Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Bone metabolism
Role of immune cells in bone...
Role of immune cytokines in bone...
Future prospects of applied...
Conclusions and prospects
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(16):3697-3707. doi:10.7150/ijms.61080 This issue Cite
Review
The Role of the Immune Microenvironment in Bone Regeneration
Ning Yang1,2, Yao Liu1,2 ![]()
1. Department of Pediatric Dentistry, School and Hospital of Stomatology, China Medical University, Shenyang, China.
2. Liaoning Provincial Key Laboratory of Oral Diseases, Shenyang, China.
Received 2021-3-30; Accepted 2021-9-3; Published 2021-9-21
Abstract

Bone is an active tissue, being constantly renewed in healthy individuals with participation of the immune system to a large extent. Any imbalance between the processes of bone formation and bone resorption is linked to various inflammatory bone diseases. The immune system plays an important role in tissue formation and bone resorption. Recently, many studies have demonstrated complex interactions between the immune and skeletal systems. Both of immune cells and cytokines contribute to the regulation of bone homeostasis, and bone cells, including osteoblasts, osteoclasts, osteocytes, also influence the cellular functions of immune cells. These crosstalk mechanisms between the bone and immune system finally emerged, forming a new field of research called osteoimmunology. Therefore, the immune microenvironment is crucial in determining the speed and outcome of bone healing, repair, and regeneration. In this review, we summarise the role of the immune microenvironment in bone regeneration from the aspects of immune cells and immune cytokines. The elucidation of immune mechanisms involved in the process of bone regeneration would provide new therapeutic targets for improving the curative effects of bone injury treatment.
Keywords: immune microenvironment, bone regeneration, immune cell, immune cytokine, osteoimmunology
Introduction
Bone is composed of osteoblasts, osteoclasts, osteocytes, and other basic elements. As a dynamic organ, there is a balance between bone formation and bone resorption and there are complex interactions between the immune and skeletal systems. Bone and immune cells not only co-exist in the bone marrow (BM) cavity, but also have common progenitor cells and share a variety of regulatory molecules. Immune cells participate in the regulation of bone homeostasis; conversely, bone cells also influence the proliferation and differentiation of immune cells. Therefore, the term 'osteoimmunology' was coined to highlight the two-way communication between the bone and immune systems [1]. Additionally, the immune system plays an important role in tissue repair and regeneration. The immune microenvironment is crucial in the healing, repair, and regeneration of bone tissue, which determines the ability of bone tissue to regenerate [2]. In this review, we summarise the role of the immune microenvironment in bone regeneration from the aspect of immune cells and cytokines, which is helpful in elucidating the immune mechanisms involved in the bone regeneration process and provides a new therapeutic target for improving the curative effect of bone injury treatment.
Bone metabolism
The skeleton is an extremely dynamic organ that undergoes remodeling throughout life to maintain bone strength and mineral homeostasis by constantly renewing the bone matrix [3]. Physiological bone remodelling is a dynamic equilibrium occurring as a result of a series of well-orchestrated biological events, all regulated by complex interactions between the various cell types that are resident in bone. The main cells involved in bone remodelling are osteoblasts and osteoclasts [4]. The functions of these cells are closely coupled, sequentially carrying out the absorption of old damaged bone and the formation of new bone [4]. In addition, osteocytes, acted as mechanical sensors, also play an important role in the process of bone remodelling, while bone lining cells initiate bone remodelling through matrix degradation [5, 6].
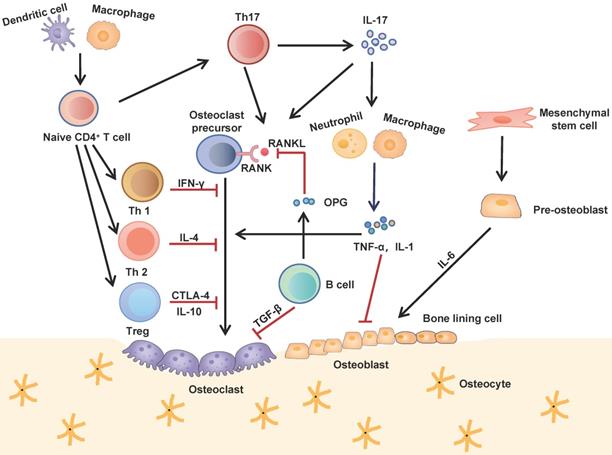
Interactions between the immune and skeletal systems. The T cell subsets (Th1, Th2, Th17 and Treg) play an important role in bone repair and regeneration. Th17 cells, known as osteoclastogenesis-supporting T cells, which secret IL-17 to upregulate RANKL expression and induce inflammatory cytokines such as TNF-α and IL-1 from innate immune cells. These cytokines further activate osteoclast precursor cells and inhibite osteoblast function. Contrarily, Th1, Th2 and Treg inhibit osteoclastogenesis by secreting cytokines INF-γ, IL-4, CTLA-4 and IL-10 respectively. B cells antagonistically block the effect of RANKL by secreting OPG and induce osteoclast apoptosis by secreting TGF-β.
The bone remodelling cycle consists of five sequential phases: activation, resorption, reversal, formation, and termination. In the activation phase, unknown triggering signals are induced on the cell surfaces of osteoblasts and/or bone lining cells, providing the site of bone remodelling onset, where osteoclasts are recruited and fused to form mature osteoclasts. In the following resorption phase, osteoclasts dissolve the inorganic matrix by creating an acidic microenvironment and degrade the organic matrix with specific enzymes. Next is the reversal phase, in which, with the completion of bone resorption, osteoclasts undergo apoptosis, macrophage-like reverse cells migrate to the absorbed lacuna and clear the debris left by the osteoclasts; subsequently, osteoblasts are summoned to the resorption lacuna to synthesise new bone matrix and then mineralise it to fill the resorption lacuna in the formation phase. Once mineralisation is complete, osteoblasts undergo apoptosis, change into bone lining cells, or are buried within the bone matrix and eventually differentiate into osteocytes in the termination phase. The resting bone surface environment is maintained until the next wave of remodelling is initiated [7, 8].
Therefore, bone coupling involves the interaction of a wide range of cell types and control mechanisms, including signals from immune cells and signaling taking place within the osteoblast lineage. It has been shown, that macrophages maintain osteoblast differentiation, which may be related to the Oncostatin M secreted by activated macrophage [9, 10]. T cells can mediate the anabolic action of parathyroid hormone, which stimulates bone formation and triggers osteoblast's secretion of pro-hematopoietic factors like IL-6 and IL-6R [11, 12]. And the recent identification of Wnt1 as a B-cell product in bone marrow that promotes bone formation [13].
Role of immune cells in bone regeneration
Tissue damage triggers an immediate immune response, which usually lasts for days or even weeks. The initial immune response is mainly composed of the innate immune system, including neutrophils, mast cells, monocytes, and macrophages. The later immune response is mainly composed of an adaptive immune system, including T and B cells. The impact of immune cells on the bone regeneration have two main aspects: The anabolic function of acute inflammation during bone regeneration and the catabolic function of chronic inflammation during bone resorption. For example, acute inflammation leading to the production of chemokines that accelerates bone formation in early bone fracture healing by stimulating the proliferation and osteoblastic differentiation of mesenchymal progenitor cells [14, 15]. In contrast, chronic inflammation suppresses the production of factors that stimulates osteoblastic cells to form new bone, thereby inhibiting osseous coupling and promoting osteolytic lesion formation during periodontitis [15, 16].
Neutrophils
As the most abundant leukocytes in mammalian blood, neutrophils are well recognised as one of the major players of the innate immune system. Upon tissue injury, neutrophils are usually the first to be recruited and migrated to the injured site. Neutrophils clear invasive pathogenic microorganisms, initiate an acute inflammatory response, and enhance host defence [17]. A large number of clinical cases and animal experiments have confirmed that neutrophil aggregation and infiltration is seen in bone injury and plays a key role in the early stages of bone repair. In a mouse model of bone fractures, the authors noted that depletion of neutrophils led to impaired bone healing after fracture [18]. Furthermore, they also observed increased levels of inflammatory cytokines as well as altered monocyte and macrophage recruitment, indicating a crucial role for neutrophils in initiating the bone repair process [18]. However, some studies have reported that bacteria produce endotoxin-lipopolysaccharide during trauma to stimulate neutrophils to express receptor activator of nuclear factor kappa B ligand (RANKL), which activates osteoclasts to exert bone resorption [19, 20]. Moreover, in contrast to CD3+ T lymphocytes, activated neutrophils only express the membrane protein RANKL and do not secrete soluble RANKL, suggesting that neutrophils activate osteoclast function only through inter-cellular contact [20, 21]. In osteoporosis induced by long-term glucocorticoid treatment and chronic gout, neutrophils directly inhibit bone formation by affecting the function of osteoblasts [22, 23]. Additionally, neutrophils have also been confirmed to play an important role in bone lesions in animal experiments of osteoarthritis and aggressive periodontitis [24, 25]. In summary, neutrophils not only perform a function as immune cell, but also regulate bone homeostasis in the initial stage of bone injury.
Macrophages
Macrophages, derived from monocytes, are ubiquitous in tissues, act as phagocytes to prevent pathogens from invading, and effectively remove necrotic tissue and cell debris. Upon bone injury, macrophages secrete release various cytokines, chemokines, and growth factors to initiate the recruitment of fibroblasts, MSCs, and osteoprogenitor cells from their local niches [26]. Additional macrophage-derived inflammatory mediators and growth factors guide the proliferation, differentiation and extracellular matrix production of recruited MSCs and osteoprogenitor cells with additional growth factors being released from the remodeling extracellular matrix, which plays an important role in tissue repair and regeneration [27]. Under the stimulation of endogenous or exogenous factors, macrophages are transformed into two phenotypes: M1 and M2. M1 macrophages are generally pro-inflammatory, whereas M2 macrophages are anti-inflammatory [28]. In some cases, the M1 phenotype can be reversed to M2, in vitro and in vivo, and vice versa [29]. A growing number of reports have demonstrated macrophages' central role in osteogenesis. Macrophages support osteoblast differentiation and proliferation through the release of cytokines including BMP-2, BMP-4, and TGF-β1 [30, 31]. In contrast to other tissue regeneration, macrophages participate in every phase of bone injury, healing, and regeneration, but the specific mechanism is not clear. Using a mouse model of femoral fracture, it has been found that macrophages are not involved in the early stage of fracture healing, but prevent the ultimate endochondral ossification by inhibiting the formation of hard callus [32]. Further studies have shown that M2 macrophages are crucial in the ossification phase of bone regeneration. By increasing the ratio of M2 macrophages through interleukin 4 (IL-4) and IL-13, bone regeneration can be significantly enhanced [32]. Vi et al. found that compared with wild-type mice, macrophage-deficient mice display decreased numbers of trabeculae, and reduced bone regeneration ability after fracture, suggesting that M2 macrophages promote osteoblast differentiation [33]. M1 macrophages play an important pro-inflammatory role in the early stage of bone injury, which helps to resist the invasion of bacteria, viruses, and other pathogens, promotes antigen presentation, activates the immune response, and initiates tissue regeneration.
In addition, studies have also shown that all macrophage subtypes have the capacity to promote MSC mediated osteogenesis but their relevance may change during different physiological conditions. Loi et al. examined the impact of sequential modulation of macrophage phenotype on the osteogenic ability of pre-osteoblasts. All of the macrophage phenotypes M0, M1 and M2 increased the osteogenic ability of MC3T3 cells. Furthermore, modulation of macrophage polarization from M1 to M2 via IL-4 treatment after 72 h of M1-MC3T3 co-culture further enhanced bone formation, suggesting that transient inflammatory phase followed by M2 dominated regenerative phase might lead to optimal bone formation [34]. However, sustained, chronic, M1-macrophage mediated inflammation reduce the curative effect of bone fracture healing by inhibiting neovascularisation and prolonging the initial inflammatory response [35]. And the continued production of pro-inflammatory cytokines of M1 results in bone resorption via increasing osteoclast activity and suppression of bone formation by osteoblasts [36]. Thus, the transformation ratio of M1 to M2 macrophages in different stages of bone regeneration is particularly important, with brief acute inflammation being beneficial and chronic inflammation highly detrimental to bone healing.
Dendritic Cells
Currently, dendritic cells (DCs) are the most functional antigen-presenting cells known. Major histocompatibility complex II (MHC II) are highly expressed on the surface of DC membranes, which effectively activates the immune function of T cells and initiates the immune response [37]. In the physiological state, DCs do not exist in bone tissue or adjacent BM matrix, and there is no obvious abnormality of the skeletal system in DC-deficient mice [38]. However, in a study of rheumatoid arthritis (RA), it was found that a large number of immature and mature DCs gathered in the active lesion site of the synovium and distributed around the adjacent bone tissue [39]. Chemotactically aggregated DCs interact with T cells through the RANK-RANKL signalling pathway to activate Th1 inflammatory-type responses and improve osteoclast activity [40]. DCs are present in oral gingival tissue. In chronic periodontitis, DCs aggregate into the periodontal lesion tissue to destroy periodontal bone tissue by forming aggregates with T cells [41]. Therefore, it is believed that DCs may indirectly affect inflammation-related bone loss by activating and regulating T cell function.
Both DCs and osteoclasts are derived from monocyte/macrophage precursor cells. Granulocyte macrophage colony-stimulating factor (GM-CSF) promotes monocyte/macrophage precursor cells to differentiate into DCs and inhibit osteoclast formation. The specific molecular mechanism is related to the c-Fos signal [42]. Co-stimulation of macrophage colony-stimulating factor (M-CSF) and RANKL induces human peripheral blood-derived DCs to differentiate into osteoclasts under in vitro inductive conditions [43], suggesting that M-CSF and GM-CSF have opposite effects on the regulation of DC and osteoclast differentiation. Alnaeeli et al. have found that mouse CD11c+ DCs co-cultured with CD4+ T cells differentiate into functional osteoclasts and that systematic infusion of CD11c+ DCs delays the repair and regeneration of mouse parietal bone defects [44]. These results clearly indicate that DCs have the potential to differentiate into osteoclasts, directly regulate the bone resorption process, and then affect bone remodelling and regeneration.
Innate lymphoid cells
Innate lymphoid cells (ILCs) are the most recently identified subset of innate lymphocytes. The members of the ILC family, mainly composed of natural killer cells, ILC1, ILC2, ILC3, and lymphoid tissue inducer cells. ILCs can be classified in three principal groups according to their transcription factor expression and cytokine secretion [45]. Group 1 ILCs (ILC1s) consist of NK cells and ILC1, which produce IFN-γ. ILC2s secrete IL-13 and IL-5, while ILC3s consists of lymphoid tissue inducer cells and ILC3 which secrete IL-22 and IL-17 [46]. In the last few years, different reports highlighted the important contribution of ILCs to bone homeostasis. The research shows that ILCs are present in periodontal tissues of mice [47]. Further, periodontal tissues of the ligature-induced murine model of periodontitis display significant increases in each subset of ILCs with the effect more marked for ILC2s [47]. Importantly, periodontal tissues of human subjects obtained from sites affected by periodontitis show remarkable similarities to those of the murine model [47, 48]. Furthermore, RANKL expression was detected on a fraction of the ILC1s suggesting a possible role of ILCs to directly contribute to inflammatory osteolysis [48]. However, Omata et al. found that ILC2 suppressed osteoclast formation through the release of cytokines, such as IL-4 and IL-13, and adoptive transfer of ILC2 completely abrogated ovariectomy-induced bone loss [49]. To sum up, the role of ILCs in bone homeostasis is controversial and needs further study.
T Cells
T cells are important lymphocytes in the adaptive immune system, are derived from hematopoietic stem cells, develop and mature in the thymus, and then migrate to the peripheral lymphoid organs. Many studies have confirmed that T cells produce a series of cytokines and growth factors, and play a key role in the process of bone remodelling and regeneration. According to different T cell receptors, T cells are divided into two categories: αβ T cells and γδ T cells. αβ T cells are further subcategorised into CD4+ and CD8+ T cells, which have dual functions of promoting and inhibiting regeneration. γδ T cells are a small subset of T cells, which are considered to promote regeneration, but the mechanism is not clear [50, 51]. Therefore, the regulatory role of T cells in bone regeneration may be related to T cell subsets, cytokines, and local factors.
T help cell 1 (Th1)/Th2 Cells
Under antigen stimulation, naive T cells differentiate into Th1 and Th2 cells. Th1 cells mainly secrete interferon γ (IFN-γ), IL-2, and tumour necrosis factor α (TNF-α) to eradicate intracellular pathogens. Th2 cells are involved in the activation of B cells and elimination of extracellular pathogens by the secretion of IL-4, IL-5, IL-6, and IL-10 [52]. At present, the role of Th1 and Th2 cells in bone regeneration is still controversial. Sato et al. found that IFN-γ and IL-4 produced by Th1 and Th2 cells inhibit osteoclast differentiation and promote bone formation by degrading TNF receptor associated factor 6 (TRAF-6) [53]. Previous studies have demonstrated that Th1 cells are the main source of RANKL. Th1 cells mainly promote osteoclast precursor cell differentiation and activate osteoclast function through TNF-α, which indirectly leads to bone tissue absorption and destruction [54]. In addition, under conditions of oestrogen deficiency or inflammation, the osteoclastogenic effect of Th1 cells is dominant [55].
Th17 Cell
Th17 cells are a type of CD4+ T cells that have been discovered in recent years. Under the stimulation of transforming growth factor β (TGF-β) and IL-6, naive T cells differentiate into Th17 cells, which mainly secrete IL-17, IL-22, and IL-26, and promote bone resorption [56]. IL-17 produced by Th17 cells induces the expression of RANKL on the surface of osteoclast precursors, bone marrow stromal cells, and osteoblasts, thus promoting osteoclast formation and inhibiting osteoblast differentiation and function [57]. Th17 cells directly promote the bone resorption process by secreting increasing amounts of active RANKL compared to Th1 cells [53]. IL-17 also indirectly enhances bone resorption by recruiting and activating other immune cells to up-regulate levels of TNF-α and IL-1 in bone tissue [58]. In summary, Th17 cells, known as osteoclastogenesis-supporting T cells, and their pro-inflammatory cytokine IL-17, play a negative regulatory role in bone regeneration.
T regulatory Cells (Treg)
T regulatory cells (Treg) are a subset of T cells with immunosuppressive functions, which secrete cytokines, such as IL-4 and TGF-β, maintain autoantigen immune tolerance, and homeostasis of the immune system. Treg play an important positive regulatory role in bone wound healing and regeneration. Zaiss et al. found that transgenic mice with elevated level of Treg display higher bone mineral density and lower bone resorption in their long bones than wild-type mice [59]. In a study of the repair of calvarial defects in mice, systematic injection of Treg was found to effectively reduce the levels of inflammatory factors in the local area of trauma, improve the osteogenic ability of transplanted stem cells, and finally achieve functional bone regeneration [54]. In vitro studies have also found that Treg cells directly up-regulate the function of osteoblasts and improve the osteogenic differentiation ability of osteoblast precursor cells, bone marrow mesenchymal stem cells (BMMSCs) [60]. This mechanism may be related to the CD39-CD93-adenosine receptor (AdoR) signalling pathway [60]. Other studies have discovered that Treg interact with CD8+ T cells to up-regulate the production of WNT10b, which acts on stromal cells and osteoblasts to promote bone formation [61]. Moreover, Treg are able to downregulate the differentiation and function of osteoclasts: Treg directly inhibit the differentiation of peripheral blood-derived monocytes into osteoclasts [62]; and they also inhibit osteoclast function by secreting cytotoxic T-lymphocyte associated protein 4 (CTLA4) to activate the CD80/CD86 receptor of osteoclast precursors [63]. Uncontrolled inflammation after tissue injury can lead to impaired healing and tissue remodeling. In the process of bone repair, Tregs are recruited to the damaged site to facilitate inflammation resolution and to regulate immunopathologic reactions after injury [64]. For instance, Tregs indirectly promote tissue regeneration by controlling neutrophil behavior and macrophage polarization [65, 66]. Moreover, as immunosuppressive cells, Treg effectively inhibit over-activated T cells in inflammatory lesions, such as tissue trauma, inflammation, and tumours, induce immune tolerance, and reduce levels of Th1, Th17, and pro-inflammatory cytokines in local tissue, thus promoting bone regeneration. For instance, it has been demonstrated that Tregs facilitate MSC-based bone regeneration by inhibiting CD4+ T-cells, which secrete IFN-γ and TNF-α [54].
B Cells
B cells are derived from hematopoietic stem cells in the BM and enter the spleen and lymph nodes after maturation. B cells proliferate and differentiate into plasma cells under the stimulation of antigens. As an important source of antibody synthesis and secretion, B cells are an important part of the adaptive immune system. In addition to immune function, the differentiation and maturation of B cells are closely related to bone cells. B cells differentiate and mature in the BM cavity, where BMMSCs provide a stable microenvironment for B cell differentiation. An abnormal BM phenotype leads to B cell differentiation and dysfunction [67]. Although the role of B cells during normal bone remodelling appears minimal, activated B cells play an important role in inflammation-related bone diseases. In multiple myeloma, B cells promote bone resorption by directly up-regulating the expression of RANKL and blocking the activity of runt-related transcription factor 2 (RUNX2)/core-binding factor alpha 1 [68, 69]. Plasma cells derived from malignant B cells secrete proteases such as sclerostin and Dickkopf WNT signalling pathway inhibitor 1 to inhibit osteoblast differentiation [70]. B cells in RA mice express high levels of the osteoblast inhibitors C-C motif chemokine ligand 3 and TNF, and inhibit mesenchymal precursor cell differentiation into osteoblasts by activating extracellular signal-regulated kinase and NF-κB signalling pathways, while B cell depletion attenuates bone erosion and osteoblast inhibition in RA mice [71]. In addition, in osteoporosis caused by oestrogen deficiency, B cells secrete RANKL to promote osteoclast function, which is one of the important mechanisms of decreased bone mineral density [72].
B cells and plasma cells are the main sources of osteoprotegerin (OPG) in bone tissue. B cells antagonistically block the effect of RANKL by secreting OPG and promoting bone tissue regeneration. In an inflammatory environment, activated T cells promote B cell to product OPG via the CD40/CD40L signalling pathway, which has a promoting effect on bone tissue regeneration [73]. In an in vitro osteoclast culture system, TGF-β secreted by B cells induces osteoclast apoptosis; TGF-β further promotes B cells to produce OPG, suggesting that B cells inhibit osteoclast differentiation [74]. In summary, the role of B cells in bone regeneration is still controversial, and its specific role may depend on local pathological conditions.
Role of immune cytokines in bone regeneration
Immune cells play a key role in the process of tissue injury, healing, and regeneration, and cytokines are important effectors of immune cells. Cytokines may play different roles in promoting or inhibiting bone tissue regeneration, which may be related to the type, concentration, expression time of cytokines, as well as local environment.
TNF-α
TNF-α is a transmembrane protein mainly produced by monocytes, binding to its receptor (TNFR1, TNFR2): TNF-α binds to TNFR1 to initiate NF-κB signalling pathways, as well as binds to TNFR2 to activate T cells via mitogen-activated protein kinase (MAPK) [75]. At present, TNF-α is believed to inhibit osteogenesis, promote bone resorption, and negatively regulate the bone homeostasis of many chronic and inflammatory diseases [76]. Recent studies indicate that osteoblast lineage cells, including PDL cells, that have osteoblastic characteristics are the primary source of RANKL [77]. RANKL produced by periodontal ligament and bone lining cells is required for orthodontic tooth movement and formation of osteoclast [78]. Also RANKL produced by osteocytes is critical for inflammation induced bone loss in a periodontitis model [79]. The increased expression of TNF-α leads to nuclear factor-kappa B activation in periodontal ligament cells, osteoblasts, and osteocytes, which has negative impact on bone formation and expression of bone-producing factors such as fibroblast growth factor-2, TGF-β1, bone morphogenetic protein-2, and bone morphogenetic protein [16, 80]. However, recent studies have indicated that vesicular RANK, which is secreted from the maturing osteoclasts, binds osteoblastic RANKL and promotes bone formation by triggering RANKL reverse signaling, indicating that osteoblastic RANKL functions also as a coupling signal acceptor that recognizes vesicular RANK [81]. Additional, TNF-α suppresses osteoblast differentiation by inhibiting classical WNT signalling, and inhibits osteogenic process by up-regulating the expression of the SMAD ubiquitin regulatory factor in osteoblasts [82, 83]. In addition, TNF-α induces osteoclast precursor cells to chemotaxis to bone resorption sites and differentiate into mature osteoclasts [84].
Interestingly, some researchers believe that TNF-α promotes bone healing and regeneration. The level of TNF-α reached its peak 24 h after fracture, which was helpful in recruiting BMMSCs involved in bone regeneration into the injured tissue. The second peak appears 4 weeks after fracture, which is essential for endochondral ossification, suggesting that TNF-α may be a cytokine that marks the beginning of tissue healing [85]. Thus, the role of TNF-α in bone healing and regeneration is regulated by many factors, and its effect mainly depends on the concentration and the differentiation state of the osteocytes, as well as on the exposure time [86].
IL-1
IL-1 is produced by activated monocytes-macrophages and exists in the form of IL-1α and IL-1β. IL-1 is a key factor in osteoclast differentiation and formation, and its mechanism is related to the RANKL/RANK signalling pathway. IL-1α may promote osteoclast differentiation by stimulating the secretion of prostaglandin E2 (PGE2) [87]. Systematic injection of IL-1α into mice causes hypercalcemia and increases the number of osteoclast precursors and osteoclasts in the bone marrow [88]. IL-1β enhances bone resorption by promoting osteoclast adherence to the surface of calcified bone trabeculae [89]. In addition, IL-1β induces peripheral blood mononuclear cells to pass through the vascular endothelium, gather and differentiate into osteoclasts [90]. A large amount of IL-1β is released during bone injury and binds to IL-1R1/MyD88 receptors, which promotes osteoclast function while reducing MSC proliferation, migration, and differentiation, thus inhibiting bone regeneration [91].
In the process of fracture healing, IL-1 not only has a synergistic effect with TNF-α, but also serves as the basis for the function of TNF-α [92]. In the mouse fracture healing model, the concentration of IL-1 increases sharply after fracture and peaks after 24 h, during which time IL-1 promotes the release of pro-inflammatory factors, such as IL-6 and PGE2, and promotes angiogenesis and cartilage callus formation to stabilise the fracture site [93]. The second peak appears 3 weeks after fracture, when IL-1 promotes protease activity to degrade injured tissue and promotes bone remodelling and regeneration [93].
IL-6
IL-6 is a pleiotropic immune cytokine expressed and secreted by osteoblast cell lines and osteoclasts, which is essential for normal bone development and bone regeneration. Compared with wild-type mice, IL-6 knockout mice show symptoms, such as decreased bone mineral density, a reduced number of bone trabeculae, decreased fracture healing ability, and significantly delayed mineralisation and callus maturation [94]. The specific mechanism is related to IL-6, which regulates the differentiation of osteoblasts and osteoclasts, and stimulates the release of vascular endothelial growth factor to promote vascularisation. In addition, in vitro experiments show that IL-6 treatment promotes the expression of the osteogenic proteins RUNX2 and osteocalcin, and promotes osteoblast differentiation [95]. All the above studies suggest that IL-6 plays an important role in promoting bone regeneration.
In contrast to other cytokines, such as IL-1 and TNF-α, IL-6 does not play a regulatory role in the whole process of bone tissue injury, healing, and regeneration. The expression level of IL-6 peaked on the first day after fracture and decreased significantly after operation or seven days after fracture. Volpin et al. also confirmed that IL-6 levels in patients with different types of fracture returns to normal, six months after injury, which suggests that IL-6 is crucial in promoting tissue healing and osteogenic regeneration in the early stage of bone tissue injury and can thus be used as an early marker of fracture healing [96, 97].
IL-17
IL-17 is a pro-inflammatory cytokine that has been discovered in recent years, and is mainly secreted by Th17 cells. After binding to its receptor, IL-17 promotes inflammatory development and activates the immune response, which plays an important role in inflammation and autoimmune diseases [98]. Although IL-17 as the main effector of “osteoclastogenesis supporting Th17 cells”, however its exact effect on osteoclasts is not clear. Some studies have reported that IL-17 up-regulates the activity of osteoclasts in animal models of arthritis, but the specific mechanism may be related to the indirect effects of other cytokines, such as IL-1, TNF-α, and RANKL [99]. Recent studies have found that IL-17 is crucial in the regulation of MSCs. Huang et al. found that IL-17 stimulates the formation of BMMSC clones in humans and mice, suggesting that IL-17 derived from T cells may act as a growth factor for MSCs [100]. Other studies have also shown that IL-17 effectively stimulates the proliferation, migration, and osteogenic differentiation of MSCs, but inhibits adipogenic differentiation [101, 102]. Interestingly, IL-17 inhibits the differentiation and maturation of mouse myoblast line cells into muscle cells and promotes differentiation into osteoblasts [103]. Moreover, IL-17 acts synergistically with bone morphogenetic protein 2 (BMP-2) to enhance new bone formation [104]. In summary, the role of IL-17 in bone regeneration is controversial and needs further study.
IFN-γ
IFN-γ was first found in RA, and then entered the field of osteoimmunology; it inhibits the excessive formation of osteoclasts and has an osteoprotective effect [105]. At present, there is no consensus regarding the effect of IFN-γ on osteoclasts. IFN-γ inhibits RANKL signalling by activating the ubiquitin proteasome to accelerate TRAF-6 degradation, directly affects osteoclast precursors, and inhibits osteoclast formation and maturation [106], suggesting that IFN-γ is the central link of T cells inhibiting osteoclast formation induced by RANKL. Although there is evidence that IFN-γ inhibits osteoclasts in vitro, its effect in vivo is more complex. In the treatment of osteosclerosis with IFN-γ, IFN-γ induces the expression of MHC II molecules in vivo, promotes antigen presentation, and directly inhibits the formation of osteoclasts by targeting mature osteoclasts, leading to the process of osteoclast formation and bone absorption [107].
In addition, IFN-γ has a regulatory effect on osteoblasts and their progenitor cells. MSCs cultured in osteo-inductive conditions in vitro secrete a large amount of IFN-γ to promote osteogenic differentiation [108]. Compared with wild-type mice, IFN-γ receptor knockout mice exhibit lower structural parameters of cortical and trabecular bone, showing an osteoporotic phenotype [109]. On the other hand, the synergistic effect of IFN-γ and TNF-α inhibits the osteogenic and adipogenic differentiation of MSCs through the NF-κB signalling pathway [110]. IFN-γ activates indoleamine, which regulates the metabolism of MSCs through the tryptophan/kynurenine pathway, thus inhibiting the differentiation of MSCs into osteoblasts [111]. In summary, IFN-γ plays an important role in the regulation of bone homeostasis and bone regeneration.
Additional Cytokines
In addition to the inflammatory cytokines described above, there are other cytokines that exert an effect on MSCs during the process of bone regeneration and remodeling. Recent studies have identified that the IL-12 cytokine family, consisting of pro-inflammatory IL-12 and IL-23 and the anti-inflammatory IL-27 and IL-35 cytokines, play an important role in bone regeneration [112]. The proinflammatory cytokines of IL-12 family, namely, IL-12 and IL-23, indirectly inhibited BMMSC differentiation by stimulating CD4+ T cells to increase IFN-γ and IL-17 [113]. Anti-inflammatory cytokine IL-27, was found to have inhibitory effects on osteoclastogenesis and osteoblast apoptosis, thus promotes bone formation [114]. Moreover, IL-35, the most recently discovered IL-12 family member, is a direct inhibitor of osteoclastogenesis [115].
Future prospects of applied osteoimmunology
Achieving satisfactory reconstruction of bone remains an important goal in orthopedic and dental conditions such as bone trauma, osteoporosis, arthritis and periodontitis. An acute inflammatory response is crucial at the onset of bone repair, while an adaptive immune response has important implications during late bone remodeling. During the initial phase of bone healing, macrophages and neutrophils respond by releasing a variety of cytokines, chemokines, and growth factors. As part of the adaptive immune response, T and B cells and their inflammatory cytokines play an important role in regulating bone regeneration [116]. A well-controlled inflammatory response can be beneficial to bone repair, but can be destructive to bone tissue when the inflammation is unregulated [117]. For example, reduced levels of bone matrix molecules and the osteoblast transcription factors in peri-implantitis [118]. In another model of rheumatoid arthritis, resolution of inflammation was accompanied by bone regeneration [119]. Taken together, the control of inflammation is indispensable for an anabolic microenvironment that supports bone formation and repair.
The challenge remains as to how these cellular mechanisms can be modulated to the benefit of the patient. From a clinical point of view, by modifying the surface qualities of biomaterials while maintaining stability and longevity, it may cause favorable responses of immune cells [120], thus giving immunomodulatory properties to transplanted materials, which is expected to provide new strategies for future orthopedic surgery, trauma surgery and implant repair.
In addition, the monoclonal antibody therapy has marked a milestone in the treatment of bone damage caused by immune-related diseases such as rheumatoid arthritis. For example, Tocilizumab (target: IL-6R) and Adalimumab (target: TNF-α) are approved very successful therapeutic drugs for rheumatoid arthritis [121, 122]. However, in view of the complex relationship between bone and immune system, it seems meaningful to find the best therapeutic target so as to open up new strategies for therapies based on molecular and cellular principles of osteoimmunology.
Conclusions and prospects
The repair and regeneration of bone tissue is a complex process involving the participation of a variety of cells and is regulated by many factors. Immune cells and immune cytokines play critical roles in regulating the balance of bone formation and bone resorption. However, the immunomodulatory mechanism of bone regeneration is still unclear. Currently, increasing evidence indicates that regulating the immune microenvironment is a promising therapeutic target to promote bone tissue functional regeneration. At the same time, we still face many problems, such as how to select correct immune cells and immune factors as targets for bone regeneration therapy to establish effective immune microenvironment, and how to combine immune microenvironment regulation with bone tissue engineering. It is believed that further studies on the roles of immunology in bone regeneration will provide new and effective targets for the treatment of bone tissue defects.
Abbreviations
BM: bone marrow; RANKL: receptor activator of nuclear factor kappa B ligand; IL-4: interleukin 4; DCs: dendritic cells; MHC II: major histocompatibility complex II; RA: rheumatoid arthritis; GM-CSF: granulocyte macrophage colony-stimulating factor; M-CSF: macrophage colony-stimulating factor; ILCs: Innate lymphoid cells; ILC1s: Group 1 ILCs; Th1: T help cell 1; IFN-γ: interferon γ; TNF-α: tumour necrosis factor α; TRAF-6: TNF receptor associated factor 6; TGF-β: transforming growth factor β; Treg: T regulatory Cells; BMMSCs: bone marrow mesenchymal stem cells; AdoR: adenosine receptor; CTLA4: cytotoxic T-lymphocyte associated protein 4; RUNX2: runt-related transcription factor 2; OPG: osteoprotegerin; MAPK: mitogen-activated protein kinase; MSCs: mesenchymal stem cells; PGE2: prostaglandin E2; BMP-2: bone morphogenetic protein 2.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81600825 to Y.L.), and High-Level Innovative Talents Project of Shenyang City (RC190480 to Y.L.). The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article. The authors thank Prof. Yan Li, No. 1 English Department, School of Fundamental Sciences, China Medical University, for her careful language editing.
Author Contributions
Yao Liu had the idea for the article and revised the article critically. She is guarantor. Ning Yang performed the literature search and wrote the first draft of the article. All authors approved the final version to be published and agree to be accountable for the work and to ensure that any questions relating to the accuracy and integrity of the paper are investigated and properly resolved.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408:535-536
2. Weitzmann MN. Bone and the immune system. Toxicol Pathol. 2017;45:911-924
3. Siddiqui JA, Partridge NC. Physiological bone remodeling: Systemic regulation and growth factor involvement. Physiology (Bethesda). 2016;31:233-245
4. Furuya M, Kikuta J, Fujimori S. et al. Direct cell-cell contact between mature osteoblasts and osteoclasts dynamically controls their functions in vivo. Nat Commun. 2018;9:300
5. Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606-615
6. Rauner M, Sipos W, Pietschmann P. Osteoimmunology. Int Arch Allergy Immunol. 2007;143:31-48
7. Katsimbri P. The biology of normal bone remodelling. Eur J Cancer Care (Engl). 2017 26. doi: 10.1111/ecc.12740
8. Lerner UH, Kindstedt E, Lundberg P. The critical interplay between bone resorbing and bone forming cells. J Clin Periodontol 46 Suppl. 2019;21:33-51
9. Guihard P, Danger Y, Brounais B. et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells. 2012;30:762-772
10. Nicolaidou V, Wong MM, Redpath AN. et al. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PLoS One. 2012;7:e39871
11. Mirosavljevic D, Quinn JM, Elliott J. et al. T-cells mediate an inhibitory effect of interleukin-4 on osteoclastogenesis. J Bone Miner Res. 2003;18:984-993
12. Allan EH, Häusler KD, Wei T. et al. EphrinB2 regulation by PTH and PTHrP revealed by molecular profiling in differentiating osteoblasts. J Bone Miner Res. 2008;23:1170-1181
13. Laine CM, Joeng KS, Campeau PM. et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368:1809-1816
14. Ono T, Takayanagi H. Osteoimmunology in bone fracture healing. Curr Osteoporos Rep. 2017;15:367-375
15. Huang Z, Pei X, Graves DT. The interrelationship between diabetes, IL-17 and bone loss. Curr Osteoporos Rep. 2020;18:23-31
16. Graves DT, Ding Z, Yang Y. The impact of diabetes on periodontal diseases. Periodontol 2000. 2020;82:214-224
17. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159-175
18. Kovtun A, Bergdolt S, Wiegner R. et al. The crucial role of neutrophil granulocytes in bone fracture healing. Eur Cell Mater. 2016;32:152-162
19. Poubelle PE, Chakravarti A, Fernandes MJ. et al. Differential expression of RANK, RANK-L and osteoprotegerin by synovial fluid neutrophils from patients with rheumatoid arthritis and by healthy human blood neutrophils. Arthritis Res Ther. 2007;9:R25
20. Chakravarti A, Raquil MA, Tessier P. et al. Surface RANKL of Toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood. 2009;114:1633-1644
21. Kanamaru F, Iwai H, Ikeda T. et al. Expression of membrane-bound and soluble receptor activator of NF-kappa B ligand (RANKL) in human T cells. Immunol Lett. 2004;94:239-246
22. Allaeys I, Rusu D, Picard S. et al. Osteoblast retraction induced by adherent neutrophils promotes osteoclast bone resorption: Implication for altered bone remodeling in chronic gout. Lab Invest. 2011;91:905-920
23. Brunetti G, Faienza MF, Piacente L. et al. High dickkopf-1 levels in sera and leukocytes from children with 21-hydroxylase deficiency on chronic glucocorticoid treatmen. Am J Physiol Endocrinol Metab. 2013;304:E546-554
24. Kantarci A, Oyaizu K, Van Dyke TE. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: Findings from localized aggressive periodontitis. J Periodontol. 2003;74:66-75
25. Tanaka D, Kagari T, Doi H. et al. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology. 2006;119:195-202
26. Wu AC, Raggatt LJ, Alexander KA. et al. Unraveling macrophage contributions to bone repair. Bonekey Rep. 2013;2:373
27. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445-455
28. Murray PJ, Allen JE, Biswas SK. et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity. 2014;41:14-20
29. Kong L, Wang Y, Smith W. et al. Macrophages in bone homeostasis. Curr Stem Cell Res Ther. 2019;14:474-481
30. Fromigué O, Marie PJ, Lomri A. Bone morphogenetic protein-2 and transforming growth factor-beta2 interact to modulate human bone marrow stromal cell proliferation and differentiation. J Cell Biochem. 1998;68:411-426
31. Blom AB, van Lent PL, Holthuysen AE. et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage. 2004;12:627-635
32. Schlundt C, El Khassawna T, Serra A. et al. Macrophages in bone fracture healing: Their essential role in endochondral ossification. Bone. 2018;106:78-89
33. Vi L, Baht GS, Whetstone H. et al. Macrophages promote osteoblastic differentiation in-vivo: Implications in fracture repair and bone homeostasis. J Bone Miner Res. 2015;30:1090-1102
34. Loi F, Córdova LA, Zhang R. et al. The effects of immunomodulation by macrophage subsets on osteogenesis in vitro. Stem Cell Res Ther. 2016;7:15
35. Schmidt-Bleek K, Schell H, Kolar P. et al. Cellular composition of the initial fracture hematoma compared to a muscle hematoma: A study in sheep. J Orthop Res. 2009;27:1147-1151
36. Bastian O, Pillay J, Alblas J. et al. Systemic inflammation and fracture healing. J Leukoc Biol. 2011;89:669-673
37. Collin M, Bigley V. Human dendritic cell subsets: An update. Immunology. 2018;154:3-20
38. McKenna HJ, Stocking KL, Miller RE. et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95:3489-3497
39. Page G, Miossec P. RANK and RANKL expression as markers of dendritic cell-T cell interactions in paired samples of rheumatoid synovium and lymph nodes. Arthritis Rheum. 2005;52:2307-2312
40. Santiago-Schwarz F, Anand P, Liu S. et al. Dendritic cells (DCs) in rheumatoid arthritis (RA): Progenitor cells and soluble factors contained in RA synovial fluid yield a subset of myeloid DCs that preferentially activate Th1 inflammatory-type responses. J Immunol. 2001;167:1758-1768
41. Cirrincione C, Pimpinelli N, Orlando L. et al. Lamina propria dendritic cells express activation markers and contact lymphocytes in chronic periodontitis. J Periodontol. 2002;73:45-52
42. Miyamoto T, Ohneda O, Arai F. et al. Bifurcation of osteoclasts and dendritic cells from common progenitors. Blood. 2001;98:2544-2554
43. Rivollier A, Mazzorana M, Tebib J. et al. Immature dendritic cell transdierentiation into osteoclasts: A novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. 2004;104:4029-4037
44. Alnaeeli M, Penninger JM, Teng YT. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. J Immunol. 2006;177:3314-3326
45. Vivier E, Artis D, Colonna M. et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054-1066
46. Spits H, Artis D, Colonna M. et al. Innate lymphoid cells-a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145-149
47. Qin X, Hoda MN, Susin C. et al. Increased innate lymphoid cells in periodontal tissue of the murine model of periodontitis: The role of AMP-activated protein kinase and relevance for the human condition. Front Immunol. 2017;8:922
48. Kindstedt E, Koskinen Holm C, Palmqvist P. et al. Innate lymphoid cells are present in gingivitis and periodontitis. J Periodontol. 2019;90:200-207
49. Omata Y, Frech M, Lucas S. et al. Type 2 innate lymphoid cells inhibit the differentiation of osteoclasts and protect from ovariectomy-induced bone loss. Bone. 2020;136:115335
50. Ramirez K, Witherden DA, Havran WL. All hands on DE(T)C: Epithelial-resident γδ T cells respond to tissue injury. Cell Immunol. 2015;296:57-61
51. Ono T, Okamoto K, Nakashima T. et al. IL-17-producing γδ T cells enhance bone regeneration. Nat Commun. 2016;7:10928
52. Lai LW, Yong KC, Lien YH. Pharmacologic recruitment of regulatory T cells as a therapy for ischemic acute kidney injury. Kidney Int. 2012;81:983-992
53. Sato K, Suematsu A, Okamoto K. et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673-2682
54. Liu Y, Wang L, Kikuiri T. et al. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nat Med. 2011;17:1594-1601
55. Liu Y, Wang L, Liu S. et al. Transplantation of SHED prevents bone loss in the early phase of ovariectomy-induced osteoporosis. J Dent Res. 2014;93:1124-1132
56. Volpe E, Servant N, Zollinger R. et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650-657
57. Adamopoulos IE, Chao CC, Geissler R. et al. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res Ther. 2010;12:R29
58. Adamopoulos IE, Bowman EP. Immune regulation of bone loss by Th17 cells. Arthritis Res Ther. 2008;10:225
59. Zaiss MM, Frey B, Hess A. et al. Regulatory T cells protect from local and systemic bone destruction in arthritis. J Immunol. 2010;184:7238-7246
60. Lei H, Schmidt-Bleek K, Dienelt A. et al. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front Pharmacol. 2015;6:184
61. Tyagi AM, Yu M, Darby TM. et al. The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of Wnt10b expression. Immunity. 2018;49:1116-1131.e7
62. Luo CY, Wang L, Sun C. et al. Estrogen enhances the functions of CD4(+)CD25(+)Foxp3(+) regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol. 2011;8:50-58
63. Axmann R, Herman S, Zaiss M. et al. CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis. 2008;67:1603-1609
64. Murphy TJ, Ni Choileain N, Zang Y. et al. CD4+CD25+regulatory T cells control innate immune reactivity after injury. J Immunol. 2005;174:2957-2963
65. Lavine KJ, Epelman S, Uchida K. et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111:16029-16034
66. Aurora AB, Porrello ER, Tan W. et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382-1392
67. Horowitz MC, Fretz JA, Lorenzo JA. How B cells influence bone biology in health and disease. Bone. 2010;47:472-479
68. Giuliani N, Colla S, Morandi F. et al. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood. 2005;106:2472-2483
69. Giuliani N, Colla S, Sala R. et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: A potential role in multiple myeloma bone disease. Blood. 2002;100:4615-4621
70. Colucci S, Brunetti G, Oranger A. et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. 2011;1:e27
71. Sun W, Meednu N, Rosenberg A. et al. B cells inhibit bone formation in rheumatoid arthritis by suppressing osteoblast differentiation. Nat Commun. 2018;9:5127
72. Kanematsu M, Sato T, Takai H. et al. Prostaglandin E2 induces expression of receptor activator of nuclear factor-kappa B ligand/osteoprotegrin ligand on pre-B cells: Implications for accelerated osteoclastogenesis in estrogen deciency. J Bone Miner Res. 2000;15:1321-1329
73. Könnecke I, Serra A, EI Khassawna T. et al. T and B cells participate in bone repair by infiltrating the fracture callus in a two-wave fashion. Bone. 2014;64:155-165
74. Hughes DE, Dai A, Tiffee JC. et al. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2:1132-1136
75. Faustman D, Davis M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat Rev Drug Discov. 2010;9:482-493
76. Zhao B. TNF and bone remodeling. Curr Osteoporos Rep. 2017;15:126-134
77. Tsukasaki M, Komatsu N, Nagashima K. et al. Host defense against oral microbiota by bone-damaging T cells. Nat Commun. 2018;9:701
78. Yang CY, Jeon HH, Alshabab A. et al. RANKL deletion inperiodontal ligament and bone lining cells blocks orthodontic tooth movement. Int J Oral Sci. 2018;10:3
79. Graves DT, Alshabab A, Albiero ML. et al. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J Clin Periodontol. 2018;45:285-292
80. Pacios S, Kang J, Galicia J. et al. Diabetes aggravates periodontitis by limiting repair through enhanced inflammation. FASEB J. 2012;26:1423-1430
81. Ikebuchi Y, Aoki S, Honma M. et al. Coupling of bone resorption and formation by RANKL reverse signaling. Nature. 2018;561:195-200
82. Sang C, Zhang Y, Chen F. et al. Tumor necrosis factor alpha suppresses osteogenic differentiation of MSCs by inhibiting semaphorin 3B via Wnt/ β-catenin signaling in estrogen-deficiency induced osteoporosis. Bone. 2016;84:78-87
83. Kaneki H, Guo R, Chen D. et al. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem. 2006;281:4326-4333
84. Boyce BF, Schwarz EM, Xing L. Osteoclast precursors: Cytokine-stimulated immunomodulators of inflammatory bone disease. Curr Opin Rheumatol. 2006;18:427-432
85. Mountziaris PM, Mikos AG. Modulation of the inflammatory response for enhanced bone tissue regeneration. Tissue Eng Part B Rev. 2008;14:179-186
86. Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-α on bone homeostasis. Front Immunol. 2014;5:48
87. Lader CS, Flanagan AM. Prostaglandin E2, interleukin 1alpha, and tumor necrosis factor-alpha increase human osteoclast formation and bone resorption in vitro. Endocrinology. 1998;139:3157-3164
88. Haynes DR, Atkins GJ, Loric M. et al. Bidirectional signaling between stromal and hemopoietic cells regulates interleukin-1 expression during human osteoclast formation. Bone. 1999;25:269-278
89. Tsutsumimoto T, Kawasaki S, Ebara S. et al. TNF-alpha and IL-1beta suppress N-cadherin expression in MC3T3-E1 cells. J Bone Miner Res. 1999;14:1751-1760
90. Tokukoda Y, Takata S, Kaji H. et al. Interleukin-1beta stimulates transendothelial mobilization of human peripheral blood mononuclear cells with a potential to differentiate into osteoclasts in the presence of osteoblasts. Endocr J. 2001;48:443-452
91. Martino MM, Maruyama K, Kuhn GA. et al. Inhibition of IL-1R1/MyD88 signalling promotes mesenchymal stem cell-driven tissue regeneration. Nat Commun. 2016;7:11051
92. Kobayashi K, Takahashi N, Jimi E. et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191:275-286
93. Lange J, Sapozhnikova A, Lu C. et al. Action of IL-1beta during fracture healing. J Orthop Res. 2010;28:778-784
94. Yang X, Ricciardi BF, Hernandez-Soria A. et al. Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone. 2007;41:928-936
95. Li Y, Bäckesjö CM, Haldosén LA, Lindgren U. IL-6 receptor expression and IL-6 effects change during osteoblast differentiation. Cytokine. 2008;43:165-173
96. Volpin G, Cohen M, Assaf M. et al. Cytokine levels (IL-4, IL-6, IL-8 and TGFβ) as potential biomarkers of systemic inlammatory response in trauma patients. Int Orthop. 2014;38:1303-1309
97. Pesic G, Jeremic J, Nikolic T. et al. Interleukin-6 as possible early marker of stress response after femoral fracture. Mol Cell Biochem. 2017;430:191-199
98. Moseley TA, Haudenschild DR, Rose L. et al. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155-174
99. Yago T, Nanke Y, Kawamoto M. et al. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res Ther. 2007;9:R96
100. Huang W, La Russa V, Alzoubi A. et al. Interleukin-17A: A T-cell-derived growth factor for murine and human mesenchymal stem cells. Stem Cells. 2006;24:1512-1518
101. Huang H, Kim HJ, Chang EJ. et al. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: Implications for bone remodeling. Cell Death Differ. 2009;16:1332-1343
102. Ahmed M, Gaffen SL. IL-17 in obesity an adipogenesis. Cytokine Growth Factor Rev. 2010;21:449-453
103. Kocić J, Santibañez JF, Krstić A. et al. Interleukin 17 inhibits myogenic and promotes osteogenic differentiation of C2C12 myoblasts by activating ERK1,2. Biochim Biophys Acta. 2012;1823:838-849
104. Croes M, Kruyt MC, Groen WM. et al. Interleukin 17 enhances bone morphogenetic protein-2-induced ectopic bone formation. Sci Rep. 2018;8:7269
105. Takayanagi H, Sato K, Takaoka A. et al. Interplay between interferon and other cytokine systems in bone metabolism. Immunol Rev. 2005;208:181-193
106. Takayanagi H, Ogasawara K, Hida S. et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600-605
107. Key LL Jr, Rodriguiz RM, Willi SM. et al. Long-term treatment of osteopetrosis with recombinant human interferon gamma. N Engl J Med. 1995;332:1594-1599
108. Duque G, Huang DC, Macoritto M. et al. Autocrine regulation of interferon gamma in mesenchymal stem cells plays a role in early osteoblastogenesis. Stem Cells. 2009;27:550-558
109. Duque G, Huang DC, Dion N. et al. Interferon-γ plays a role in bone formation in vivo and rescues osteoporosis in ovariectomized mice. J Bone Miner Res. 2011;26:1472-1483
110. Wang L, Zhao Y, Liu Y. et al. IFN-γ and TNF-α synergistically induce mesenchymal stem cell impairment and tumorigenesis via NFκB signaling. Stem Cells. 2013;31:1383-1395
111. Croitoru-Lamoury J, Lamoury FM, Caristo M. et al. Interferon-γ regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2,3 dioxygenase (IDO). PLoS One. 2011;6:e14698
112. Vignali DA, Kuchroo VK. IL-12 family cytokines: Immunological playmakers. Nat Immunol. 2012;13:722-728
113. Xu J, Wang Y, Li J. et al. IL-12p40 impairs mesenchymal stem cell-mediated bone regeneration via CD4+ T cells. Cell Death Differ. 2016;23:1941-1951
114. Shukla P, Mansoori MN, Kakaji M. et al. Interleukin 27 (IL-27) alleviates bone loss in estrogen-deficient conditions by induction of early growth response-2 gene. J Biol Chem. 2017;292:4686-4699
115. Peng M, Wang Y, Qiang L. et al. Interleukin-35 inhibits TNF-α-induced osteoclastogenesis and promotes apoptosis via shifting the activation from TNF receptor-associated death domain (TRADD)-TRAF2 to TRADD-Fas-associated death domain by JAK1/STAT1. Front Immunol. 2018;9:1417
116. Loi F, Córdova LA, Pajarinen J. et al. Inflammation, fracture and bone repair. Bone. 2016;86:119-130
117. O'Keefe RJ, Mao J. Bone tissue engineering and regeneration: From discovery to the clinic-an overview. Tissue Eng Part B Rev. 2011;17:389-392
118. Schminke B, Vom Orde F, Gruber R. et al. The pathology of bone tissue during peri-implantitis. J Dent Res. 2015;94:354-361
119. Matzelle MM, Gallant MA, Condon KW. et al. Resolution of inflammation induces osteoblast function and regulates the Wnt signaling pathway. Arthritis Rheum. 2012;64:1540-1550
120. Sridharan R, Cameron AR, Kelly DJ. et al. Biomaterial based modulation of macrophage polarization: A review and suggested design principles. Materials Today. 2015;18:313-325
121. Scott LJ. Tocilizumab: A review in rheumatoid arthritis. Drugs. 2017;77:1865-1879
122. Weinblatt ME, Keystone EC, Furst DE. et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: The ARMADA trial. ArthritisRheum. 2003;48:35-45
Author contact
![]() Corresponding author: Yao Liu, Department of Pediatric Dentistry, School and Hospital of Stomatology, China Medical University, 117 Nanjing North Street, Shenyang 110002, China (Tel.: 86-24-3192-7717; E-mail: liuyaoedu.cn).
Corresponding author: Yao Liu, Department of Pediatric Dentistry, School and Hospital of Stomatology, China Medical University, 117 Nanjing North Street, Shenyang 110002, China (Tel.: 86-24-3192-7717; E-mail: liuyaoedu.cn).