive dysfunction [41]. Furthermore, previous studies have found that oligomeric Aβ peptides are present in the brain cells of AD patients during the induction of Tau protein hyperphosphorylation [62]. In addition, Derkinderen and colleagues pointed out that c-Abl phosphorylates Tau protein at Y394 [63]. It was also found in AD brain that activated ABL is present in granular structures of hippocampal neurons.
Neuroinflammation
Inflammatory mediators are detected in brain sections from AD and PD patients, and neuroinflammation may be one of the causes of these neurodegenerative diseases [64]. Numerous studies have also found that the etiology of AD and PD may be related to chronic neuroinflammation [65-66]. In the study of two transgenic mice (AblPP/tTA mice and ArgPP/tTA mice), it was found that c-Abl overexpression can lead to neuronal loss and neuroinflammatory response [65]. Neuroinflammation can be triggered by a variety of biological mechanisms, including OS and glial response. Neuroinflammatory mediators, such as cytokines and prostaglandins, play an important role in the development of neurodegenerative diseases [67-69]. In the early stage of neuroinflammation in AD, a vicious cycle of microglial activation, proinflammatory factor release and neuronal damage may occur [70]. Some authors have pointed out that c-Abl activation by neuronal cells can lead to neurodegenerative changes and neuroinflammatory changes [66]. Treatment of AD model mice with a c-Abl blocker led to the clearance of Aβ peptides, reduced the number of astrocytes and dendritic cells, and regulated the distribution of cytokines and chemokines [71]. It has also been indicated that continuous overexpression of c-Abl in neurons can cause the degeneration of neuronal cells in the hippocampal region. In a follow-up study, it was found that this pathophysiological process is mainly caused by transient and obvious changes in the cell cycle that are associated with protein and DNA replication in the olfactory bulb and activation of transcription 1 (STAT1) signaling pathway, which is crucial for the regulation of c-Abl-induced neuroinflammation and neurodegenerative diseases [66], in the hippocampal region. Wu et al. found that c-Abl can abnormally activate P38 and lead to neuronal death in the neuroinflammatory environment [72]. It is known that P38 is a member of mitogen activated protein kinases (MAPKs) family and plays an important role in inflammation, neurodegeneration and cell death. Therefore, according to all above these findings, we speculate that the activation of c-Abl in neurons leads to pathological changes related to neuroinflammation.
Roles of c-Abl blockers
Imatinib, nilotinib, and bosutinib have been shown to inhibit proinflammatory cytokines (TNF-α, IL-1β, IL-6, iNOS, COX-2, NLRP3) production, thereby reducing the recruitment of inflammatory cells to central nervous system [71-74]. AD is the most common neurodegenerative disease and the main cause of dementia. Its main neuropathological hallmarks are mainly the accumulation of extracellular neurotrophic plaques comprising Aβ peptides and the formation of intracellular and extracellular NFTs in brain regions. Neuroinflammation also plays an important role in the pathogenesis and progression of AD [74]. Infection, trauma, ischemia, and toxins increase levels of proinflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-18, and chemokines such as C-C motif chemokine ligand 1 (CCL1), CCL5 and C-X-C motif chemokine ligand 1 (CXCL1). The release of pro-inflammatory molecules can lead to synaptic dysfunction, neuronal death and neurogenesis inhibition. During the progression of AD, memory loss and cognitive decline are accompanied by the degradation of specific neurons in the hippocampus and cerebral cortex. Studies have shown that the phosphorylation of tyrosine kinases at T412 is significantly increased in the hippocampus and entorhinal cortex in the brains of AD patients [75]. Moreover, Aβ can enhance the activity of c-Abl, and this kinase is involved in regulating the death of neurons [57]. c-Abl blockers have also been shown to significantly reduce Tau protein phosphorylation in transgenic AD animal models. It has also been found in vitro that Aβ can significantly increase c-Abl expression and phosphorylation of Tau protein in nerve cells [76]. This indicates that the expression of c-Abl is significantly increased during the pathogenesis of AD and that kinase blockers can significantly reduce the accumulation and expression of abnormal proteins.
In addition, similar studies have also shown that c-Abl blocker imatinib can significantly reduce plasma Aβ protein levels [77]. Intraperitoneal injection of the drug into AD model mice can not only ameliorate cognitive decline but also reduce neuronal apoptosis and Tau protein phosphorylation [78]. In addition, a study on AD-related diseases in humans showed that the level of c-Abl in the brains of AD patients was obviously higher than that in the brains of controls. The Abl-py412, an activated form of c-Abl phosphorylated on tyrosine residue 412, has an increased level in the early stage of AD. In the late stage of the disease, the level of abl-py412 is mainly increased in the hippocampal area. The Abl-pt735, an alternative phosphorylated form of c-Abl on threonine residue 735, has an increased level specifically in the hippocampal region. Furthermore, c-Abl and phosphorylated Tau protein interact in the brains of AD patients, and there is a certain correlation between the levels of these two proteins. The above results show that functional state of c-Abl is different at different stages of AD and that phosphorylated c-Abl and Tau protein interact in the pathogenesis of AD [54].
PD is the second most common neurodegenerative disease in the elderly. Studies have found that Parkin is a substrate of c-Abl and that c-Abl can specifically phosphorylate Parkin at Y143 [79-80]. The main pathway of this disease is the direct phosphorylation of α-synuclein at Y39 by c-Abl, which leads to abnormal aggregation of α-synuclein and a reduction in Parkin expression. In addition, c-Abl can phosphorylate E3 ubiquitin ligase at Y143 and inactivate it [81-82]. These changes can lead to an abnormal increase in the level of toxic protein zinc finger protein 746 (PARIS) and aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2) [81-82]. In addition, previous studies have demonstrated that virus induced PARIS transgenic mice can lead to c-Abl activity dependent on PD characteristics such as dyskinesia, dopaminergic neuron loss and neuroinflammation [83]. Nilotinib, a c-Abl blocker, can significantly reduce c-Abl activity and Parkin levels, and improve neuronal apoptosis and cognitive function [84]. Wu et al. [85] found that in the study of BV2 microglia and PD model of mouse brain neuroinflammation induced by lipopolysaccharide (LPS), nilotinib can reduce TNF-α, IL-1β, IL-6, iNOS, COX-2, and other proinflammatory factors in BV2 cells. And it can significantly inhibit LPS induced neuroinflammation. In addition, tyrosine hydroxylase (TH) is an important player in PD, and nilornib can increase the number of TH- and Nissl-positive neurons in PD patients. It has also been found that c-Abl blockers can significantly reduce dopaminergic neuron loss in c-Abl knockout animals [80]. In vitro studies, ladotinib can protect against mitochondrial function impairment caused by α-synuclein and reduce the formation of α-synuclein inclusions. Furthermore, in vivo studies, the drug can effectively reduce dopaminergic neuron loss and neuroinflammation and improve cognitive function [86]. The intracellular inflammasome complex is involved in the recognition and execution of host inflammatory responses. Studies using pharmacological inhibition of c-Abl found that dasatinib reduced inflammasome activation, mitochondrial oxidative stress and LPS induced microglial activation [87]. Besides, the relate vitro study also indicated that c-Abl mediated microglia activation may be an important source of inflammatory mediators [88]. In addition, many studies have shown that the activity and expression of c-Abl are significantly increased in the brains of PD patients. Kinase blockers can improve brain function, reduce neuron death, inhibit CDK5 phosphorylation, regulate α-synuclein elimination, and inhibit Parkin phosphorylation [89-93]. In addition to animal experiments, clinical studies have also found that the treatment of patients with moderate or severe PD with nilotinib for 6 months can significantly alleviate cognitive symptoms [94]. In addition, some studies have found that c-Abl kinase inhibitor PD180970 can reduce Toll like receptor-4 mediated NF-κB and inhibits the release of pro-inflammatory cytokines such as IL-6 and monocyte chemoattractant protein-1 (MCP-1) [95].
In addition to the above common neurodegenerative diseases, c-Abl is also associated with Niemann-Pick C (NPC), a fatal autosomal recessive disease characterized by the accumulation of free cholesterol and sphingolipids in the endolysosomal system. c-Abl is activated and triggers neuronal apoptosis in vitro and in vivo nasopharyngeal carcinoma models [96-97]. Klein et al. found that the c-Abl/p73 pathway is related to neurodegeneration in NPC and that c-Abl blocker can delay neurodegeneration in this disease [96]. In addition, in other studies of NPC models in neurons, the c-Abl/Histone deacetylases (HDAC2) signaling pathway was found to be involved in the regulation of neurons. Inhibition of c-Abl may be a pharmacological strategy for preventing the adverse effects of elevated HDAC2 levels in nasopharyngeal carcinoma patients [98]. In addition, c-Abl is involved in ALS, which is characterized by neuron death. In ALS, c-Abl signaling is triggered through mitochondrial alteration-mediated ROS production [99]. Some researches suggest that c-Abl is a treatment target for ALS, and it has been found that the c-Abl blocker dasatinib has neuroprotective effects against this disease in vitro and in vivo [100]. Small interfering RNA (siRNA)-mediated c-Abl gene knockout attenuated the production of proinflammatory mediators in LPS induced glial cell culture [101].
Conclusions
The expression and activation of c-Abl are abnormally elevated in various neurodegenerative diseases (AD, PD, NPC, ALS, etc.), mainly as a result of neuroinflammation, OS, and abnormal Aβ and Tau protein. In addition, various c-Abl blockers (nilotinib, imatinib, dasatinib, ladotinib and other drugs) can effectively reduce abnormal protein levels and ameliorate cognitive dysfunction. The main mechanism of anesthesia-induced POCD is very similar to the mechanism by which c-Abl is involved in the pathogenesis of neurodegenerative diseases. Therefore, we hypothesized that anesthesia-induced POCD may also be related to abnormal activation or increased expression of c-Abl following the exposure to anesthetic drugs. These drugs, which may have certain preventive effects against POCD, may be new options for the treatment of postoperative neurological dysfunction caused by surgery and anesthesia. However, high-quality studies confirming specific role and potential mechanism of c-Abl in dementia and cognitive decline after anesthesia are lacking. c-Abl is a very promising target for perioperative intervention and treatment of POCD.
Abbreviations
ABL: Abelson; c-Abl: Abelson nonreceptor tyrosine kinase; CNS: central nervous system; POCD: postoperative cognitive dysfunction; PD: Parkinson's disease; AD: Alzheimer's disease; CDK5: cyclin-dependent kinase 5; GVD: granulovacuolar degeneration; ATM: ataxia-telangiectasia mutated; NFT: neurofibrillary tangle; PARIS: zinc finger protein 746; AIMP2: aminoacyl tRNA synthetase complex-interacting multifunctional protein type 2; TH: tyrosine hydroxylase; NPC: Niemann-Pick C; ALS: amyotrophic lateral sclerosis; MAPKs: mitogen activated protein kinases; LPS: lipopolysaccharide; MCP-1: monocyte chemoattractant protein-1; siRNA: small interfering RNA; E3: ubiquitin protein ligase; H2O2: hydrogen peroxide; JIP1: c-Jun interacting protein-1; E9: embryos day9; MTL: medial temporal lobe; WML: white matter lesions.
Facts
- c-Abl plays a crucial role in neurodegenerative diseases, mainly through mechanisms such as neuroinflammation, oxidative stress (OS) and Tau protein phosphorylation.
- Blockers of c-Abl may have a good preventive and treatment effects on postoperative neurodegeneration.
- This article summarizes the current understanding and research progress on the mechanisms by which c-Abl may be related to postoperative neurodegeneration.
Open questions
- Does c-Abl inhibition have anti-inflammatory and neuroprotective effects?
- Does c-Abl inhibition alleviate postoperative cognitive dysfunction (POCD)?
- Through which mechanisms is c-Abl related to POCD?
Acknowledgements
Funding
This work was supported by grants from the Military Medical Science and Technology Youth Incubation Program (20QNPY110, 19QNP060), the National Natural Science Foundation of China (81900357, 81941021, 81903392, 81901252, 82001476, 81802804, 81801251), the Excellent Youth Incubation Program of Chinese People's Liberation Army General Hospital (2020-YQPY-007), the Natural Science Foundation of Hainan Province (821QN389, 821MS112, 822MS198, 820MS126, 820QN383), the National Key R&D Program of China (2018YFC2000400), the National S&T Resource Sharing Service Platform Project of China (YCZYPT[2018]07), the Specific Research Fund of Innovation Platform for Academicians of Hainan Province (YSPTZX202216), the Medical Big Data R&D Project of Chinese People's Liberation Army General Hospital (MBD2018030), the China Postdoctoral Science Foundation Funded Project (2019M650359, 2020M682816, 2021T140298), the Heatstroke Treatment and Research Center of Chinese People's Liberation Army, the Simulation Training for Treatment of Heat Stroke, and the Major Science and Technology Programme of Hainan Province (ZDKJ2019012). The sponsors had no role in the design, conduct, interpretation, review, approval or control of this article.
Author contributions
LF, SF, YY, YL, LX, YZ and LL contributed to the design of study, the review of literature and drifting of the manuscript. All authors have read and approved the manuscript.
Availability of supporting data
The datasets used and/or analysed during the current study are not publicity available. All data are available from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777-783
2. Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83(2):266-282
3. Lardenoije R, Iatrou A, Kenis G, Kompotis K, M Steinbusch HW, Mastroeni D. et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21-64
4. Kovacs GG, Adle-Biassette H, Milenkovic I, Cipriani S, van Scheppingen J, Aronica E. Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience. 2014;269:152-172
5. Leicht H, König HH, Stuhldreher N, Bachmann C, Bickel H, Fuchs A. et al. Predictors of costs in dementia in a longitudinal perspective. PLoS One. 2013;8(7):e70018
6. Kok WF, Koerts J, Tucha O, Scheeren TW, Absalom AR. Neuronal damage biomarkers in the identification of patients at risk of long-term postoperative cognitive dysfunction after cardiac surgery. Anaesthesia. 2017;72(3):359-369
7. Reitman E, Flood P. Anaesthetic considerations for non-obstetric surgery during pregnancy. Br J Anaesth. 2011;107(Suppl 1):i72-i78
8. Bosnjak ZJ, Logan S, Liu Y, Bai X. Recent insights into molecular mechanisms of propofol-induced developmental neurotoxicity: implications for the protective strategies. Anesth Analg. 2016;123(5):1286-1296
9. Evered L, Scott DA, Silbert B, Maruff P. Postoperative cognitive dysfunction is independent of type of surgery and anesthetic. Anesth Analg. 2011;112(5):1179-1185
10. Paredes S, Cort.nez L, Contreras V, Silbert B. Post-operative cognitive dysfunction at 3 months in adults after non-cardiac surgery: a qualitative systematic review. Acta Anaesthesiol Scand. 2016;60(8):1043-1058
11. Ghaffary S, Ghaeli P, Talasaz AH, Karimi A, Noroozian M, Salehiomran A. et al. Effect of memantine on post-operative cognitive dysfunction after cardiac surgeries: A randomized clinical trial. Daru. 2017;25(1):24
12. Klinger RY, James OG, Borges-Neto S, Bisanar T, Li YJ, Qi Wj. et al. 18F-florbetapir positron emission tomography-determined cerebral beta-amyloid deposition and neurocognitive performance after cardiac surgery. Anesthesiology. 2018;128(4):728-44
13. Feng L, Chu Z, Quan X, Zhang Y, Yuan W, Yao Y. et al. Malnutrition is positively associated with cognitive decline in centenarians and oldest-old adults: A cross-sectional study. EClinicalMedicine. 2022;47:101336
14. Polunina AG, Golukhova EZ, Guekht AB, Lefterova NP, Bokeria LA. Cognitive dysfunction after on-pump operations: neuropsychological characteristics and optimal core battery of tests. Stroke Res Treat. 2014;2014:302824
15. Wang C, Zhang X, Liu F, Paule MG, Slikker W Jr. Anesthetic-induced oxidative stress and potential protection. Scientific World Journal. 2010;10:1473-1482
16. Blinkouskaya Y, Caçoilo A, Gollamudi T, Jalalian S, Weickenmeier J. Brain aging mechanisms with mechanical manifestations. Mech Ageing Dev. 2021;200:111575
17. Alberghina L, Colangelo AM. The modular systems biology approach to investigate the control of apoptosis in Alzheimer's disease neurodegeneration. BMC Neurosci. 2006 7 Suppl 1(Suppl 1):S2
18. Nakao S, Yamamoto T, Kimura S, Mino T, Iwamoto T. Brain white matter lesions and postoperative cognitive dysfunction: a review. J Anesth. 2019;33(2):336-340
19. Zhang Q, Li Y, Bao Y, Yin C, Xin X, Guo Y. et al. Pretreatment with nimodipine reduces incidence of POCD by decreasing calcineurin mediated hippocampal neuroapoptosis in aged rats. BMC Anesthesiol. 2018;18(1):42
20. Wang Y, Yin CP, Tai YL, Zhao ZJ, Hou ZY, Wang QJ. Apoptosis inhibition is involved in improvement of sevoflurane-induced cognitive impairment following normobaric hyperoxia preconditioning in aged rats. Exp Ther Med. 2021;21(3):203
21. Zhang Y, Fu S, Ding D, Lutz MW, Zeng Y, Yao Y. Leisure activities, APOE ε4, and cognitive decline: a longitudinal cohort study. Front Aging Neurosci. 2021;13:736201
22. Coutu JP, Goldblatt A, Rosas HD, Salat DH; Alzheimer's Disease Neuroimaging Initiative (ADNI). White matter changes are associated with ventricular expansion in aging, mild cognitive impairment, and Alzheimer's disease. J Alzheimers Dis. 2016;49(2):329-342
23. Lassmann H. Multiple Sclerosis Pathology. Cold spring harb perspect med. 2018;8(3):a028936
24. Fu S, Ping P, Luo L, Ye P. Deep analyses of the associations of a series of biomarkers with insulin resistance, metabolic syndrome, and diabetes risk in nondiabetic middle-aged and elderly individuals: results from a Chinese community-based study. Clin Interv Aging. 2016;11:1531-1538
25. Zhu X, Raina AK, Perry G, Smith MA. Apoptosis in Alzheimer disease: a mathematical improbability. Curr Alzheimer Res. 2006;3(4):393-396
26. Fu S, Guo Y, Zhang Z, Luo L, Ye P. Single-marker and multi-marker approaches to appraise the relationships between biomarkers and microalbuminuria in Chinese middle-aged and elderly from communities: a cross-sectional analysis. BMC Nephrol. 2018;19(1):93
27. Maekawa K, Baba T, Otomo S, Morishita S, Tamura N. Low pre-existing gray matter volume in the medial temporal lobe and white matter lesions are associated with postoperative cognitive dysfunction after cardiac surgery. PLoS One. 2014;9(1):e87375
28. Fu S, Ping P, Li Y, Li B, Zhao Y, Yao Y. et al. Centenarian longevity had inverse relationships with nutritional status and abdominal obesity and positive relationships with sex hormones and bone turnover in the oldest females. J Transl Med. 2021;19(1):436
29. Lin JQ, Wang JX, Yu S, Fu SH, Zhang YJ. Newly discovered molecules associated with trimetazidine on improvement of skeletal muscle function in aging: evidence from myoblasts and mice. Exp Gerontol. 2022;161:111733
30. Su W, Xie M, Li Y, Gong X, Li J. Topiramate reverses physiological and behavioral alterations by postoperative cognitive dysfunction in rat model through inhibiting TNF signaling pathway. Neuromolecular Med. 2020;22(2):227-238
31. Ozanne B, Wheeler T, Zack J, Smith G, Dale B. Transforming gene of a human leukaemia cell is unrelated to the expressed tumour virus related gene of the cell. Nature. 1982;299(5885):744-747
32. Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5(1):33-44
33. Meltser V, Ben-Yehoyada M, Shaul Y. c-Abl tyrosine kinase in the DNA damage response: cell death and more. Cell Death Differ. 2011;18(1):2-4
34. Brahmachari S, Ge P, Lee SH, Kim D, Karuppagounder SS, Kumar M. et al. Activation of tyrosine kinase c-Abl contributes to α-synuclein-induced neurodegeneration. J Clin Invest. 2016;126(8):2970-2988
35. Jones SB, Lu HY, Lu Q. Abl tyrosine kinase promotes dendrogenesis by inducing actin cytoskeletal rearrangements in cooperation with Rho family small GTPases in hippocampal neurons of article. J Neurosci. 2004;24(39):8510-8521
36. Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM. et al. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation,kinase upregulation, and neurite outgrowth. Neuron. 2000;26(3):633-646
37. Martin LJ, Liu Z, Pipino J, Chestnut B, Landek MA. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb Cortex. 2009;19(6):1273-1293
38. Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399(6738):809-813
39. Rhee J, Mahfooz NS, Arregui C, Lilien J, Balsamo J, VanBerkum MF. Activation of the repulsive receptor Roundabout inhibits N-cadherin-mediated cell adhesion. Nat Cell Biol. 2002;4(10):798-805
40. Brahmachari S, Karuppagounder SS, Ge P, Lee S, Dawson VL, Dawson TM. et al. c-Abl and Parkinson's Disease: Mechanisms and Therapeutic Potential. J Parkinsons Dis. 2017;7(4):589-601
41. Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci. 2016;129(1):9-16
42. Cancino GI, Toledo EM, Leal NR, Hernandez DE, Yévenes LF, Inestrosa NC. et al. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer's beta-amyloid deposits. Brain. 2008:131(Pt9):2425-2442.
43. Estrada LD, Chamorro D, Yañez MJ, Gonzalez M, Leal N, von Bernhardi R. et al. Reduction of blood Amyloid-β Oligomers in Alzheimer's disease transgenic mice by c-Abl Kinase inhibition. J Alzheimers Dis. 2016;54(3):1193-1205
44. Zhou L, Zhang Q, Zhang P, Sun L, Peng C, Yuan Z. et al. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 2017;8(10):e3117
45. Feng L, Sun ZG, Liu QW, Ma T, Xu ZP, Feng ZG. et al. Propofol inhibits the expression of Abelson nonreceptor tyrosine kinase without affecting learning or memory function in neonatal rats. Brain Behav. 2020;10(11):e01810
46. Courtney KD, Grove M, Vandongen H, Vandongen A, LaMantia AS, Pendergast AM. Localization and phosphorylation of Abl-interactor proteins, Abi-1 and Abi-2, in the developing nervous system. Mol Cell Neurosci. 2000;16(3):244-257
47. Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA. et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21(6):1259-1272
48. Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell. 2008;100(11):617-631
49. Dajas-Bailador F, Jones EV, Whitmarsh AJ. The JIP1 scaffold protein regulates axonal development in cortical neurons. Curr Biol. 2008;18(3):221-226
50. Miller HL, Lee Y, Zhao J, Chong MJ, McKinnon PJ. Atm and c-Abl cooperate in the response to genotoxic stress during nervous system development. Brain Res Dev Brain Res. 2003;145(1):31-38
51. Whang YE, Tran C, Henderson C, Syljuasen RG, Rozengurt N, McBride WH. et al. c-Abl is required for development and optimal cell proliferation in the context of p53 deficiency. Proc Natl Acad Sci U S A. 2000;97(10):5486-5491
52. Sawyers CL, McLaughlin J, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell. 1994;77(1):121-131
53. Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65(7):1153-1163
54. Jing Z, Caltagarone J, Bowser R. Altered subcellular distribution of c-Abl in Alzheimer's disease. J Alzheimers Dis. 2009;17(2):409-422
55. Fu S, Hu J, Chen X, Li B, Shun H, Deng J. et al. Mutant single nucleotide polymorphism rs189037 in Ataxia-Telangiectasia mutated gene is significantly associated with ventricular wall thickness and human lifespan. Front Cardiovasc Med. 2021;8:658908
56. Herrup K. Reimagining Alzheimer's disease-an age-based hypothesis. J Neurosci. 2010;30(50):16755-16762
57. Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS. Activation of the neuronal c-Abl tyrosine kinase by amyloid-beta-peptide and reactive oxygen species. Neurobiol Dis. 2004;17(2):326-336
58. Hoshi Y, Uchida Y, Tachikawa M, Ohtsuki S, Couraud PO, Suzuki T. et al. Oxidative stress-induced activation of Abl and Src kinases rapidly induces P-glycoprotein internalization via phosphorylation of caveolin-1 on tyrosine-14, decreasing cortisol efflux at the blood-brain barrier. J Cereb Blood Flow Metab. 2020;40(2):420-436
59. Gonfloni S, Maiani E, Di Bartolomeo C, Diederich M, Cesareni G. Oxidative stress, DNA damage, and c-Abl signaling: at the crossroad in neurodegenerative diseases? Int J Cell Biol. 2012;2012:683097
60. Yaribeygi H, Panahi Y, Javadi B, Sahebkar A. The underlying role of oxidative stress in neurodegeneration: a mechanistic review. CNS Neurol Disord Drug Targets. 2018;17(3):207-215
61. Yang Y, Wang JZ. Nature of Tau-associated neurodegeneration and the molecular mechanisms. J Alzheimers Dis. 2018;62(3):1305-1317
62. De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol Aging. 2008;29(9):1334-1347
63. Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA. Tyrosine 394 is phosphorylated in Alzheimer's paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci. 2005;25(28):6584-6593
64. Rogers J, Webster S, Lue LF, Brachova L, Civin W H, Emmerling M. et al. Inflammation and Alzheimer's disease pathogenesis. Neurobiol Aging. 1996;17(5):681-686
65. Tremblay MA, Acker CM, Davies P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J Alzheimers Dis. 2011;25(1):119-133
66. Schlatterer SD, Suh HS, Conejero-Goldberg C, Chen S, Acker CM, Lee SC. et al. Neuronal c-Abl activation leads to induction of cell cycle and interferon signaling pathways. J Neuroinflammation. 2012;9:208
67. Aktas O, Ullrich O, Infante-Duarte C, Nitsch R, Zipp F. Neuronal damage in brain inflammation. Arch Neurol. 2007;64(2):185-189
68. Alcendor DJ, Charest AM, Zhu WQ, Vigil HE, Knobel SM. Infection and upregulation of proinflammatory cytokines in human brain vascular pericytes by human cytomegalovirus. J Neuroinflammation. 2012;9:95
69. Alexander JK, Popovich PG. Neuroinflammation in spinal cord injury: therapeutic targets for neuroprotection and regeneration. Prog Brain Res. 2009;175:125-137
70. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement. 2016;12(6):719-732
71. Lonskaya I, Hebron ML, Selby ST, Turner RS, Moussa CE. Nilotinib and bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in Alzheimer's disease models. Neuroscience. 2015;304:316-327
72. Wu R, Chen H, Ma J, He Q, Huang Q, Liu Q. et al. c-Abl-p38α signaling plays an important role in MPTP-induced neuronal death. Cell Death Differ. 2016;23(3):542-552
73. Azizi G, Haidari M.R, Khorramizadeh M. Effects of imatinib mesylate in mouse models of multiple sclerosis and in vitro determinants. Iran J Allergy Asthma Immunol. 2014;13(3):198-206
74. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157-172
75. Schlatterer SD, Acker CM, Davies P. c-Abl in neurodegenerative disease. J Mol Neurosci. 2011:45(3):445-52.
76. Cancino GI, Perez de Arce K, Castro PU, Toledo EM, von Bernhardi R, Alvarez AR. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol Aging. 2011;32(7):1249-1261
77. Fu S, Yao Y, Lv F, Zhang F, Zhao Y, Luan F. Associations of immunological factors with metabolic syndrome and its characteristic elements in Chinese centenarians. J Transl Med. 2018;16(1):315
78. Feng L, Wu D, Lin J, Li Y, Zhao Y, Zhang P. et al. Associations between age-related hearing loss, cognitive decline, and depression in Chinese centenarians and oldest-old adults. Ther Adv Chronic Dis. 2022;13:20406223221084833
79. Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ. et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci USA. 2010;107(38):16691-16696
80. Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M. et al. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci. 2011;31(1):157-163
81. Shin JH, Ko HS, Kang H, Lee YJ, Lee Y-Il, Pletinkova O. et al. PARIS(ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144(5):689-702
82. Lee Y, Karuppagounder SS, Shin JH, Lee Yun-Il, Ko HS, Swing D. et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci. 2013;16(10):1392-1400
83. Kim H, Shin JY, Jo A, Kim JH, Park S, Choi JY. et al. Parkin interacting substrate phosphorylation by c-Abl drives dopaminergic neurodegeneration. Brain. 2021;144(12):3674-3691
84. Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson's disease. Sci Rep. 2014;4:4874
85. Wu J, Xu X, Zheng L, Mo J, Jin X, Bao Y. Nilotinib inhibits microglia-mediated neuroinflammation to protect against dopaminergic neuronal death in Parkinson's disease models. Int Immunopharmacol. 2021;99:108025
86. Lee S, Kim S, Park YJ, Yun SP, Kwon SH, Kim Dhoon. et al. The c-Abl inhibitor, radotinib HCl, is neuroprotective in a preclinical Parkinson's disease mouse model. Hum Mol Genet. 2018;27(13):2344-2356
87. Lawana V, Singh N, Sarkar S, Charli A, Jin H, Anantharam V. et al. Involvement of c-Abl kinase in microglial activation of NLRP3 inflammasome and impairment in autolysosomal system. J Neuroimmune Pharmacol. 2017;12(4):624-660
88. Gordon R, Singh N, Lawana V, Ghosh A, Harischandra DS, Jin H. et al. Protein kinase Cdelta upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson's disease. Neurobiol Dis. 2016;93:96-114
89. Mahul-Mellier AL, Fauvet B, Gysbers A, Dikiy I, Oueslati A, Georgeon S. et al. c-Abl phosphorylates alpha-synuclein and regulates its degradation: implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson's disease. Hum Mol Genet. 2014;23(11):2858-2879
90. Sun Z, Ping P, Li Y, Feng L, Liu F, Zhao Y. et al. Relationships between traditional Chinese medicine constitution and age-related cognitive decline in Chinese centenarians. Front Aging Neurosci. 2022;14:870442
91. Brahmachari SGP, Lee SH, Kim D, Kim Dhoon, Karuppagounder SS, Kumar M. et al. Activation of tyrosine kinase c-Abl contributes to a-synuclein-induced neurodegeneration. J Clin Invest. 2016;126(8):2970-2988
92. Tanabe A, Yamamura Y, Kasahara J, Morigaki R, Kaji R, Goto S. A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson's disease. Front Cell Neurosci. 2014;8:50
93. Lonskaya I, Hebron ML, Desforges NM, Schachter JB, Moussa CE. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med. 2014;92(4):373-386
94. Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR. et al. Nilotinib effects in Parkinson's disease and Dementia with Lewy bodies. J Parkinsons Dis. 2016;6(3):503-517
95. Sn S, Pandurangi J, Murumalla R, Dj V, Garimella L, Acharya A. et al. Small molecule modulator of aggrephagy regulates neuroinflammation to curb pathogenesis of neurodegeneration. EBioMedicine. 2019;50:260-273
96. Klein A, Maldonado C, Vargas LM, Gonzalez M, Robledo F, Perez de Arce K. et al. Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiol Dis. 2011;41(1):209-18
97. Alvarez AR, Klein A, Juan C, Cancino GI, Amigo J, Mosqueira M. et al. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 2008;22(10):3617-27
98. Contreras PS, Gonzalez-Zuñiga M, González-Hódar L, Yáñez MJ, Dulcey A, Marugan J. et al. Neuronal gene repression in Niemann-Pick type C models is mediated by the c-Abl/HDAC2 signaling pathway. Biochim Biophys Acta. 2016;1859(2):269-279
99. Rojas F, Gonzalez D, Cortes N, Ampuero E, Hernández DE, Fritz E. et al. Reactive oxygen species trigger motoneuron death in non-cell-autonomous models of ALS through activation of c-Abl signaling. Front Cell Neurosci. 2015;9:203
100. Ryu K, Shinsuke I, Masahisa K, Kawai K, Sone J, Huang Z. et al. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS ONE. 2012;7(9):e46185
101. Song GJ, Rahman MH, Jha MK, Gupta DP, Park SH, Kim JH. et al. A Bcr-Abl Inhibitor GNF-2 attenuates inflammatory activation of Glia and chronic pain. Front Pharmacol. 2019;10:543
Author contact
![]() Corresponding authors: Shihui Fu, Department of Cardiology, Hainan Hospital of Chinese People's Liberation Army General Hospital, Sanya 572013, China. E-mail: fushihuicom.cn; Longhe Xu, Department of Anesthesiology, The Third Medical Center of Chinese People's Liberation Army General Hospital, Beijing 100027, China. E-mail: Longhexucom; Yali Zhao, Central Laboratory, Hainan Hospital of Chinese People's Liberation Army General Hospital, Sanya 572013, China. E-mail: zhaoyl301com; Leiming Luo, Department of Geriatric Cardiology, Chinese People's Liberation Army General Hospital, Beijing 100856, China. Email: luoleimingsina.com.
Corresponding authors: Shihui Fu, Department of Cardiology, Hainan Hospital of Chinese People's Liberation Army General Hospital, Sanya 572013, China. E-mail: fushihuicom.cn; Longhe Xu, Department of Anesthesiology, The Third Medical Center of Chinese People's Liberation Army General Hospital, Beijing 100027, China. E-mail: Longhexucom; Yali Zhao, Central Laboratory, Hainan Hospital of Chinese People's Liberation Army General Hospital, Sanya 572013, China. E-mail: zhaoyl301com; Leiming Luo, Department of Geriatric Cardiology, Chinese People's Liberation Army General Hospital, Beijing 100856, China. Email: luoleimingsina.com.