Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
T cells and traumatic brain...
T cells and their derived...
The Th1/Th2 Balance
The Th17/T-Regulatory cells...
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(16):3644-3651. doi:10.7150/ijms.46834 This issue Cite
Review
The Peripheral Immune System and Traumatic Brain Injury: Insight into the role of T-helper cells
Wangxiao Bao, Yajun Lin, Zuobing Chen ![]()
Department of Rehabilitation Medicine, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Received 2020-4-9; Accepted 2021-8-17; Published 2021-9-9
Abstract

Emerging evidence suggests that immune-inflammatory processes are key elements in the physiopathological events associated with traumatic brain injury (TBI). TBI is followed by T-cell-specific immunological changes involving several subsets of T-helper cells and the cytokines they produce; these processes can have opposite effects depending on the disease course and cytokine concentrations. Efforts are underway to identify the T-helper cells and cytokine profiles associated with prognosis. These predictors may eventually serve as effective treatment targets to decrease morbidity and mortality and to improve the management of TBI patients. Here, we review the immunological response to TBI, the possible molecular mechanisms of this response, and therapeutic strategies to address it.
Keywords: traumatic brain injury, cytokines, T-helper cell
T cells and traumatic brain injury
Traumatic brain injury (TBI), whose proximate cause is mechanical trauma, is the leading specific cause of death and disability worldwide [1]. It is generally accepted that the majority of brain damage caused by TBI is inflicted by secondary effects of the injury, rather than by the primary injury itself [2]. Secondary injury, which is progressive and lasts for a long time, contributes significantly to several post-TBI pathological events, including an exacerbated inflammatory response with subsequent brain edema, neuronal apoptosis, and activation of local immune cells, including microglia and astrocytes [3]. Additionally, breakdown of the blood-brain barrier (BBB) allows immune cells and molecules to enter the injured brain tissue, where acute and chronic inflammatory reactions to TBI are aggravated [4, 5]. Elevated circulating levels of inflammatory cytokines lead to multiple organ dysfunction syndrome and death [6]. Immune-inflammatory processes are integral to secondary brain damage [7], in which intracerebral and peripheral immune cells are activated [4, 8] and inflammatory cytokines are recruited [9]. Studies TBI models also reveal that TBI can result in immunosuppression. Immune cells, especially lymphocytes, decreased within several hours after TBI, indicating the possible pathophysiological effects [10]. The crosstalk between the immune and neurological systems was closely correlated with clinical outcome [11]. The presence of concomitant symptoms such as non-neurologic organ injury, neuropsychiatric symptoms and infections make TBI a systematic injury. Ongoing research to reveal post-traumatic immune process may aid in developing effective therapeutic strategies for patients with TBI [12]. Sex and age were reported to influence the immune response after TBI. Researchers demonstrate that aged rats exhibited more robust microglial responses, exaggerated secondary neuroinflammation, and worsens neurological outcomes after TBI [13, 14]. And TBI leads to a more aggressive neuroinflammatory profile in male compared to female mice, suggesting a rapid and pronounced peripheral inflammatory response and cortical microglia/macrophage activation [15, 16].
Increasing evidence indicates that the immune system is targeted following TBI [17, 18]. Neutrophils are first recruited to the site of the damaged brain [19], followed by local activation of microglia and astrocytes as well as the recruitment of other peripheral immune cells, including monocytes, natural killer cells, dendritic cells, and T lymphocytes [20]. T lymphocytes, critical constituents of the peripheral immune system, include many subsets, including CD3+, CD4+, and CD8+. In TBI models, CD4+ T cells first increase and then decrease, while CD8+ T cells have the opposite tendency [21, 22]. Previous data suggest that autoreactive T cells have beneficial effects on tissue repair following brain injury [23-25]. Regarded as T-helper (Th) cells, CD4+ T cells play a central role between antigen presenting cells and B cells. Although Th cells were previously thought to be detrimental [26], several studies have reported a beneficial effect after traumatic injury [27, 28]. Evidence shows both potentially destructive (causing autoimmune disease [29]) and beneficial (resisting post-traumatic degeneration [30]) effects of Th cells in the peripheral immune system after trauma. However, no clear relationship has been established between the levels of T cells and the clinical outcome following TBI.
Possible mechanism and the interactions between brain and systemic immunity response after traumatic brain injury (TBI).
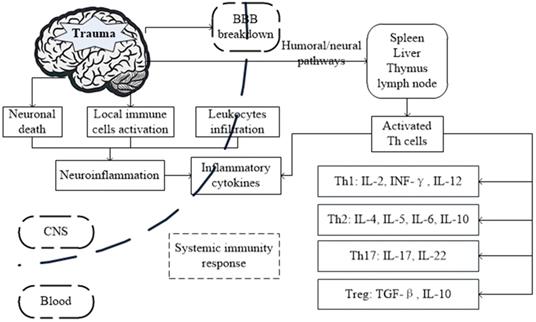
In this review, we summarize the distinct cellular and molecular events in TBI and highlight the role of Th cells and their cytokines involved in the immune-inflammatory processes associated with brain damage and recovery.
T cells and their derived cytokines
Th cell subsets, which express CD4 and MHC class II molecules on their surface, begin as naive, uncommitted Th precursors (Th0). Once stimulated by antigen presenting cells, Th cells appear to specifically differentiate into T cell subsets, including Th type 1 cells (Th1), Th type 2 cells (Th2), Th type 17 cells (Th17), and regulatory T (Treg) cells [1, 31]. For example, Th0 cells develop into Th1 cells when exposed primarily to interleukin (IL)-12 and interferon (IFN)-γ, whereas they differentiate into Th2 cells when stimulated primarily by IL-4 [32]. IL-6, transforming growth factor (TGF)-β, and IL-1β are vital factors in Th17 cell development [33] while IL-2 is responsible for Treg cell development [34]. Cytokines, a group of messengers released by Th cells, are involved in the subsequent pathophysiological processes that occur in the injured brain [35, 36]. Cytokines have pro- and anti-inflammatory effects and play dual roles in secondary brain damage. Both animal and clinical studies have suggested a correlation between TBI and pro- and anti-inflammatory cytokines [37].
Alterations in various T cell subsets as well as their own signature cytokines have been shown to influence immune-inflammatory responses and are associated with the pathogenesis of TBI. Infiltrating T-lymphocytes, cross the BBB via distinct mechanisms, are likely to be associated with brain edema and other acute responses to TBI [38], while activated CD4+T cells may exacerbate the acute damage [39]. Studies on the novel immunosuppressive agent FTY720 showed that FTY720 can significantly reduce the number of circulating lymphocytes and attenuate the invasion of immune cells to damaged brain parenchyma [40-42], decrease infiltrating T cells and NK cells but increase the percentage of Treg cells and IL-10 concentration [43]. Previous studies have reported the central and peripheral imbalance [44] of Th cells during acute and chronic phases [45] caused by different severities of TBI [21, 46, 47]. Many inflammatory mediators in the peripheral immune system have been investigated in TBI patients to identify early biomarkers with diagnostic and prognostic value. Although non-specific inflammatory markers have been extensively studied and reviewed, less attention has been given to the T-cell-specific immunological responses after trauma. Table 1 lists the various T cell subsets and their signature cytokines in the pathogenesis of TBI, including the Th1 cytokines IL-2, IL-12, and IFN-γ, the Th2 cytokines IL-4, IL-5, IL-6, and IL-10, and Th17 and Treg cytokines. However, many of these cytokines are also expressed and released from other cellular sources such as monocytes, microglia, astroglia and neuronal cells [48], which may be reviewed in the future study.
The role and function of T-helper cells and their cytokines in traumatic brain injury
| T cell subsets | Cytokines | Peripheral level | Role | Function in TBI |
|---|---|---|---|---|
| Th 1 | IL-2 | diminished | Pro-inflammatory | Immunosuppression of IL-2-regulated response in TBI patients |
| Th 1 | INF-γ | elevated | Pro-inflammatory | Interfere with TBI patients' cognitive functioning |
| Th 1 | IL-12 | elevated/diminished | Pro-inflammatory | A contributing factor to TBI-induced cognitive impairments in rats |
| Th 2 | IL-4 | elevated | Anti-inflammatory | Beneficial for TBI animal models and patients |
| Th 2 | IL-5 | elevated | Pleiotropic | Marking TBI patients more susceptible to undesirable complications |
| Th 2 | IL-6 | elevated | Pro-inflammatory | Neurotrophic and neuroprotective effects in TBI animal models and patients |
| Th 2 & Treg | IL-10 | elevated | Anti-inflammatory | Beneficial and detrimental effects in TBI animal models |
| Th 17 | IL-17 | diminished | Pro-inflammatory | Induce the production and recruitment of pro-inflammatory cytokines after TBI |
| Treg | TGF-β | elevated | Anti-inflammatory | Improve the neurobehavioral deficits in brain-damaged rats |
The Th1/Th2 Balance
The most prominent components of Th cells are the Th1 and Th2 subtypes. Th1 cells are potent activators of macrophages and mediate delayed-type hypersensitivity reactions (also termed cell-mediated immunity), whereas Th2 cells promote antibodies secreted by B cells and immediate-type hypersensitivity reactions (also termed humoral immunity). Cytokines such as IL-2, IL-12, and INF-γ have been characterized as the Th1-associated group of cytokines, whereas cytokines such as IL-4, IL-5, IL-6, and IL-10 have been assigned to the Th2-associated group of cytokines [49].
TBI is accompanied by a severe shift from a Th1- to a Th2-associated response, which may further act as yet-to-be identified risk factor for sepsis, systemic inflammatory response syndrome, and multiple organ failure [50]. Shifts in the Th1/Th2 balance also appear in cerebrovascular [51] and neurodegenerative diseases [52], accompanied by various complex interactions and cell signals, suggesting a profound immunological dysfunction. Under normal circumstances, Th0 cells proportionally differentiate into Th1 and Th2 cells. However, a bias toward the Th2 response and Th1 suppression can be induced by TBI [53], which could be associated with a poor clinical outcome [54]. Tan et al. [47] reported that administering probiotics improved recovery in TBI patients by adjusting the Th1/Th2 imbalance. The balance between Th1 and Th2 cytokines may be decisive for the progression of TBI. Our discussion will focus on Th1 and Th2 cytokines in peripheral blood.
IL-2 is a pleiotropic cytokine with a complex signaling cascade [55, 56]. Among its many actions, IL-2 is a potent Th1 cell growth factor, and an essential factor for the cellular immune response [29]. IL-2 is more broadly involved with Th1 [57], Th2 [58], and Th17 [59] cells by regulating the expression of corresponding cytokine receptors [60, 61]. Julita et al. [62] demonstrated a significant reduction in serum IL-2 and its soluble receptor (sIL-2R) in TBI patients 10-50 days after trauma, suggesting immunosuppression of IL-2-regulated responses during the post-injury period. He et al. [63] revealed that the serum IL-2/sIL-2R level in trauma patients is low. The decrease of serum IL-2 level and increase of serum sIL-2R level may be involved in the post-traumatic complications and survival, suggesting the prognostic value [64]. As an aspect of the cascade of immunological defects after TBI, this decrease in IL-2 may be induced by inhibitory monocytes and immature lymphocytes [65].
In addition to IL-2, IFN-γ, and IL-12 are pro-inflammatory cytokines. IFN-γ is expressed predominantly by Th1 cells, and is an activator of the Th1 immune response and stimulator of IL-12 [66]. The expression of IFN-γ in the circulating peripheral blood mononuclear cells was thought to decrease in trauma patients because of immune defects [54], but recent evidence suggests that IFN-γ remains persistently high during the acute [67] and chronic phase [68] of TBI. IL-12 had been defined as a promotor of IFN-γ expression and natural killer cell activity [69, 70]. IL-12 signaling, related to the development of Th1 [71, 72], is governed by the transcription factor signal transducer and activator of transcription 4 through the IL-12 receptor [73]. Stahel et al. [74] reported that IL-12 was significantly elevated 14 days after trauma in TBI patients, whereas Schwulst et al. [75] showed that IL-12 expression was subsequently diminished in TBI patients 14 days later. Evidence also shows that peripheral IFN-γ and IL-12 levels are significantly associated with poorer cognitive recovery. Furthermore, high levels of IFN-γ and IL-12 interfere with TBI-induced cognitive impairment [68, 76], thus affecting the magnitude of the behavioral change [76].
IL-4 is an anti-inflammatory Th2 cytokine that downregulates the Th1 response. While it has been generally accepted that Th1 and Th2 cytokines are mutually inhibitory, IL-4 enhances the expression of IL-12 [77]. Majetschak et al. [78] reported that increased IL-4 levels are more prominent in trauma patients with favorable outcomes than that in those with an unexpected recovery. Kipnis et al. [79] suggested that IL-4 production is induced by T cells after central nervous system (CNS) injury in a MyD88-dependent manner and promotes neuronal survival and recovery through neurotrophic signaling. Although IL-4 levels increase after trauma, they may be protective as well as predictive. Administering IL-4 may be beneficial for patients with TBI by regulating a dysregulated inflammatory response [80, 81].
IL-5 was initially identified to activate B cells, but it exerts pleiotropic functions on various target cells via a high-affinity receptor [82]. Trauma patients may exhibit early elevations in plasma IL-5 levels, making them more susceptible to undesirable complications [83, 84].
IL-6 and -10 perform pro- and anti-inflammatory functions, respectively [85, 86] and are contributing factors to the inflammatory response following TBI. In a rat TBI model, IL-6 peaked at 6 h after trauma, while IL-10 peaked at 24 h [35]. The IL-6 response is more related to the type of brain damage than the IL-10 response [87]. Previous studies have indicated that although increasing IL-6 leads to exaggerated brain damage, IL-6 plays a neuroprotective role by improving post-traumatic healing [88, 89]. Kumar et al. [90] reported that elevated IL-6 is associated with an increased inflammatory response, thus leading to an unfavorable global outcome in TBI patients. However, Ley et al. [91] indicated that an IL-6 deficiency in a TBI animal model was associated with poor behavioral performance, suggesting neurotrophic and neuroprotective roles for IL-6. A plasma IL-6 level with a cut-off of 100 pg/mL has been identified to be a predictor for prognosis during the acute phase of brain-injured patients [92].
Apart from a higher pro-inflammatory burden due to IL-6, plasma levels of the anti-inflammatory cytokine IL-10 are significantly higher in TBI patients [93]. Elevated serum levels of IL-10 imply a poor outcome after TBI and are positively correlated with injury severity [94, 95]. Thus, serum IL-10 at the early phase may have significant prognostic value in TBI patients [96]. Administering IL-10 to rat models results in increased neuronal survival by suppressing several inflammatory events [97]. Intravenous and subcutaneous, but not intracerebroventricular, administration of IL-10 improves recovery [98]. IL-10 also plays an important role in the neuroprotection of hyperbaric oxygen therapy against TBI in mice [99]. Contrary to the results from animal experiments, administering IL-10 suppresses the beneficial effects in TBI patients [100]. Although IL-10 is consistently elevated during the acute phase of TBI, the contradictory effects of IL-10 occur as a result of different pre-clinical or clinical conditions [101]. Kumar et al. [102] reported that an elevated serum IL-6/IL-10 ratio was associated with outcome in TBI patients. The predictive value of IL-6 and -10 in trauma patients remains to be fully elucidated [103].
The Th17/T-Regulatory cells balance
Th17 cells, characterized by the production of IL-17A, IL-17F, and IL-22, were identified as a new lineage of Th cells in 2005 [104]. IL-17A is also called IL-17 because it is secreted in the greatest quantities and contributes to most of the Th-17 effects [105]. Studies have shown that IL-17 was significantly upregulated after TBI, which may be related to the pathogenesis of TBI [106]. As IL-17 is induced with subsequent pro-inflammatory cytokines, Th17 cells have major functions in tissue inflammation. However, recent experiments in Rag1- / - mice have demonstrated that IL-17 is also produced via a RAG-independent cellular source [107]. Treg cells are another lineage of Th cells but present a totally different picture compared with Th17 [108]. Treg cells downregulate the inflammatory response by maintaining peripheral immune tolerance [34], and preventing autoimmunity and chronic inflammation [109]. Treg cells are known to be neuroprotective by modulating the function of effector T cells [110] and secreting anti-inflammatory molecules such as IL-10 and TGF-β [111, 112]. Similar to IL-10, TGF-β is an anti-inflammatory cytokine that modulates immune-inflammatory processes [113, 114].
The balance between Th17 and Treg cells is critical for the health of the host by controlling inflammatory and autoimmune disorders [33]. Besides sharing a similar development pathway, Th17 and Treg trans-differentiate into each other under some conditions [115]. A Th17/Treg imbalance has been reported to be associated with severity of injury in trauma patients [116]. Gupta et al. have previously showed that the higher the ratio of Th17 cells to Treg, the worse the post-traumatic complications [116]. Besides, the imbalance of Th17/Treg cells is believed to be a key factor in the progression of inflammatory response [117, 118]. Therefore, adjusting Th17/Treg balance may be an effective way to manage the secondary damage of TBI. Propofol, an intravenous anesthetic drug, maintained Th17/Treg balance and reduced inflammation when injected into the TBI models [119]. Emerging findings suggest that the level of circulating Treg cells is positively correlated with neurological recovery in animal models [120] and TBI patients [121]. Kipnis et al. [122, 123] reported that transferring exogenous Treg cells into an immune-deficient animal host following CNS injury leads to neuroprotection. Increasing the number of Treg cells and their signature cytokines IL-10 and TGF-β by inhibiting mTOR signaling improves the neurobehavioral performance in brain-injured rats [124].
Conclusions
Secondary brain injury after trauma is a complex process involving central and peripheral immune responses [4]. Immune-inflammatory processes play a vital part in the pathophysiology of TBI. BBB dysfunction allows the passage of immune cells and inflammatory molecules that trigger a systemic inflammatory response [125]. Immune-inflammatory processes play a vital part in the pathophysiology of TBI. Recent evidences have established the role of Th cells and their derived cytokines in TBI. Cytokines play a dual role in TBI depending on different time courses and concentrations. A more comprehensive understanding of the cytokines in TBI is needed to develop diagnostic and therapeutic products. Modulating the immunological balance between Th1/Th2, Th17, and Treg cells may also represent a promising therapeutic strategy. Additional investigations are needed to elucidate the basic pathological mechanism of Th cells and their cytokines in the pathogenesis of TBI, and to open up new possible avenues for treating secondary brain injury.
Acknowledgements
This work was supported by grants from Natural Science Foundation of Zhejiang Province (LGF18H170002) and Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2020370189).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997;349:1436-42
2. Sahuquillo J, Poca MA, Amoros S. Current aspects of pathophysiology and cell dysfunction after severe head injury. Current pharmaceutical design. 2001;7:1475-503
3. Osthoff M, Walder B, Delhumeau C, Trendelenburg M, Turck N. Association of Lectin Pathway Protein Levels and Genetic Variants Early after Injury with Outcomes after Severe Traumatic Brain Injury: A Prospective Cohort Study. Journal of neurotrauma. 2017;34:2560-6
4. Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acute traumatic brain injury. Journal of neuroinflammation. 2012;9:236
5. Utagawa A, Truettner JS, Dietrich WD, Bramlett HM. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Experimental neurology. 2008;211:283-91
6. Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Frontiers in bioscience. 2009;14:3795-813
7. Kadhim HJ, Duchateau J, Sebire G. Cytokines and brain injury: invited review. Journal of intensive care medicine. 2008;23:236-49
8. Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS. et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic science international. 2004;146:97-104
9. Ferreira LC, Regner A, Miotto KD, Moura S, Ikuta N, Vargas AE. et al. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain injury. 2014;28:1311-6
10. McDonald SJ, Sharkey JM, Sun M, Kaukas LM, Shultz SR, Turner RJ. et al. Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. Journal of neurotrauma. 2020;37:770-81
11. Sharma R, Shultz SR, Robinson MJ, Belli A, Hibbs ML, O'Brien TJ. et al. Infections after a traumatic brain injury: The complex interplay between the immune and neurological systems. Brain, behavior, and immunity. 2019;79:63-74
12. Sun M, McDonald SJ, Brady RD, O'Brien TJ, Shultz SR. The influence of immunological stressors on traumatic brain injury. Brain, behavior, and immunity. 2018;69:618-28
13. Ritzel RM, Doran SJ, Glaser EP, Meadows VE, Faden AI, Stoica BA. et al. Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiology of aging. 2019;77:194-206
14. Sun M, Brady R, Casillas-Espinosa P, Wright D, Semple B, Kim H. et al. Aged rats have an altered immune response and worse outcomes after traumatic brain injury. Brain, behavior, and immunity. 2019;80:536-50
15. Doran SJ, Ritzel RM, Glaser EP, Henry RJ, Faden AI, Loane DJ. Sex Differences in Acute Neuroinflammation after Experimental Traumatic Brain Injury Are Mediated by Infiltrating Myeloid Cells. Journal of neurotrauma. 2019;36:1040-53
16. Villapol S, Loane DJ, Burns MP. Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia. 2017;65:1423-38
17. Plesnila N. The immune system in traumatic brain injury. Current opinion in pharmacology. 2016;26:110-7
18. McKee CA, Lukens JR. Emerging Roles for the Immune System in Traumatic Brain Injury. Frontiers in immunology. 2016;7:556
19. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature reviews Immunology. 2013;13:159-75
20. Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA neurology. 2015;72:355-62
21. Bai R, Gao H, Han Z, Huang S, Ge X, Chen F. et al. Flow Cytometric Characterization of T Cell Subsets and Microglia After Repetitive Mild Traumatic Brain Injury in Rats. Neurochemical research. 2017;42:2892-901
22. Bai R, Gao H, Han Z, Ge X, Huang S, Chen F. et al. Long-Term Kinetics of Immunologic Components and Neurological Deficits in Rats Following Repetitive Mild Traumatic Brain Injury. Medical science monitor: international medical journal of experimental and clinical research. 2017;23:1707-18
23. Hofstetter HH, Sewell DL, Liu F, Sandor M, Forsthuber T, Lehmann PV. et al. Autoreactive T cells promote post-traumatic healing in the central nervous system. Journal of neuroimmunology. 2003;134:25-34
24. Jones TB, McDaniel EE, Popovich PG. Inflammatory-mediated injury and repair in the traumatically injured spinal cord. Current pharmaceutical design. 2005;11:1223-36
25. Popovich PG, Jones TB. Manipulating neuroinflammatory reactions in the injured spinal cord: back to basics. Trends in pharmacological sciences. 2003;24:13-7
26. Kielar ML, Rohan Jeyarajah D, Lu CY. The regulation of ischemic acute renal failure by extrarenal organs. Current opinion in nephrology and hypertension. 2002;11:451-7
27. Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nature medicine. 1999;5:49-55
28. Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A. et al. Protective autoimmunity is a physiological response to CNS trauma. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:3740-8
29. Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunology today. 1995;16:34-8
30. Kipnis J, Mizrahi T, Yoles E, Ben-Nun A, Schwartz M. Myelin specific Th1 cells are necessary for post-traumatic protective autoimmunity. Journal of neuroimmunology. 2002;130:78-85
31. Toor D, Sharma N. T cell subsets: an integral component in pathogenesis of rheumatic heart disease. Immunologic research. 2017
32. Sigal LH. Basic science for the clinician 28: T-helper cell subtypes. Journal of clinical rheumatology: practical reports on rheumatic & musculoskeletal diseases. 2004;10:222-6
33. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annual review of pathology. 2013;8:477-512
34. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nature immunology. 2005;6:345-52
35. Maegele M, Sauerland S, Bouillon B, Schafer U, Trubel H, Riess P. et al. Differential immunoresponses following experimental traumatic brain injury, bone fracture and "two-hit"-combined neurotrauma. Inflammation research: official journal of the European Histamine Research Society [et al]. 2007;56:318-23
36. Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165-77
37. Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ. The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:658-70
38. Czigner A, Mihaly A, Farkas O, Buki A, Krisztin-Peva B, Dobo E. et al. Kinetics of the cellular immune response following closed head injury. Acta neurochirurgica. 2007;149:281-9
39. Fee D, Crumbaugh A, Jacques T, Herdrich B, Sewell D, Auerbach D. et al. Activated/effector CD4+ T cells exacerbate acute damage in the central nervous system following traumatic injury. Journal of neuroimmunology. 2003;136:54-66
40. Mencl S, Hennig N, Hopp S, Schuhmann MK, Albert-Weissenberger C, Siren AL. et al. FTY720 does not protect from traumatic brain injury in mice despite reducing posttraumatic inflammation. Journal of neuroimmunology. 2014;274:125-31
41. Zhang Z, Zhang Z, Fauser U, Artelt M, Burnet M, Schluesener HJ. FTY720 attenuates accumulation of EMAP-II+ and MHC-II+ monocytes in early lesions of rat traumatic brain injury. J Cell Mol Med. 2007;11:307-14
42. Zhang Z, Fauser U, Schluesener HJ. Early attenuation of lesional interleukin-16 up-regulation by dexamethasone and FTY720 in experimental traumatic brain injury. Neuropathology and applied neurobiology. 2008;34:330-9
43. Gao C, Qian Y, Huang J, Wang D, Su W, Wang P. et al. A Three-Day Consecutive Fingolimod Administration Improves Neurological Functions and Modulates Multiple Immune Responses of CCI Mice. Mol Neurobiol. 2017;54:8348-60
44. Braun M, Vaibhav K, Saad N, Fatima S, Brann DW, Vender JR. et al. Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. Journal of immunology. 2017;198:3615-26
45. Quattrocchi KB, Frank EH, Miller CH, Amin A, Issel BW, Wagner FC Jr. Impairment of helper T-cell function and lymphokine-activated killer cytotoxicity following severe head injury. Journal of neurosurgery. 1991;75:766-73
46. Ritzel RM, Doran SJ, Barrett JP, Henry RJ, Ma EL, Faden AI. et al. Chronic Alterations in Systemic Immune Function after Traumatic Brain Injury. Journal of neurotrauma. 2018
47. Tan M, Zhu JC, Du J, Zhang LM, Yin HH. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Critical care. 2011;15:R290
48. Brett BL, Gardner RC, Godbout J, Dams-O'Connor K, Keene CD. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol Psychiatry. 2021
49. Amick JE, Yandora KA, Bell MJ, Wisniewski SR, Adelson PD, Carcillo JA. et al. The Th1 versus Th2 cytokine profile in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatric critical care medicine: a journal of the Society of Critical Care Medicine and the World Federation of Pediatric Intensive and Critical Care Societies. 2001;2:260-4
50. De AK, Kodys KM, Pellegrini J, Yeh B, Furse RK, Bankey P. et al. Induction of global anergy rather than inhibitory Th2 lymphokines mediates posttrauma T cell immunodepression. Clinical immunology. 2000;96:52-66
51. Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T. et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. The Journal of experimental medicine. 2003;198:725-36
52. Gold SM, Mohr DC, Huitinga I, Flachenecker P, Sternberg EM, Heesen C. The role of stress-response systems for the pathogenesis and progression of MS. Trends in immunology. 2005;26:644-52
53. Decker D, Schondorf M, Bidlingmaier F, Hirner A, von Ruecker AA. Surgical stress induces a shift in the type-1/type-2 T-helper cell balance, suggesting down-regulation of cell-mediated and up-regulation of antibody-mediated immunity commensurate to the trauma. Surgery. 1996;119:316-25
54. Miller AC, Rashid RM, Elamin EM. The "T" in trauma: the helper T-cell response and the role of immunomodulation in trauma and burn patients. The Journal of trauma. 2007;63:1407-17
55. Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene. 2000;19:2566-76
56. Doersch KM, DelloStritto DJ, Newell-Rogers MK. The contribution of interleukin-2 to effective wound healing. Experimental biology and medicine. 2017;242:384-96
57. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nature immunology. 2011;12:551-9
58. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY. et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nature immunology. 2008;9:1288-96
59. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z. et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371-81
60. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557-69
61. Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM. et al. The Interleukin-2-mTORc1 Kinase Axis Defines the Signaling, Differentiation, and Metabolism of T Helper 1 and Follicular B Helper T Cells. Immunity. 2015;43:690-702
62. Teodorczyk-Injeyan JA, McRitchie DI, Peters WJ, Lalani S, Girotti MJ. Expression and secretion of IL-2 receptor in trauma patients. Annals of surgery. 1990;212:202-8
63. He Z, Wang S, Su Z, Huang Y, Yang J. [Changes of interleukin-2 and soluble interleukin-2 receptor serum level and multiple organ dysfunction syndrome in severely traumatic patients]. Zhonghua wai ke za zhi [Chinese journal of surgery]. 1999;37:492-3
64. Jobin N, Garrel D, Bernier J. Increased serum-soluble interleukin-2 receptor in burn patients: characterization and effects on the immune system. Hum Immunol. 2000;61:233-46
65. Faist E, Mewes A, Baker CC, Strasser T, Alkan SS, Rieber P. et al. Prostaglandin E2 (PGE2)-dependent suppression of interleukin alpha (IL-2) production in patients with major trauma. The Journal of trauma. 1987;27:837-48
66. Tannenbaum CS, Hamilton TA. Immune-inflammatory mechanisms in IFNgamma-mediated anti-tumor activity. Seminars in cancer biology. 2000;10:113-23
67. Nwachuku EL, Puccio AM, Adeboye A, Chang YF, Kim J, Okonkwo DO. Time course of cerebrospinal fluid inflammatory biomarkers and relationship to 6-month neurologic outcome in adult severe traumatic brain injury. Clinical neurology and neurosurgery. 2016;149:1-5
68. Licastro F, Hrelia S, Porcellini E, Malaguti M, Di Stefano C, Angeloni C. et al. Peripheral Inflammatory Markers and Antioxidant Response during the Post-Acute and Chronic Phase after Severe Traumatic Brain Injury. Frontiers in neurology. 2016;7:189
69. Roquilly A, David G, Cinotti R, Vourc'h M, Morin H, Rozec B. et al. Role of IL-12 in overcoming the low responsiveness of NK cells to missing self after traumatic brain injury. Clinical immunology. 2017;177:87-94
70. Chan SH, Perussia B, Gupta JW, Kobayashi M, Pospisil M, Young HA. et al. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. The Journal of experimental medicine. 1991;173:869-79
71. Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE Jr. et al. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. The Journal of experimental medicine. 1995;181:1755-62
72. Szabo SJ, Jacobson NG, Dighe AS, Gubler U, Murphy KM. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665-75
73. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547-9
74. Stahel PF, Kossmann T, Joller H, Trentz O, Morganti-Kossmann MC. Increased interleukin-12 levels in human cerebrospinal fluid following severe head trauma. Neuroscience letters. 1998;249:123-6
75. Schwulst SJ, Trahanas DM, Saber R, Perlman H. Traumatic brain injury-induced alterations in peripheral immunity. The journal of trauma and acute care surgery. 2013;75:780-8
76. Vonder Haar C, Martens KM, Riparip LK, Rosi S, Wellington CL, Winstanley CA. Frontal Traumatic Brain Injury Increases Impulsive Decision Making in Rats: A Potential Role for the Inflammatory Cytokine Interleukin-12. Journal of neurotrauma. 2017;34:2790-800
77. Gor DO, Rose NR, Greenspan NS. TH1-TH2: a procrustean paradigm. Nature immunology. 2003;4:503-5
78. Majetschak M, Christensen B, Obertacke U, Waydhas C, Schindler AE, Nast-Kolb D. et al. Sex differences in posttraumatic cytokine release of endotoxin-stimulated whole blood: relationship to the development of severe sepsis. The Journal of trauma. 2000;48:832-9 discussion 9-40
79. Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR. et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. The Journal of clinical investigation. 2015;125:699-714
80. Messingham KA, Heinrich SA, Schilling EM, Kovacs EJ. Interleukin-4 treatment restores cellular immunity after ethanol exposure and burn injury. Alcoholism, clinical and experimental research. 2002;26:519-26
81. DiPiro JT, Howdieshell TR, Goddard JK, Callaway DB, Hamilton RG, Mansberger AR Jr. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Archives of surgery. 1995;130:1159-62 discussion 62-3
82. Takatsu K. Interleukin-5 and IL-5 receptor in health and diseases. Proceedings of the Japan Academy Series B, Physical and biological sciences. 2011;87:463-85
83. Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R. et al. Temporal Patterns of Circulating Inflammation Biomarker Networks Differentiate Susceptibility to Nosocomial Infection Following Blunt Trauma in Humans. Annals of surgery. 2016;263:191-8
84. Xu J, Guardado J, Hoffman R, Xu H, Namas R, Vodovotz Y. et al. IL33-mediated ILC2 activation and neutrophil IL5 production in the lung response after severe trauma: A reverse translation study from a human cohort to a mouse trauma model. PLoS medicine. 2017;14:e1002365
85. Bell MJ, Kochanek PM, Doughty LA, Carcillo JA, Adelson PD, Clark RS. et al. Comparison of the interleukin-6 and interleukin-10 response in children after severe traumatic brain injury or septic shock. Acta neurochirurgica Supplement. 1997;70:96-7
86. Bell MJ, Kochanek PM, Doughty LA, Carcillo JA, Adelson PD, Clark RS. et al. Interleukin-6 and interleukin-10 in cerebrospinal fluid after severe traumatic brain injury in children. Journal of neurotrauma. 1997;14:451-7
87. Mellergard P, Aneman O, Sjogren F, Saberg C, Hillman J. Differences in cerebral extracellular response of interleukin-1beta, interleukin-6, and interleukin-10 after subarachnoid hemorrhage or severe head trauma in humans. Neurosurgery. 2011;68:12-9 discussion 9
88. Penkowa M, Giralt M, Lago N, Camats J, Carrasco J, Hernandez J. et al. Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury. Experimental neurology. 2003;181:130-48
89. Penkowa M, Quintana A, Carrasco J, Giralt M, Molinero A, Hidalgo J. Metallothionein prevents neurodegeneration and central nervous system cell death after treatment with gliotoxin 6-aminonicotinamide. Journal of neuroscience research. 2004;77:35-53
90. Kumar RG, Diamond ML, Boles JA, Berger RP, Tisherman SA, Kochanek PM. et al. Acute CSF interleukin-6 trajectories after TBI: associations with neuroinflammation, polytrauma, and outcome. Brain, behavior, and immunity. 2015;45:253-62
91. Ley EJ, Clond MA, Singer MB, Shouhed D, Salim A. IL6 deficiency affects function after traumatic brain injury. The Journal of surgical research. 2011;170:253-6
92. Woiciechowsky C, Schoning B, Cobanov J, Lanksch WR, Volk HD, Docke WD. Early IL-6 plasma concentrations correlate with severity of brain injury and pneumonia in brain-injured patients. The Journal of trauma. 2002;52:339-45
93. Maskin B, Gammella D, Solari L, Videta W, Barboza MF, Geliz L. et al. [Early release of the antiinflammatory cytokine IL-10 in traumatic brain injury]. Medicina. 2001;61:573-6
94. Schneider Soares FM, Menezes de Souza N, Liborio Schwarzbold M, Paim Diaz A, Costa Nunes J, Hohl A. et al. Interleukin-10 is an independent biomarker of severe traumatic brain injury prognosis. Neuroimmunomodulation. 2012;19:377-85
95. Kirchhoff C, Buhmann S, Bogner V, Stegmaier J, Leidel BA, Braunstein V. et al. Cerebrospinal IL-10 concentration is elevated in non-survivors as compared to survivors after severe traumatic brain injury. European journal of medical research. 2008;13:464-8
96. Dziurdzik P, Krawczyk L, Jalowiecki P, Kondera-Anasz Z, Menon L. Serum interleukin-10 in ICU patients with severe acute central nervous system injuries. Inflammation research: official journal of the European Histamine Research Society [et al]. 2004;53:338-43
97. Brewer KL, Bethea JR, Yezierski RP. Neuroprotective effects of interleukin-10 following excitotoxic spinal cord injury. Experimental neurology. 1999;159:484-93
98. Knoblach SM, Faden AI. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Experimental neurology. 1998;153:143-51
99. Chen X, Duan XS, Xu LJ, Zhao JJ, She ZF, Chen WW. et al. Interleukin-10 mediates the neuroprotection of hyperbaric oxygen therapy against traumatic brain injury in mice. Neuroscience. 2014;266:235-43
100. Kline AE, Bolinger BD, Kochanek PM, Carlos TM, Yan HQ, Jenkins LW. et al. Acute systemic administration of interleukin-10 suppresses the beneficial effects of moderate hypothermia following traumatic brain injury in rats. Brain research. 2002;937:22-31
101. Garcia JM, Stillings SA, Leclerc JL, Phillips H, Edwards NJ, Robicsek SA. et al. Role of Interleukin-10 in Acute Brain Injuries. Frontiers in neurology. 2017;8:244
102. Kumar RG, Boles JA, Wagner AK. Chronic Inflammation After Severe Traumatic Brain Injury: Characterization and Associations With Outcome at 6 and 12 Months Postinjury. The Journal of head trauma rehabilitation. 2015;30:369-81
103. Jastrow KM 3rd, Gonzalez EA, McGuire MF, Suliburk JW, Kozar RA, Iyengar S. et al. Early cytokine production risk stratifies trauma patients for multiple organ failure. Journal of the American College of Surgeons. 2009;209:320-31
104. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM. et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature immunology. 2005;6:1123-32
105. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051-7
106. Li T, Zhang YM, Han D, Hua R, Guo BN, Hu SQ. et al. Involvement of IL-17 in Secondary Brain Injury After a Traumatic Brain Injury in Rats. Neuromolecular Med. 2017;19:541-54
107. Sparber F, Dolowschiak T, Mertens S, Lauener L, Clausen BE, Joller N. et al. Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS pathogens. 2018;14:e1007069
108. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annual review of immunology. 2004;22:531-62
109. Debnath M, Berk M. Th17 pathway-mediated immunopathogenesis of schizophrenia: mechanisms and implications. Schizophrenia bulletin. 2014;40:1412-21
110. Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S. et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nature medicine. 2009;15:192-9
111. Suri-Payer E, Fritzsching B. Regulatory T cells in experimental autoimmune disease. Springer seminars in immunopathology. 2006;28:3-16
112. O'Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nature medicine. 2004;10:801-5
113. Qiao YC, Shen J, Hong XZ, Liang L, Bo CS, Sui Y. et al. Changes of regulatory T cells, transforming growth factor-beta and interleukin-10 in patients with type 1 diabetes mellitus: A systematic review and meta-analysis. Clinical immunology. 2016;170:61-9
114. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC. et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:770-4
115. Sun L, Fu J, Zhou Y. Metabolism Controls the Balance of Th17/T-Regulatory Cells. Frontiers in immunology. 2017;8:1632
116. Gupta DL, Bhoi S, Mohan T, Galwnkar S, Rao DN. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with post traumatic sepsis. Cytokine. 2016;88:214-21
117. Luo T, Ji WJ, Yuan F, Guo ZZ, Li YX, Dong Y. et al. Th17/Treg Imbalance Induced by Dietary Salt Variation Indicates Inflammation of Target Organs in Humans. Scientific reports. 2016;6:26767
118. Shen Y, Tang XY, Yang YC, Ke X, Kou W, Pan CK. et al. Impaired balance of Th17/Treg in patients with nasal polyposis. Scand J Immunol. 2011;74:176-85
119. Cui C, Zhang D, Sun K, Li H, Xu L, Lin G. et al. Propofol maintains Th17/Treg cell balance and reduces inflammation in rats with traumatic brain injury via the miR1453p/NFATc2/NFkappaB axis. Int J Mol Med. 2021 48
120. Yu Y, Cao F, Ran Q, Sun X. Regulatory T cells exhibit neuroprotective effect in a mouse model of traumatic brain injury. Molecular medicine reports. 2016;14:5556-66
121. Li M, Lin YP, Chen JL, Li H, Jiang RC, Zhang JN. Role of regulatory T cell in clinical outcome of traumatic brain injury. Chinese medical journal. 2015;128:1072-8
122. Walsh JT, Kipnis J. Regulatory T cells in CNS injury: the simple, the complex and the confused. Trends in molecular medicine. 2011;17:541-7
123. Kipnis J, Avidan H, Caspi RR, Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(Suppl 2):14663-9
124. Lu Q, Gao L, Huang L, Ruan L, Yang J, Huang W. et al. Inhibition of mammalian target of rapamycin improves neurobehavioral deficit and modulates immune response after intracerebral hemorrhage in rat. Journal of neuroinflammation. 2014;11:44
125. Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Current opinion in critical care. 2002;8:101-5
Author contact
![]() Corresponding author: Zuobing Chen, E-mail: czb1971edu.cn.
Corresponding author: Zuobing Chen, E-mail: czb1971edu.cn.