Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(13):3039-3049. doi:10.7150/ijms.60399 This issue Cite
Research Paper
Antitumor Activity of Small Activating RNAs Induced PAWR Gene Activation in Human Bladder Cancer Cells
Kai Yang1 ![]() *, Jie Shen2*, Fu-Qing Tan1, Xiang-Yi Zheng1, Li-Ping Xie1
*, Jie Shen2*, Fu-Qing Tan1, Xiang-Yi Zheng1, Li-Ping Xie1
1. Department of Urology, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310003, P.R. China.
2. Department of Pharmacy, Traditional Chinese Medical Hospital of Zhejiang Province, Hangzhou, Zhejiang 310006, P.R. China.
* These authors contributed equally to this work.
Received 2021-3-13; Accepted 2021-5-30; Published 2021-6-16
Abstract

Small double-stranded RNAs (dsRNAs) have been proved to effectively up-regulate the expression of particular genes by targeting their promoters. These small dsRNAs were also termed small activating RNAs (saRNAs). We previously reported that several small double-stranded RNAs (dsRNAs) targeting the PRKC apoptosis WT1 regulator (PAWR) promoter can up-regulate PAWR gene expression effectively in human cancer cells. The present study was conducted to evaluate the antitumor potential of PAWR gene induction by these saRNAs in bladder cancer. Promisingly, we found that up-regulation of PAWR by saRNA inhibited the growth of bladder cancer cells by inducing cell apoptosis and cell cycle arrest which was related to inhibition of anti‑apoptotic protein Bcl-2 and inactivation of the NF-κB and Akt pathways. The activation of the caspase cascade and the regulation of cell cycle related proteins also supported the efficacy of the treatment. Moreover, our study also showed that these saRNAs cooperated with cisplatin in the inhibition of bladder cancer cells. Overall, these data suggest that activation of PAWR by saRNA may have a therapeutic benefit for bladder cancer.
Keywords: RNA activation, small activating RNA, PAWR, bladder cancer, cell cycle arrest, apoptosis.
Introduction
Bladder cancer is the fourth most common cancer in men in United States. In 2020, there are estimated 62,100 new cases and 13,050 deaths of bladder cancer in United States [1]. Although chemotherapy has revolutionized the treatment of advanced tumors [2, 3], cisplatin-containing combination chemotherapy for metastatic disease only achieve a prolonged median survival of up to 14 months [4] and the associated side effects induced by lack of specificity to tumor cells remain a challenging problem. Therefore, novel therapeutic strategies for the treatment of advanced bladder cancer are urgently required.
Therapeutics based on RNA interference (RNAi) have become powerful and ideal methods for the treatment of many diseases including cancer which are mainly caused by overactive oncogenes because of the high specificity, high efficacy and low toxicity of the RNAi trigger - small dsRNA [5-7]. However, there are still many cancers mainly caused by complete inactivation or reduced expression of tumor suppressor genes (TSGs). Recently, new evidence has emerged that synthetic small double-strand RNAs (dsRNAs) could induce sequence-specific transcriptional gene activation of E-cadherin, p21WAF1/CIP1 and VEGF by targeting specific regions in their gene promoters [8]. This phenomenon was termed RNA-induced gene activation (RNAa) and such dsRNA molecules were termed small activating RNAs (saRNAs) [8]. Their observation was supported by another group reporting similar findings [9] and subsequent studies suggests RNAa may be a general and conserved phenomenon of gene regulation [10-17]. Moreover, several studies demonstrate that restoration of p21 expression by saRNAs in different cancer cells has been shown to inhibit cell proliferation and tumor growth [18-23]. Thus, RNAa holds great promise as an alternative to traditional vector-based systems and would supplement RNA-mediated gene silencing to broaden the gene pool susceptible to therapeutic regulation by small RNAs.
Human PAWR (PRKC apoptosis WT1 regulator) gene, whose other aliases include PAR4 and Par-4, is located in chromosome 12q21 and encodes a leucine zipper domain protein first identified in prostate cancer cells undergoing apoptosis induced by an exogenous insult [24, 25]. Functional PAWR protein is essential for apoptosis via diverse cell death pathways [26-28]. More importantly, ectopic PAWR over-expression is sufficient to induce apoptosis in most cancer cells in vitro and growth inhibition of prostate cancer xenografts in nude mice, but not in normal or immortalized cells [26, 29]. Therefore, PAWR is an ideal target and a candidate TSG for RNAa.
Our previous study has reported that dsPAWR-435, a small dsRNA targeting PAWR promoter, can up-regulate PAWR gene expression effectively in human cancer cells [17]. In this study, we investigated the antitumor effects of dsPAWR-435 on the bladder cancer cells and found that up-regulation of PAWR by saRNA could not only inhibit cell growth by inducing cell apoptosis and G1-phase arrest, but also cooperate with cisplatin against the growth of bladder cancer cells.
Materials and Methods
dsRNA Design and Synthesis
The sequence of dsPAWR-435 (S, 5'- AUA AUA CGG UCU UGU ACU U [dT][dT]-3'; AS, 5'- AAG UAC AAG ACC GUA UUA U [dT][dT]-3') was designed as previously described [17] and the control dsRNA (dsCon: S, 5'-ACU ACU GAG UGA CAG UAG A[dT][dT]-3'; AS, 5'-UCU ACU GUC ACU CAG UAG U[dT][dT]-3') is the same as the dsCon-2 which was specifically designed by Li to lack homology to all known human sequences [8]. All dsRNAs were chemically synthesized by GeneChem (Shanghai, China) with dTdT 3' overhangs.
Cell Culture and Transfection
The human bladder cancer cell line T24 and 5637 were obtained from the Shanghai Institute of Cell Biology, Chinese Academy of Science. The cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 mg/L) in a humidified atmosphere containing 5% CO2 maintained at 37oC. The day before transfection cells were plated in growth medium without antibiotics at a density of 30-40%. Transfections of dsRNAs were carried out by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol and lasted for 24, 48 or 72 hours. Cell images were taken by using a phase-contrast microscope at 100× magnification (Olympus, Japan).
Cell growth/viability assay
Proliferation of cells was determined by the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Approximately 3000 to 8000 (depending on how long they would be cultured) cells were plated in each well of a 96-well plate. After overnight incubation, the cells were treated with the appropriate dsRNA (mock, 50 nmol/l dsCon, or 1-50 nmol/l dsPAWR-435) for 24, 48, or 72 hours, or with the dsRNA (50 nmol/l dsCon or dsPAWR-435) for 24 hours, followed by incubation for 48 hours with or without 1 μg/ml cisplatin. At the various times after treatment, the medium was removed and MTT (20 μl of a 5 mg/ml solution) was added to each well, and plates were incubated at 37°C for 4 hours. After that, the plates were spun in a centrifuge, and the purple-colored formazan precipitate in each well was dissolved in 150 μl of dimethyl sulfoxide. Absorbance was measured at 490 nm in an absorbance reader (MRX II; DYNEX Technologies, Chantilly, VA, USA). The reduction in viability of each group was expressed as a percentage of the mock or dsCon cells, which were considered to be 100% viable.
Real-time quantitative RT-PCR
Total RNA was extracted from cells using TRIzol (Invitrogen, Carlsbad, CA, USA) and reverse transcribed using oligo(dT) primers. For real-time quantitative RT-PCR, the resulting cDNA was amplified in a real-time PCR system (ABI Prism 7500, Applied Biosystems, CA, USA) using the DNA-binding dye Sybergreen I (Invitrogen, Carlsbad, CA, USA) for detection of PCR products. Values are expressed as fold-difference compared with Mock. Primer sequences for PAWR are 5'-GCCGCAGAGTGCTTAGATGAG-3' (forward) and 5'-GCAGATAGGAACTGCCTGGATC-3' (reverse) and, for GAPDH are 5'-AAGAAGGTGGTGAAGCAGGC-3' (forward) and 5'-TCCACCACCCTGTTGCTGTA-3' (reverse).
Western blotting analysis
Briefly, cells were harvested at 48 or 72 hours following dsRNAs treatment as described above, washed and lysed with lysis buffer. Protein concentration in the resulting lysate was determined using the bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL, USA). Appropriate amounts of protein (30-50 µg) were resolved by electrophoresis in 10-15% Tris-glycine polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked and then incubated overnight with the appropriate primary antibody at dilutions specified by the manufacturer. Primary immunoblotting antibodies were purchased from the Cell Signaling Technology, Beverly, MA, USA. The membranes were next washed three times in 15 ml TBST and incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody at 1:1000 dilution in TBST for 1 hour. After washing three times for 5 minutes each with 15 ml TBST, bound secondary antibody was detected using an enhanced chemiluminescence (ECL) system (Pierce Biotechnology Inc., Rockford, IL, USA).
To determine NF-κB cellular localization, nuclear and cytoplasmic proteins were isolated from the cells using a cell fractionation kit (KeyGen, Wuhan, China). NF-κB expression in the nuclear and cytoplasmic compartments was determined by immunoblot analysis as described above.
Detection of apoptotic cells by flow cytometry
A quantitative assessment of apoptosis was made by determining the percentage of cells with highly condensed or fragmented nuclei. Cells were plated in six-well plates and incubated overnight before treatment (Mock, 50 nmol/l dsCon and 50 nmol/l dsPAWR-435). Cells were harvested at 72 hours after dsRNAs treatment as described above, washed twice with pre-chilled PBS, and resuspended in 100 μl 1× binding buffer at a concentration of 1×106 cells/ml. Double staining with fluorescein isothiscyanate (FITC)-conjugated annexin V and propidium iodide (PI) was performed (Annexin V-FITC Apoptosis Detection Kit; BD Biosciences, San Jose, CA, USA) in accordance with the manufacturer's protocol. Cell apoptosis analysis was performed within 1 hour using flow cytometry (Beckman Coulter FC500 Flow Cytometry System with CXP Software; Beckman Coulter, Fullerton, CA, USA).
Cell cycle analysis by flow cytometry
Cell cycle analysis was performed using a commercial kit (Coulter DNA Prep™ Reagents Kit; Beckman Coulter, Fullerton, CA, USA). Cells were plated in six-well plates and incubated overnight before treatment (Mock, 50 nmol/l dsCon and 50 nmol/l dsPAWR-435). Following treatment, cells were harvested, then washed twice with pre-chilled PBS and resuspended in 100 μl PBS at a concentration of 1×106 cells/ml. Each cell sample was mixed with 100 μl DNA Prep LPR (contained in Coulter DNA Prep™ Reagents Kit), gently mixed by vortex and incubated in the dark at room temperature (25°C) for 20 minutes. Then each was mixed with 1 ml of stain (DNA Prep Stain; contained in Coulter DNA Prep™ Reagents Kit), gently mixed by vortex and again incubated in the dark at room temperature (25°C) for 20 minutes. Finally, cell cycle analysis was performed within 1 hour using flow cytometry (Beckman Coulter FC500 Flow Cytometry System with CXP Software; Beckman Coulter, Fullerton, CA, USA), and the raw data was analyzed by Multicycle for Windows (Beckman Coulter).
Statistical analysis
All values are expressed as means ± SD. Statistical significance was compared between treatment groups and controls using Student's t test. P < 0.05 was considered statistically significant.
Results
dsPAWR-435 induces PAWR gene activation in bladder cancer cells
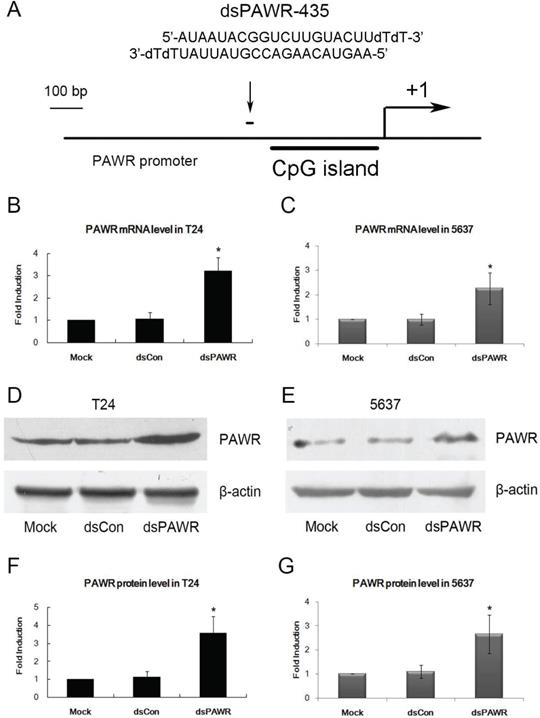
A dsRNA targeting the PAWR gene promoter at position -435 relative to the transcription start site (dsPAWR-435) was used to activate PAWR expression as described previously [17] (Fig. 1A). 50 nmol/l (nM) dsPAWR-435 and a nonspecific control dsRNA (dsCon) were transfected into T24 and 5637 human bladder cancer cells and PAWR expression levels were evaluated 48 h and 72 h later. Compared with the Mock and dsCon groups, dsPAWR-435 caused a >2-fold induction in PAWR mRNA levels in both T24 and 5637 cells, respectively (Fig. 1B and C). Induction of PAWR was also confirmed by Western blot analysis (Fig. 1D and E). Elevated levels of PAWR protein strongly correlated to the increase in PAWR mRNA expression in both cell lines (Fig. 1F and G).
dsPAWR-435 up-regulates PAWR gene expression in bladder cancer cell lines. *: P < 0.05 compared with the mock. (A) A schematic representation of the PAWR promoter with its CpG island, transcription start site, and dsRNA target. T24 (B) and 5637 (C) cells were transfected with 50 nM dsRNAs for 48 hours. mRNA expression of PAWR and GAPDH were detected by real time RT-PCR, and the results were normalized to GAPDH and presented as the mean ± SD of three independent experiments. T24 (D) and 5637 (E) cells were transfected with 50 nM dsRNAs for 72 hours. Induction of PAWR protein expression was detected by Western blot analysis. β-actin levels were also detected and served as a loading control. A representative blot is shown from three independent experiments with identical results. (F, G) Relative protein level was determined by quantifying Western blot membrane band intensity. The PAWR protein expression levels were normalized to β-actin and the results are presented as the mean ± SD of three independent experiments.
dsPAWR-435 inhibits bladder cancer cell growth and viability
Ectopic PAWR over-expression has been shown to induce growth inhibition in most cancer cells in vitro [29]. Here we investigated whether the up-regulation of PAWR by saRNA has similar effects on bladder cancer cells. T24 and 5637 bladder cancer cells were transfected by 50 nM dsPAWR-435 and dsCon for 48 or 72 h and dsPAWR-435 transfected cells gradually displayed growth inhibition and cell shrinkage (Fig. 2A & 2B). Moreover, more floated dead cells and evidently decreased cell density were observed in dsPAWR-435 treated group (Fig. 2A & 2B).
dsPAWR-435 induces growth inhibition of T24 (A) and 5637 (B) bladder cancer cells. Cells were transfected with 50 nM dsRNAs or Mock. Cell images were taken at 24, 48 and 72h after transfection at 100× magnification. dsPAWR-435 transfected cells are less dense and have more dead cells than controls. dsPAWR-435 inhibited the viability of T24 (C) and 5637 (D) cells in a dose-dependent and time-dependent manner, as assessed by the MTT assay. Reduced cell viability was seen after dsPAWR-435 treatment (1 to 50 nmol/l) at 24, 48, and 72 hours. Data are presented as means ± SD (n = 8).
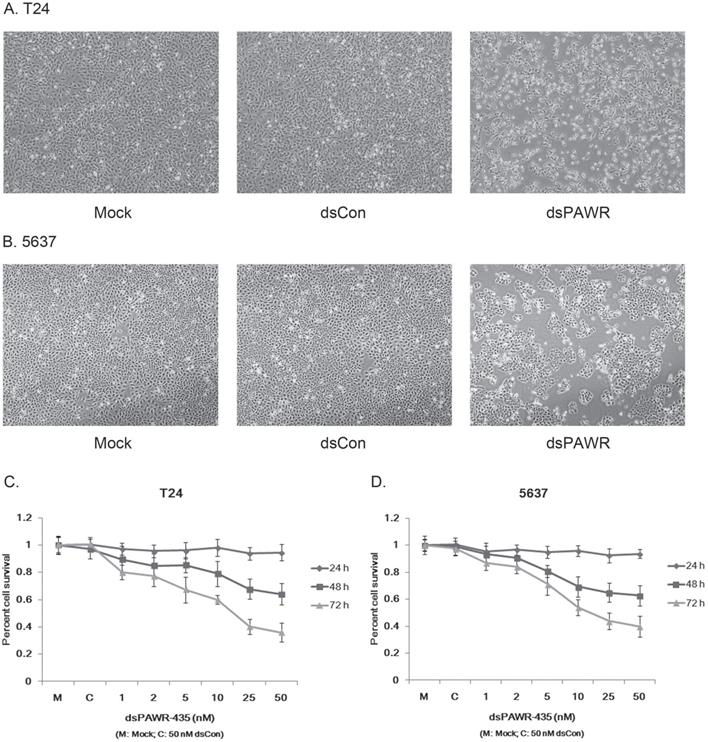
The effects of dsPAWR-435 on proliferation and viability of human bladder cancer cells were determined with varying concentrations and times (24 - 72 h) by MTT assay. As shown in Fig. 3, the effects of dsPAWR-435 on cell viability, which were dose and time dependent, occurred within 48 h and at dsRNA concentrations as low as 5 nM. Compared with dsCon transfections, reduction of viability in T24 cells with dsRNA treatment at concentrations of 1 - 50 nM after 48 h ranged from 10.5% to 36.0%, whereas after 72 h ranged from 19.9% to 64.1% (Fig. 2C). Similarly, reduction of viability in 5637 cells treated by dsPAWR-435 after 48 and 72 h ranged from 6.6% to 37.4% and 13.0% to 60.1%, respectively (Fig. 2D).
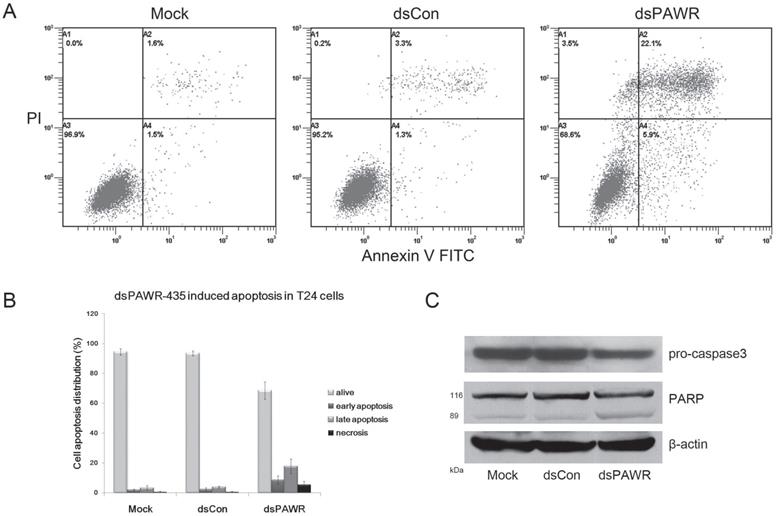
dsPAWR-435 induces cell apoptosis in T24 bladder cancer cells. Cells were transfected with 50 nM dsRNAs for 72 hours. Representative images and blots from three independent experiments with identical results are shown. (A) dsPAWR-435 resulted in apoptosis in T24 cells detected by flow cytometry using a double-staining method with fluorescein thiocyanate-conjugated annexin V and propidium iodide. Annexin V-stained cells indicates the early apoptotic cells, whereas Annexin V + propidium iodide-stained cells are the late apoptotic cells. (B) Flow cytometry data was analyzed to compare cell apoptosis populations (means ± SD from three independent experiments). Percentages of alive, apoptotic and necrotic cells are shown respectively. (C) dsPAWR-435 treatment activated caspase-3 and poly (ADP-ribose) polymerase (PARP) in T24 cells.
dsPAWR-435 induces cell apoptosis in bladder cancer cells
The antitumor ability by ectopic PAWR overexpression is related to its essential role in inducing apoptosis via diverse cell death pathways [26-28]. Thus, the relationship between dsPAWR-435-mediated loss of cell viability and cell apoptosis was investigated by flow cytometric analysis labeled with PI and Annexin V and we found that dsPAWR-435 caused evident apoptosis in the T24 cells at 72 h following treatment. As shown in Fig. 3, the ratio of late apoptotic cells (UR quadrant) increased to nearly 20% and the early apoptotic cells (LR quadrant) also increased evidently compared with controls. These data also showed that dsPAWR-435 treatment resulted in not only apoptosis but also tiny cell necrosis, which may be a secondary event in the apoptotic process.
Caspase-3 and poly(ADP-ribose) polymerase (PARP) play central roles in apoptosis. Accordingly, the level of pro-caspase-3 was observed to decrease distinctly in the 50 nM dsPAWR‑435-treated T24 cells at 72 h following treatment (Fig. 3C). Moreover, the 89 kDa cleaved PARP fragment was detected in the dsPAWR-435 treated samples. Thus, the significant changes in apoptosis-related proteins affected by dsPAWR-435 treatment confirmed the ongoing apoptosis above and the anti-tumor effects on the T24 human bladder cancer cells.
dsPAWR-435 induces cell cycle arrest in bladder cancer cells
We next investigated the relationship between dsPAWR-435 mediated inhibition of cell proliferation and cell cycle arrest by flow-cytometric analysis. As shown in Fig. 4, G1-phase arrest in 50 nM dsPAWR-435 treated cells was observed. The G1-phase population of dsPAWR-435 treated cells was about 67% at 72 h following treatment and increased more than 20% at 72 h compared to controls. The increase in cell population in the G1-phase was found to be associated with a concomitant significant decrease in the S-phase population, and less decrease in the G2 phase population was also found.
dsPAWR-435 induces G1-phase cell cycle arrest in T24 bladder cancer cells. Cells were transfected with 50 nM dsRNAs for 72 hours. Representative images and blots from three independent experiments with identical results are shown. (A) dsPAWR-435 resulted in G1-phase cell cycle arrest in T24 cells detected by flow cytometry. The sub-G0/G1 cells were not included in the calculations. (B) Flow cytometry data was analyzed to compare cell cycle distribution (means ± SD from three independent experiments). Percentages of G1, G2 and S phase cells are shown respectively. (C) The effects of dsPAWR-435 on cell cycle related proteins in T24 cells.
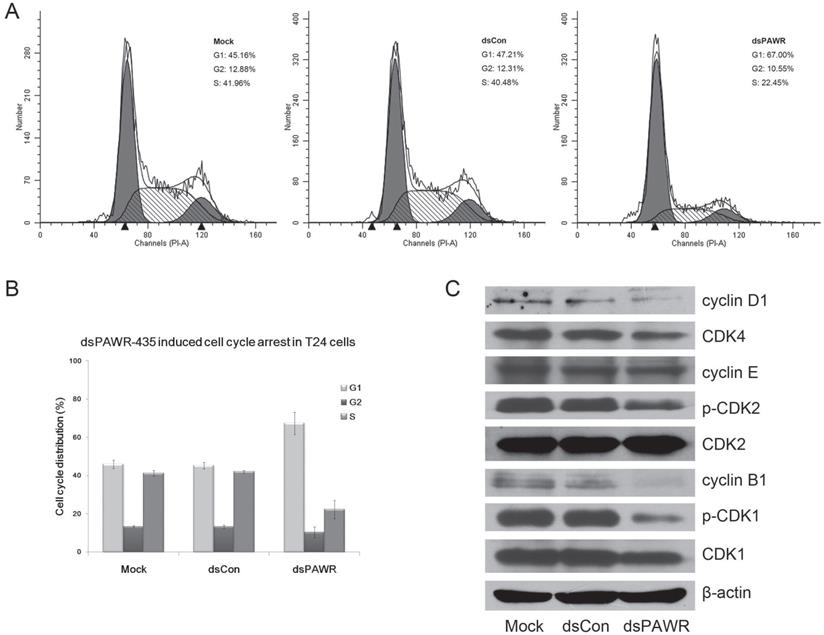
Then, we examined the expression of several cell cycle-related proteins. Accordingly, interference with the cell cycle in dsPAWR-435 treated T24 cells was associated with decreased expression of cyclin D1 and CDK4. Unexpectedly, we also found decreased expression of cyclin B1 and reduced phosphorylation of CDK2 and CDK1 in dsPAWR-435 treated cells, but no alteration in the expression of cyclin E (Fig. 4C).
The molecular mechanism related to dsPAWR-435 induced cell cycle arrest and apoptosis
Previous research has shown that PAWR is an important intersection in the network of tumor suppressors that involves the NF-κB and Akt pathways [30], which are both deregulated during tumorigenesis. Thus, we detected these proteins and found that nuclear translocation of NF-κB and phosphorylation of Akt were inhibited in the dsPAWR-435 treated cells compared with the controls (Fig. 5A), which implied inactivation of these signaling pathways.
The molecular mechanism related to dsPAWR-435 induced antitumor activity. T24 bladder cancer cells were transfected with 50 nM dsRNAs for 72 hours. Representative blots from three independent experiments with identical results are shown. (A) The nuclear translocation of NF-κB and the phosphorylation of Akt were inhibitied by dsPAWR-435 treatment. (B) The expression of pro-caspase-9 and pro-caspase-8 remarkably decreased in dsPAWR-435 treated T24 cells. (C) Bcl-2 protein was reduced and Bax protein was not affected after dsPAWR-435 treatment in T24 cells.
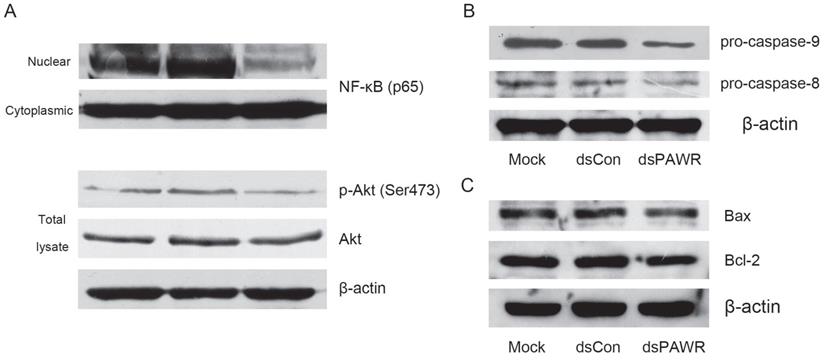
To examine which pathway plays a role in the dsPAWR-435 induced cell apoptosis, the cleavage of caspase-8 and -9 was examined. As shown in Fig. 5B, the levels of pro-caspase-8 and -9 were markedly decreased in the 50 nM dsPAWR-435 treated T24 cells at 72 h following treatment, indicating that both extrinsic and intrinsic pathways were active in the dsPAWR-435 treated cells.
PAWR-mediated apoptosis requires downregulation of Bcl-2 levels and PAWR regulates Bcl-2 gene expression through a WT1-binding site in its promoter leading to a decrease in transcription [31, 32]. Consistently, the expression of Bcl-2 was found to decrease in the dsPAWR-435 treated cells compared with the controls (Fig. 5C). However, the level of Bax, the pro-apoptotic member of the Bcl-2 family, was not altered after the treatment of dsPAWR-435.
dsPAWR-435 induced PAWR activation cooperates with cisplatin in inhibition of bladder cancer cells
Cisplatin is the main chemotherapy drug for advanced bladder cancer. Thus, we used MTT assay to determine the combined effects of dsPAWR-435 and cisplatin on T24 bladder cancer cells. After transfection with mock, dsCon, or dsPAWR-435 for 24 hours, cells were sub-divided into two groups, and treated or not with 1 μg/ml cisplatin for another 24 hours. The average reduction in cell viability with dsPAWR-435 + cispaltin treatment was 29.0% (Fig. 6A), which was much greater than the reduction obtained with the single treatment of dsPAWR-435 (48.9%) or cisplatin (75.4%).
(A) dsPAWR-435 cooperates with cisplatin in inhibiting the viability and growth of T24 bladder cancer cells detected by MTT assay. These data are presented as means ± SD (n = 8). * P < 0.05 versus single treatment with dsCon or Mock. ** P < 0.05 versus single treatment with dsPAWR-435 + cisplatin. (B) dsPAWR-435 cooperates with cisplatin in inducing apoptosis in T24 cells detected by flow cytometry. The percentage of G1, S or G2 phase cells = its number/G1+S+G2 number. The percentage of sub-G0/G1 cells = its number/the number of all cells counted. A representative image is shown from three independent experiments with identical results.
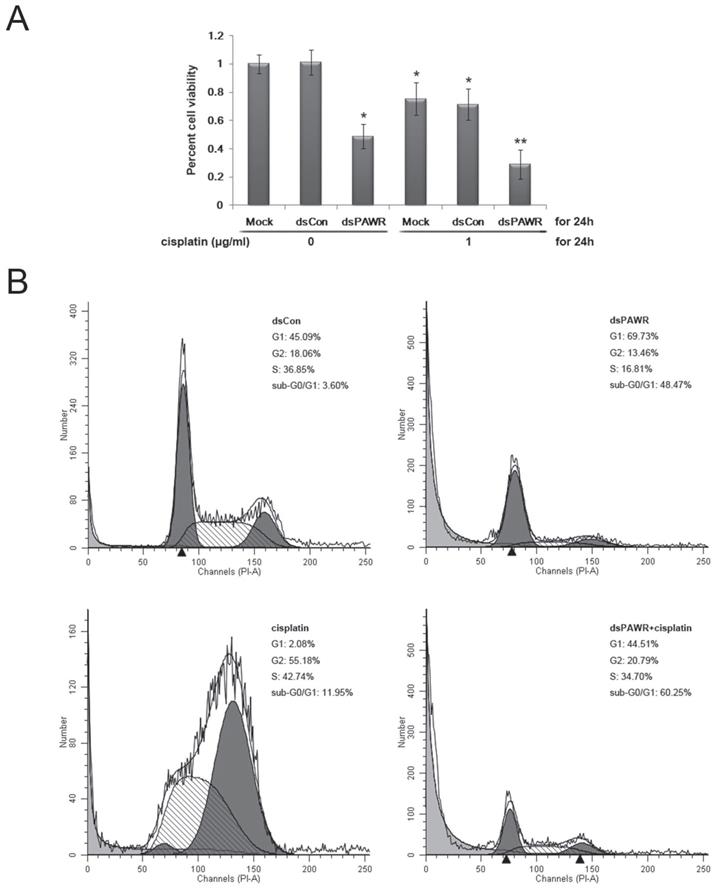
Both the live cells and the supernatant were then harvested to assess the effects of the treatment above by flow cytometry. Compared with the dsCon-treated group, G1-phase arrest was seen in the dsPAWR-435-treated group and G2-phase arrest was seen in the cisplatin-treated group (Figure 6B). Moreover, there was a significant increase in the sub-G0/G1 population in these two groups (48.47% and 11.95%, respectively), indicating apoptotic cells. When dsPAWR-435-transfected cells were subsequently treated with cisplatin for another 24 hours, the proportion of sub-G0/G1 cells increased to 60.25%, suggesting that the PAWR-targeting dsRNA can co-operate with cisplatin in the inhibition of bladder cancer cells.
Discussion
RNA activation (RNAa) is an interesting and promising discovery of small RNAs mediated gene up-regulation originally identified in several human cancer cell lines [8, 9]. It will offer an alternative to manipulate gene expression potently and specifically if this phenomenon exists in most genes as RNAi and its rules could be deciphered. RNAa thus holds great promise as therapeutics for reactivation of functionally silenced or low expressed TSGs in cancer patients. Our group and others have obtained exciting results that up-regulation of p21 by saRNA could induce cell cycle arrest and apoptosis in human bladder cancer cells [18, 19] and renal cell carcinoma cells [20] in vitro and inhibit the growth of xenograft prostate tumor [22] and orthotopic bladder tumor [23] in vivo.
Functional PAWR is a TSG essential for apoptosis and a cancer-selective target for cancer therapeutics [33]. Depending on the nature of stimulus, apoptosis can occur via two different pathways, extrinsic and intrinsic. Although the two pathways are activated by different mechanisms, they are interlinked by common signaling factors like NF-κB and proteolytic enzymes like caspases [34, 35]. Apoptosis by PAWR involves both extrinsic and intrinsic mechanisms. Specifically, PAWR can induce apoptosis by instigating the formation of death inducing signaling complex (DISC) through Fas receptor/Fas Ligand interaction with FADD and causing activation of the Fas/FasL-FADD-Caspase 8 apoptotic death pathway [36]. In parallel, the other crucial mechanisms central to apoptosis induced by PAWR is the inhibition of NF-κB transcription function [37]. NF-κB can inhibit TNF-α induced apoptosis by blocking protein kinase C (PKC) and can subsequently stall nuclear translocation of the p65 (Rel A) subunit. NF-κB regulates several pro-survival genes such as B cell lymphoma-2 (BCl-2), BCl-XL and Al/Bfl1 as well as anti-apoptotic genes such as an X-linked inhibitor of apoptosis (XIAP) [38]. PAWR translocates into the nucleus and inhibits NF-κB-mediated cell survival mechanisms. PAWR-mediated apoptosis requires downregulation of Bcl-2 levels and PAWR regulates Bcl-2 gene expression through a WT1-binding site in its promoter leading to a decrease in transcription [31, 32]. Moreover, activation of the Akt pathway is a frequent molecular event in human cancer and PAWR can also inhibit Akt and suppress Ras-induced tumorigenesis [39]. Therefore, these interacting factors place PAWR as an important part in the regulation of cell survival, implicating its potential as a candidate for RNAa.
Our attempts with dsRNAs targeting the PAWR promoter have successfully induced transcriptional activation of the PAWR gene in human cancer cells [17]. Moreover, dsPAWR-433 actually induced growth inhibition and apoptosis of prostate cancer cells, suggesting that the increased PAWR protein by saRNA is physiologically functional and has great potential for the application in cancer therapy [40]. In the present study, we demonstrated that another PAWR promoter targeted dsRNA, dsPAWR-435, could potently induce activation of PAWR gene expression in bladder cancer cells. Promisingly, MTT assay and flow cytometric analysis showed that dsPAWR-435 inhibited cell viability in a dose- and time-dependent manner and it was related to apoptotic cell death. Accordingly, activation of PAWR gene expression by dsPAWR-435 not only activated the caspase-8-dependent extracellular apoptotic pathway but also induced the caspase-9-dependent intracellular apoptosis by inhibition of Akt and NF-κB pathways and downregulation of Bcl-2 protein. Then, the activation of caspase-3 plays a central role in apoptosis by cleaving intracellular proteins vital for cell survival and growth, such as PARP [41, 42], leading to the completion of apoptosis in the dsPAWR-435-treated bladder cancer cells. Moreover, flow cytometric analysis also showed the manifestation of G1 phase arrest in dsPAWR-435-treated cells. Our analysis of cell cycle-related proteins showed that in the dsPAWR-435-treated cells, there were significantly decreased expression of both cyclin D1 and cyclin B1 and reduced phosphorylation of both CDK2 and CDK1. Although cyclin D1/CDK4 and cyclin B1/CDK1 were both inhibited, the dsPAWR-435-treated cells exhibit G1 phase cell cycle arrest.
Over the past two decades, novel therapeutic schemes containing different drug cocktails have been developed, with cisplatin occupying a central position in these regimen (for example the GC (gemcitabine + cisplatin) regimen) [43]. It is generally accepted that DNA is the preferential and cytotoxic target for cisplatin [44-46]. Cisplatin-mediated damage of genomic DNA causes severe cell cycle perturbation and arrest at certain checkpoints, and in the absence of adequate repair, the affected cells undergo cell apoptosis. In the current study, we found that upregulation of PAWR by saRNA could co-operate with cisplatin and enhance its anticancer effects additively via increased cell apoptosis in T24 bladder cancer cells.
Although dsRNA can successfully induce PAWR expression in bladder cancer cells in vitro, we have not been able to obtain good results in animal experiments yet, which might be related to the inefficient delivery and instability of dsRNA in vivo. We have tried chemical modification of dsRNA and plan to use more efficient transporter to improve in vivo experiments referring to previous literatures [22, 23].
Conclusions
In conclusion, up-regulation of PAWR by the small double strand RNA dsPAWR-435 in the bladder cancer cells could not only inhibit cell growth by inducing cell apoptosis and G1-phase arrest, but also cooperate with cisplatin against the growth of bladder cancer cells. However, it appears difficult to figure out the definite mechanisms of RNAa due to only a few genes activated and the diversity of the results from different genes. On the other hand, we still have to screen multiple targets in order to activate a particular gene. Regardless, RNAa offers a new approach to enhance endogenous gene expression and holds great promise as a therapeutic for reactivation of functionally silenced or lowly expressed TSGs in cancer patients. Despite the promise, further studies are needed to delineate the exact mechanism of RNAa and develop safe and effective in vivo saRNA delivery methods for clinical use.
Abbreviations
RNAa: RNA activation; saRNA: small activating RNA; RNAi: RNA interference; dsRNA: double-stranded RNA; siRNA: small interfering RNA; PAWR: PRKC apoptosis WT1 regulator; TSG: tumor suppressing gene; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; nM: nmol/l.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (Grant No. 81101718) and the Zhejiang Provincial Natural Science Foundation of China (Grant No. LY13H160009).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
2. Grossman HB, Natale RB, Tangen CM. et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med. 2003;349:859-66
3. Splinter TA, Scher HI, Denis L. et al. European Organization for Research on Treatment of Cancer-Genitourinary Group: The prognostic value of the pathological response to combination chemotherapy before cystectomy in patients with invasive bladder cancer. J Urol. 1992;147:606-8
4. Witjes JA, Bruins HM, Cathomas R. et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur Urol. 2020;79(1):82-104
5. Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426-33
6. Davidson BL, McCray PB Jr. Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12(5):329-40
7. Petrocca F, Lieberman J. Promise and challenge of RNA interference-based therapy for cancer. J Clin Oncol. 2011;29(6):747-54
8. Li LC, Okino ST, Zhao H. et al. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci USA. 2006;103:17337-42
9. Janowski BA, Younger ST, Hardy DB. et al. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007;3:166-73
10. Place RF, Li LC, Pookot D. et al. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105:1608-13
11. Turunen MP, Lehtola T, Heinonen SE. et al. Efficient regulation of VEGF expression by promoter-targeted lentiviral shRNAs based on epigenetic mechanism: a novel example of epigenetherapy. Circ Res. 2009;105:604-9
12. Huang V, Qin Y, Wang J. et al. RNAa is conserved in mammalian cell. PLoS ONE. 2010;5:e8848
13. Wang J, Place RF, Huang V. et al. Prognostic value and function of KLF4 in prostate cancer: RNAa and vectormediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer Res. 2010;70:10182-91
14. Matsui M, Sakurai F, Elbashir S. et al. Activation of LDL receptor expression by small RNAs complementary to a noncoding transcript that overlaps the LDLR promoter. Chem Biol. 2010;17:1344-55
15. Wang X, Wang J, Huang V. et al. Induction of NANOG expression by targeting promoter sequence with small activating RNA antagonizes retinoic acid-induced differentiation. Biochem J. 2012;443(3):821-8
16. Wang T, Li M, Yuan H. et al. saRNA guided iNOS up-regulation improves erectile function of diabetic rats. J Urol. 2013;190(2):790-8
17. Yang K, Shen J, Xie YQ. et al. Promoter-targeted double-stranded small RNAs activate PAWR gene expression in human cancer cells. Int J Biochem Cell Biol. 2013;45:1338-46
18. Chen Z, Place RF, Jia ZJ. et al. Antitumor effect of dsRNA-induced p21(WAF1/CIP1) gene activation in human bladder cancer cells. Mol Cancer Ther. 2008;7:698-703
19. Yang K, Zheng XY, Qin J. et al. Up-regulation of p21WAF1/Cip1 by saRNA induces G1-phase arrest and apoptosis in T24 human bladder cancer cells. Cancer Lett. 2008;265:206-14
20. Whitson JM, Noonan EJ, Pookot D. et al. Double stranded-RNA-mediated activation of P21 gene induced apoptosis and cell cycle arrest in renal cell carcinoma. Int J Cancer. 2009;125:446-52
21. Wei J, Zhao J, Long M. et al. p21WAF1/CIP1 gene transcriptional activation exerts cell growth inhibition and enhances chemosensitivity to cisplatin in lung carcinoma cell. BMC Cancer. 2010;10:632
22. Place RF, Wang J, Noonan EJ. et al. Formulation of small activating RNA into lipidoid nanoparticles inhibits xenograft prostate tumor growth by inducing p21 expression. Molecular Therapy-Nucleic Acids. 2012;1:e15
23. Kang MR, Yang G, Place RF. et al. Intravesical delivery of small activating RNA formulated into lipid nanoparticles inhibits orthotopic bladder tumor growth. Cancer Res. 2012;72:5069-79
24. Johnstone RW, Tommerup N, Hansen C. et al. Mapping of the human PAWR (Par-4) gene to chromosome 12q21. Genomics. 1998;53:241-3
25. Sells SF, Wood Jr DP, Joshi-Barve SS. et al. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth and Differentiation. 1994;5:457-66
26. Chakraborty M, Qiu SG, Vasudevan KM. et al. Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer Research. 2001;61:7255-63
27. Gurumurthy S, Goswami A, Vasudevan KM. et al. Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Molecular and Cellular Biology. 2005;25:1146-61
28. Goswami A, Burikhanov R, de Thonel A. et al. Binding and phosphorylation of Par-4 by Akt is essential for cancer cell survival. Molecular Cell. 2005;20:33-44
29. El-Guendy N, Zhao Y, Gurumurthy S. et al. Identification of a unique core domain of Par-4 sufficient for selective apoptosis-induction in cancer cells. Molecular and Cellular Biology. 2003;23:5516-25
30. Diaz-Meco MT, Abu-Baker S. The Par-4/PTEN connection in tumor suppression. Cell Cycle. 2009;8:2518-22
31. Qiu G, Ahmed M, Sells SF. et al. Mutually exclusive expression patterns of Bcl-2 and Par-4 in human prostate tumors consistent with downregulation of Bcl-2 by Par-4. Oncogene. 1999;18:623-31
32. Cheema SK, Mishra SK, Rangnekar VM. et al. Par-4 transcriptionally regulates Bcl-2 through a WT1-binding site on the bcl-2 promoter. J Biol Chem. 2003;278:19995-20005
33. Goswami A, Ranganathan P, Rangnekar VM. The phosphoinositide 3-kinase/Akt1/Par-4 axis: A cancer-selective therapeutic target. Cancer Res. 2006;66:2889-92
34. Burz C, Berindan-Neagoe I, Balacescu O. et al. Apoptosis in cancer: key molecular signaling pathways and therapy targets. Acta Oncol. 2009;48(6):811-21
35. Sayers TJ. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother. 2011;60(8):1173-80
36. Burikhanov R, Zhao Y, Goswami A. et al. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138(2):377-88
37. Diaz-Meco MT, Lallena MJ, Monjas A. et al. Inactivation of the inhibitory kappaB protein kinase/nuclear factor kappaB pathway by Par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. J Biol Chem. 1999;274:19606-12
38. Chendil D, Das A, Dey S. et al. Par-4, a pro-apoptotic gene, inhibits radiation-induced NFκB activity and Bcl-2 expression leading to induction of radiosensitivity in human prostate cancer cells PC-3. Cancer Biol Ther. 2002;1(2):152-60
39. Joshi J, Fernandez-Marcos PJ, Galvez A. et al. Par-4 inhibits Akt and suppresses Ras-induced lung tumorigenesis. EMBO J. 2008;27(16):2181-93
40. Yang K, Shen J, Chen SW. et al. Upregulation of PAWR by small activating RNAs induces cell apoptosis in human prostate cancer cells. Oncol Rep. 2016;35(4):2487-93
41. Salvesen GS, Dixit VM. Caspase activation: The inducedproximity model. Proc Natl Acad Sci USA. 1999;96:10964-67
42. Ivana Scovassi A, Diederich M. Modulation of poly(ADP-ribosylation) in apoptotic cells. Biochem Pharmacol. 2004;68:1041-7
43. Perabo FG, Muller SC. New agents for treatment of advanced transitional cell carcinoma. Ann Oncol. 2007;18:835-43
44. Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9-23
45. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573-84
46. Ho YP, Au-Yeung SC, To KK. Platinum-based anticancer agents: innovative design strategies and biological perspectives. Med Res Rev. 2003;23:633-55
Author contact
![]() Corresponding author: Department of Urology, the First Affiliated Hospital, Zhejiang University School of Medicine, Qingchun Road 79, Hangzhou, Zhejiang 310003, P.R. China. Tel: +86-571-8723-6133; E-mail addresses: kaiyangedu.cn (Kai Yang).
Corresponding author: Department of Urology, the First Affiliated Hospital, Zhejiang University School of Medicine, Qingchun Road 79, Hangzhou, Zhejiang 310003, P.R. China. Tel: +86-571-8723-6133; E-mail addresses: kaiyangedu.cn (Kai Yang).