Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(9):1999-2007. doi:10.7150/ijms.46474 This issue Cite
Research Paper
miR-143 is implicated in growth plate injury by targeting IHH in precartilaginous stem cells
Fu-Yong Zhang1#, Yun-Fang Zhen1#, Zhi-Xiong Guo1, Jin Dai1, Lun-Qing Zhu1, Pei-Rong Liang1, Guang-Hao Su1, Wen-Yan Zhang1, Jian-Feng Fang1, Quan-Wen Yuan1, Feng Yao1, Ya Liu1, Yi Qiao1, Ya Zhang1, Wan-Liang Guo2, Yao Liu1 ![]() , Xiao-Dong Wang1
, Xiao-Dong Wang1 ![]()
1. Department of Orthopaedics, Children's Hospital of Soochow University, Suzhou, 215000, China.
2. Department of Radiology, Children's Hospital of Soochow University, Suzhou, 215000, China.
#Co-first authors with equal contributions to this work.
Received 2020-3-30; Accepted 2021-2-2; Published 2021-3-3
Abstract

Precartilaginous stem cells (PCSCs) are able to initiate chondrocyte and bone development. The present study aimed to investigate the role of miR-143 and the underlying mechanisms involved in PCSC proliferation. In a rat growth plate injury model, tissue from the injury site was collected and the expression of miR-143 and its potential targets was determined. PCSCs were isolated from the rabbits' distal epiphyseal growth plate. Cell viability, DNA synthesis, and apoptosis were determined with MTT, BrdU, and flow cytometric analysis, respectively. Real time PCR and western blot were performed to detect the mRNA and protein expression of the indicated genes. Indian hedgehog (IHH) was identified as a target gene for miR-143 with luciferase reporter assay. Decreased expression of miR-143 and increased expression of IHH gene were observed in the growth plate after injury. miR-143 mimics decreased cell viability and DNA synthesis and promoted apoptosis of PCSCs. Conversely, siRNA-mediated inhibition of miR-143 led to increased growth and suppressed apoptosis of PCSCs. Transfection of miR-143 decreased luciferase activity of wild-type IHH but had no effect when the 3'-UTR of IHH was mutated. Furthermore, the effect of miR-143 overexpression was neutralized by overexpression of IHH. Our study showed that miR-143 is involved in growth plate behavior and regulates PCSC growth by targeting IHH, suggesting that miR-143 may serve as a novel target for PCSC-related diseases.
Keywords: miR-143, precartilaginous stem cells, IHH
Introduction
Many clinical conditions require regeneration or implantation of bone. Current therapeutic options are not sufficient to treat massive bone defects. Stem cell-based therapies have been used as an alternative to current solutions for bone-loss repair [1]. Precartilaginous stem cells (PCSCs) are a type of newly identified adult stem cells that exist in the most peripheral layer of the epiphyseal organ with a perichondrial mesenchyme in embryonic limbs. Studies have shown that PCSCs play diverse roles in endochondral ossification, limb growth, cartilage development, and repair of damaged articular cartilage [2]. However, the underlying mechanisms involved in the acquisition of PCSC properties have not been elucidated.
Hedgehog (Hh) signaling is critical for the regulation of bone development, homeostasis, and repair. Indian hedgehog (Ihh), a member of Hh proteins, is expressed in the prehypertrophic and hypertrophic chondrocytes in the growth plate. Activation of Ihh signaling synchronizes chondrogenesis and osteogenesis during endochondral ossification by promoting chondrocyte differentiation and proliferation, and specification of bone-forming osteoblasts [3,4]. Overexpression of Ihh enhances chondrogenesis and osteogenesis from mesenchymal stem cells (MSCs) [5,6]. In addition, Ihh-regulated chondrogenesis is associated with different pathways, such as Wnt/β-Catenin, parathyroid hormone-related peptide (PTHrP), and TGF-β/Smad pathways [7-10]. These findings suggest that targeting Ihh may be a novel therapeutic approach for bone-related diseases.
MicroRNAs (miRNAs) are a group of small, noncoding RNAs. By post-transcriptional regulation of their target mRNAs through complementary binding to the 3' untranslated region (UTR), miRNAs play diverse roles in cellular behaviors, including cell proliferation, apoptosis, differentiation, and angiogenesis [7]. Accumulating evidence indicates that miRNA expression is critical for cartilage differentiation and skeletal development. For example, miR-26a and miR-125b have been discovered to suppress osteoblastic differentiation, while miR-210 and miR-29b activate osteoblastic differentiation [12-14]. miR-143, a miRNA located in chromosome 5q33, was reported to suppress osteogenic differentiation [15]. Recently, Tian et al have found that overexpression of miR-143 suppressed cartilage differentiation of bone marrow mesenchymal stem cells (BMSCs) via targeting bone morphogenetic protein 2 (BMPR2) [16]. Consistently, another study confirmed that miR-143 inhibits osteogenic differentiation of dental pulp stem cells (DPSCs) by targeting tumor necrosis factor (TNF)-α [17]. However, the effect of miR-143 on PCSC physiology and the underlying mechanisms involved have not been reported yet. In the present study, we explored whether miR-143 regulates PCSC growth in an IHH-dependent manner, aiming to provide a novel therapeutic target for bone-related diseases.
Materials and Methods
Animals and growth plate injury model
All animal experiments were performed according to protocols approved by the Animal Care and Use Committee of Children's Hospital of Soochow University. Healthy 3-week-old New Zealand white rabbits were purchased from Shanghai Laboratory Animals Center (Shanghai China). Animals were housed under a 12-h light/dark cycle with unlimited access to food and water. Twelve rabbits underwent experimental drill-hole growth plate injury in the proximal tibia of both hind legs. Following anesthesia, the growth plate was made accessible through a cortical window at the center of growth plate. Three days later, animals in control and injury model groups (n=6 in each group) were euthanized and the epiphyseal plate from the proximal tibiae was harvested for later experiments. All animal experimentation was conducted in accordance with accepted standards of humane animal care in accordance with the NIH guidelines.
Cell culture
Rabbits were euthanized with a lethal dose of sodium phenobarbital. The epiphyseal plate from the proximal tibiae was harvested, washed three times with ice-cold PBS, cut into thin sections, and digested with trypsin and collagenase II. The cell suspension was filtered through a 200-mesh stainless steel filter to remove any undigested particles. Finally, cells were maintained in DMEM medium supplemented with 10% FBS, 100 U/mL ampicillin, and 100 U/mL streptomycin in a 37 ºC incubator with 5% CO2. Cells were passaged at 1:5 dilution at 30-60% confluency using trypsin. Cells from generation 2-4 were used for further experiments.
Immunofluorescence
Purified PCSCs at a density of 5×105 cells/well were plated in six-well-plates, fixed with 4% paraformaldehyde, and permeabilized with 0.2% Triton X-100. Cells were then incubated with anti-FGFR-3 (1:1000 dilution, Santa Cruz) at 4 ºC overnight. Cells were then incubated with a FITC-labeled secondary antibody (Cellular Signaling Tech, Shanghai, China) for 1 h at room temperature. Cells were then observed and images recorded under a fluorescence microscope (Olympus, Tokyo, Japan).
Cell growth assay
Cell viability was measured with MTT assay (MD Millipore, Billerica, MA, USA) at different time points (24, 48, and 72 h after transfection). Briefly, cells were cultured overnight at 3×104 cells/well in 96-well culture plates. Cells were incubated with 5 mg/mL MTT at 37 °C for 2 h. The absorbance for each well was measured at 490 nm using a scanning multi-well spectrometer (Bio-Tek instruments, Inc., Burlington, VT, USA).
BrdU incorporation assay
Cell proliferation was estimated using the BrdU Labeling and Detection Kit III (Roche Molecular Biochemicals, Indianapolis, IN, USA) following the manufacturer's instruction. Briefly, cells were plated in 96-well plates at a density of 4000 cells/well. Twenty-four hours later, cells were incubated with BrdU at 37 °C for 4 h and washed three times with PBS, followed by incubation with anti-BrdU conjugated with peroxidase at 37 °C for 30 min. The absorbance for each well was measured at 450 nm using a scanning multi-well spectrometer (Bio-Tek instruments, Inc., Burlington, VT, USA).
miRNAs transfection
miRNA transfection was performed using Lipofectamine 2000 reagents (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Briefly, 2×105 cells/well were plated in a 6-well plate overnight. After adding new medium, cells were transfected with miRNA mimics or inhibitors followed by downstream assays.
Apoptosis assay
Annexin V‑FITC/PI staining was performed to quantify the percentage of apoptotic cells. Cells were stained using the Annexin V‑FITC Apoptosis Detection kit (Invitrogen, Inc., Carlsbad, CA, USA) according to the manufacturer's protocol. After being washed with PBS, cells were resuspended in binding buffer, incubated with Annexin V‑FITC and PI, and analyzed on a BD Accuri C6 Flow Cytometer (BD Biosciences, USA).
RNA isolation and real-time PCR
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. For detection of mRNA expression, cDNA synthesis was performed from 1 µg total RNA using a SuperScript First-Standard Synthesis kit (Invitrogen Life Technologies). Real-time PCR was performed with an ABI PRISM 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The following primers were used: IHH: forward: 5'- CAAGAAGCCCGGGATCTACA-3, reverse: 5'- GCTCGGGACTTTGTTGCTTG-3' and β-actin: forward 5'- GGCTGTGCTATCCCTGTACG -3', reverse 5'- AGGTAGTCAGTCAGGTCCCG -3'. Reactions were incubated in a 96-well optical plate at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s, 60 °C for 45 s, and 72 °C for 1 min. Relative expression was calculated using the 2-ΔΔCT method.
Plasmid DNA transfection
Transfection was performed using the Lipofectamine 2000 Transfection reagent (Invitrogen) according to the manufacturer's protocol. The eukaryotic expression vector pcDNA3.1(+) (Invitrogen) was used to generate the IHH expression plasmid. The IHH genomic sequence was amplified with PCR, digested with EcoRI and BamHI (Promega, Madison, WI, USA), and subcloned into the pcDNA3.1(+) vector.
Luciferase assay
Luciferase reporter assay was performed after transient co-transfection of the psiCHECK2-report vector (Ambion, Austin, TX, USA) containing the wild type or mutant 3'UTR of the human IHH gene, which includes the putative miR-143 binding site, into HEK293T cells in 24-well plates using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. For luciferase reporter assay, 48 hours after transfection, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). Luciferase activity was monitored with SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA). Experiments were performed in triplicates and repeated three times.
Western blot
Protein samples were separated with SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. Membranes were incubated with rabbit monoclonal antibodies against IHH (dilution 1:1000, Abcam, Cambridge, UK) and β-actin (dilution 1:1000, Abcam, Cambridge, UK) overnight. Membranes were then incubated with HRP-conjugated anti-rabbit secondary antibodies (Abcam, Cambridge, UK) at room temperature for 1 h. Proteins were detected using a Super Signal Enhanced Chemiluminescence kit (Pierce, Rockford, IL, USA).
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Differences between two groups were compared using the Student's t-test, and mean values of multiple groups were compared using one-way ANOVA and Bonferroni post hoc test. P<0.05 was considered statistically significant.
Results
miR-143 is involved in the growth and DNA synthesis of PCSCs
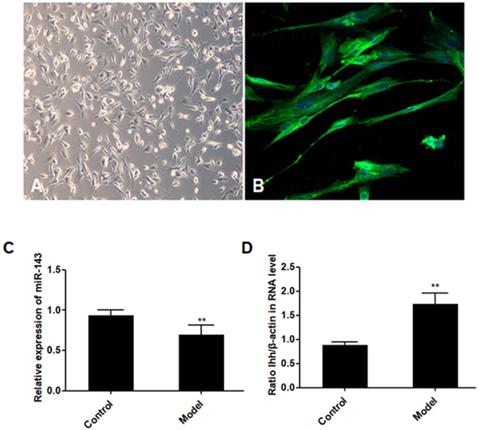
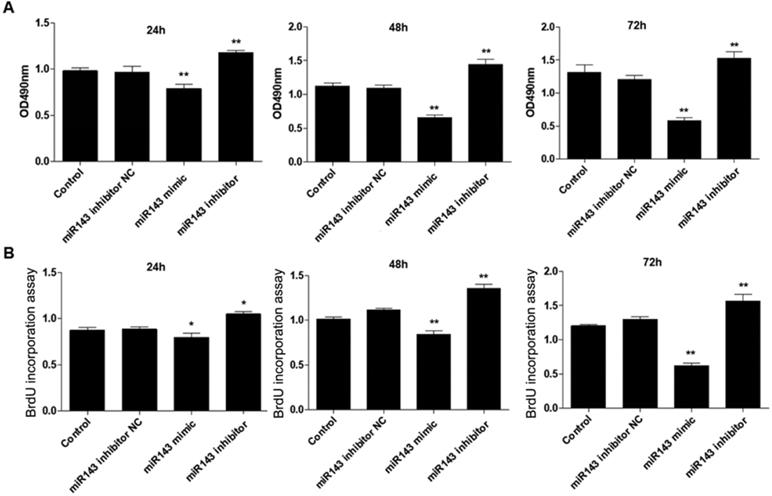
PCSCs, isolated from the neonate rabbits' distal epiphyseal growth plate, were observed using light microscopy (Fig. 1A). Immunofluorescence revealed expression of fibroblast growth factor receptor-3 (FGFR-3), which serves as a marker for PCSCs (Fig. 1B). In addition, real time PCR revealed that expression of miR-143 was reduced, while IHH levels were elevated in the injured growth plate (model group) (Fig. 1C and D). In order to investigate the role of miR-143 in cell growth, PCSCs were transfected with either miR-143 mimics or miR-143 inhibitor and their controls. After culturing for 24, 48, and 72 h, we found that transfection with miR-143 mimics for 24 h slightly inhibited PCSC growth, while the miR-143 inhibitor induced PCSC growth (Fig. 2A). At 48 h and 72 h, miR-143 further suppressed cell growth of PCSCs. In contrast, inhibition of miR-143 promoted PCSC growth (Fig. 2A). Furthermore, the antiproliferative role of miR-143 in PCSCs was confirmed with BrdU assay. Results showed that transfection with miR-143 mimics inhibited DNA synthesis of PCSCs in a time-dependent manner (Fig. 2B), which was enhanced after transfection with the miR-143 inhibitor at different time points (Fig. 2B). Collectively, these data suggest that miR-143 may play a negative role in PCSC proliferation.
Aberrant expression of miR-143 and IHH. PCSCs were isolated from the neonate rabbits' distal epiphyseal growth plate. PCSCs were observed with light microscopy (A, magnification 10×) and immunostaining with FGFR-3 (B, magnification 200×). Magnification 400x. In addition, expression of miR-143 (C) and IHH (D) was detected using real time PCR in control and injured growth plates (model group). Experiments were repeated three times with at least three replicates in each assay. Mean values were compared using Student's t-test. *P < 0.05, **P <0.01.
Effect of miR-143 on growth and DNA synthesis of PCSCs. PCSCs were transfected with miR-143 mimic, miR-143 inhibitor, and negative control (NC). MTT assay (A) and BrdU assay (B) were performed to measure cell viability and DNA synthesis of PCSCs at 24 h, 48 h, and 72 h. Experiments were repeated three times with at least three replicates for each assay. Mean values of multiple groups were compared using one-way ANOVA and Bonferroni post hoc test. *P < 0.05, **P <0.01.
miR-143 regulates apoptotic death and chondrogenesis-related genes in PCSCs
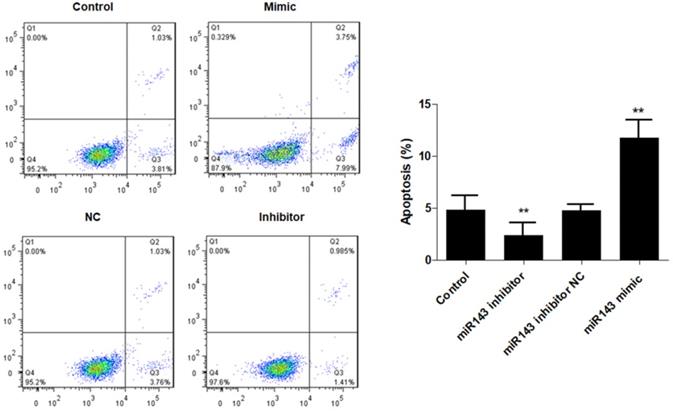
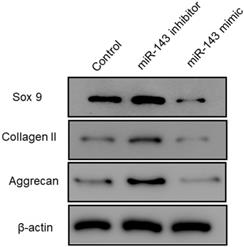
Furthermore, flow cytometry analysis was performed to determine the effect of miR-143 on PCSC apoptosis. Our data showed that overexpression of miR-143 increased the percentage of apoptotic PCSCs (11.2±2.2%) compared to the control group (4.5±1.3%). In contrast, downregulation of miR-143 decreased the percent of apoptotic PCSCs (2.1±1.1%) (Fig. 3). Taken together, these findings revealed that inhibition of miR-143 may promote PCSC growth via inhibition of apoptosis. In addition, we explored the potential role of miR-143 in PCSC differentiation. We found that the protein levels of chondrogenesis-related genes, including Sox9, collagen II, and aggrecan, were significantly upregulated after transfection with the miR-143 inhibitor but were decreased after overexpression of miR-143 in primary cultured PCSCs (Fig. 4). These data suggested that miR-143 regulated apoptotic death and chondrogenesis in PCSCs.
Effect of miR-143 on PCSCs apoptosis. PCSCs were transfected with miR-143 mimic, miR-143 inhibitor, and negative control (NC). Flow cytometric analysis was performed to measure cell apoptosis of PCSCs. Experiments were repeated three times with at least three replicates in each assay. Mean values of multiple groups were compared using one-way ANOVA and Bonferroni post hoc test. *P <0.05, **P <0.01.
IHH is a direct target of miR-143
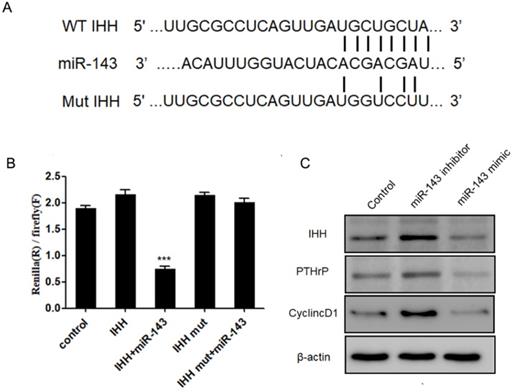
Next, we aimed to identify the potential targets of miR-143 with bioinformatic analysis using the miRNA database (http://www.mirbase.org/), miRanda (http://www.microrna.org), and TargetScan (http://www.targetscan.org/). Our data revealed that the 3'-UTR of IHH gene contained conserved binding sites for miR-143 (Fig. 5A). To validate that IHH is a target of miR-143, luciferase reporters that contained the wild-type 3'-UTR of IHH or a 3'-UTR with a miR-143 mutated biding site were generated. Consequently, the activity of wild-type IHH-3'-UTR was significantly decreased to less than half compared to control in PCSCs co-transfected with miR-143 and wild-type IHH, but remained unchanged in PCSCs transfected with the mutated IHH-3'-UTR (Fig. 5B). Moreover, the protein levels of IHH were suppressed following treatment with miR-143 mimics but were elevated by two-fold after addition of a miR-143 inhibitor (Fig. 5C). Furthermore, overexpression of miR-143 also suppressed PTHrP and Cyclin D1, which were obviously elevated in miR-143 inhibitor-treated cells (Fig. 5C). Collectively, these findings showed that IHH was a direct target gene of miR-143 in PCSCs.
Effect of miR-143 on chondrogenesis-related molecules in PCSCs. PCSCs were transfected with the miR-143 mimic and the miR-143 inhibitor and protein levels of chondrogenesis-related genes, including Sox9 and collagen II, were determined using western blot. *P <0.05, **P <0.01.
IHH is a target of miR-143. (A) Schematic diagram of the wild-type and mutant IHH. (B) Luciferase reporter assay was used to validate that IHH is a direct target of miR-143. (C) Protein levels of IHH, PTHrP, and Cyclin D1 were detected with western blot. Experiments were repeated three times with at least three replicates in each assay. Mean values of multiple groups were compared using one-way ANOVA and Bonferroni post hoc test. *P < 0.05, **P <0.01.
miR-143 mediates PCSC growth and apoptosis by targeting IHH
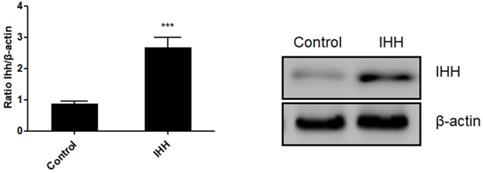
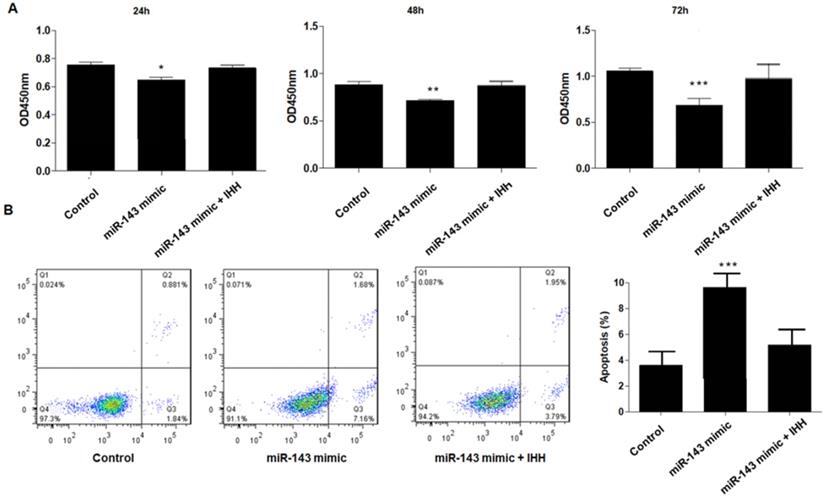
Moreover, the mechanism of miR-143 involved in PCSC growth and apoptosis was investigated. In order to validate whether miR-143 exerts its biological role via IHH, we used the pcDNA3.1 vector to direct expression of IHH. Real time PCR revealed that mRNA levels of IHH increased by about three-fold in pcDNA3.1-IHH transfected cells. Consistently, IHH protein levels were significantly elevated in PCSCs transfected with pcDNA3.1-IHH (Fig. 6). Consequently, MTT assay showed that transfection with miR-143 mimics suppressed PCSC cell viability at 24, 48, and 72 h post-transfection; in contrast, co-transfection with IHH reversed cell viability suppression by miR-143 in PCSCs (Fig. 7A). Moreover, flow cytometry analysis showed that overexpression of miR-143 increased the percentage of apoptotic PCSCs, which was reversed after co-transfection with IHH (Fig. 7B).
Ectopic expression of IHH in PCSCs. PCSCs were transfected with pcDNA3.1(+)-IHH and control plasmid. Forty-eight hours later, total RNA and protein were harvested. The mRNA and protein expression of IHH was determined with real time PCR and western blot, respectively. Experiments were repeated three times with at least three replicates in each assay. Student's t-test. ***P <0.001.
Upregulation of IHH reverses the effect of miR-143 on PCSCs. PCSCs were transfected with miR-143 mimic alone or pcDNA3.1(+)-IHH followed by MTT assay (A) to measure cell viability of PCSCs at 24 h, 48 h, and 72 h. Moreover, flow cytometry (B) was performed to measure cell apoptosis of PCSCs. Experiments were repeated three times with at least three replicates in each assay. Mean values of multiple groups were compared using one-way ANOVA and Bonferroni post hoc test. *P <0.05, **P <0.01, ***P <0.001.
Discussion
Our current findings reveal that, by targeting IHH, miR-143 downregulation could promote growth and inhibit apoptosis of PCSCs, thereby modulating chondrogenesis effects. Most of the current studies focus on the regulatory role of miR-143 in tumorigenesis. For example, miR-143 may serve as a potential target in osteosarcoma. In vitro assays have identified that miR-143 regulates proliferation, apoptosis, and migration of osteosarcoma cells via targeting multiple key molecules, such as MAPK7 and FOS-Like antigen 2 (FOSL2) [18-20]. A growing body of evidence has revealed that miRNAs play essential roles in the regulation of cartilage differentiation and repair [11-14]. Gu et al. found that transfection with miR-155 inhibited osteoblast differentiation by downregulating SMAD5 in mouse preosteoblast cells [21]. Zhang et al. demonstrated that increased miR-93-5p in patients with trauma-induced osteonecrosis of the femoral head (TIONFH) inhibited osteogenic differentiation by targeting BMP2 [22]. Several publications have suggested that miR-143 serves as a tumor suppressor and is involved in many biological processes including cell growth, apoptosis, and migration [23,24]. However, the biological function of miR-143 in PCSCs remains unknown. In the current study, the expression of miR-143 was significantly downregulated in a rabbit model of growth plate injury, together with elevated levels of IHH. Moreover, miR-143 expression was negatively correlated with IHH expression. To investigate the biological role of miR-143, we transfected either miR-143 mimics or an inhibitor into PCSCs. As a result, we found that knockdown of miR-143 significantly increased proliferation and DNA synthesis, and inhibited apoptosis in PCSCs. Conversely, upregulation of miR-143 decreased cell growth and DNA synthesis and promoted apoptosis in PCSCs. Given the association between apoptosis and proliferation [25], we evaluated the levels of apoptosis in PCSCs overexpressing or downregulating miR-143. Our data showed that knockdown of miR-143 inhibited apoptosis but overexpression of miR-143 enhanced apoptosis in PCSCs. These findings reveal that antiproliferative function of miR-143 in PCSCs is mediated by inducing apoptosis. Furthermore, we tested the potential role of miR-143 in PCSC differentiation. Data showed that protein expression of chondrogenesis-related genes, including Sox9 and collagen II, was significantly downregulated after miR-143 mimic treatment and elevated in miR-143 inhibitor-treated PCSCs, suggesting that miR-143 suppresses chondrogenic differentiation of PCSCs.
The Hh signaling pathway is important for bone development, homeostasis, and repair [26]. IHH is a transmembrane protein that forms a receptor complex for Shh together with Smoothened [27]. Based on bioinformatic analyses and anti-correlated expression of miR-143 and IHH, we hypothesized that miR-143 probably targeted the IHH gene to control PCSC growth. As expected, we identified a putative binding site of miR-143 in the 3'‑UTR region of IHH. To validate the direct interaction between miR-143 and IHH, we mutated the putative miR-143 binding sites at the 3′-UTR of IHH. Dual luciferase reporter assay revealed that the activity of wild-type IHH-3'-UTR significantly decreased in PCSCs co-transfected with miR-143. In contrast, the dual luciferase activity following co-transfection of the mutant 3'-UTR of IHH with miR-143 mimics compared with co-transfection with control plasmid showed no significant difference, indicating a direct interaction between miR-143 and the IHH gene. Data showed that levels of IHH were suppressed following treatment with miR-143 mimics but were elevated after addition of miR-143 inhibitor. IHH, which is required for the synthesis of PTHrP, interacts with PTHrP in a negative feedback loop to regulate chondrocyte differentiation [28]. Consequently, levels of PTHrP and IHH-targeted Cyclin D1 were suppressed following treatment with miR-143 mimics but were elevated after addition of a miR-143 inhibitor, showing that miR-143 inhibits the IHH pathway in PCSCs.
Most bones develop through the process of bone formation, and cartilage tissues are formed through condensation of mesenchymal cells, followed by differentiation into chondrocytes. IHH, as a member of the hedgehog family, is expressed in prehypertrophic and early hypertrophic chondrocytes [29]. IHH regulates the interaction between mesenchymal stem cells and osteoclasts for bone homeostasis and repair. Moreover, IHH stimulates precartilaginous stem cell proliferation and inhibits osteoblast development in endochondral bones [30]. In the present work, IHH was directly targeted by miR-143 and its expression was found to be negatively correlated with that of miR-143. Therefore, miR-143 may regulate the apoptosis and growth in PCSCs through targeting IHH. As expected, co-transfection with miR-143 mimics and IHH reversed the suppressive role of miR-143 in PCSCs. In addition, overexpression of miR-143 promoted apoptosis in PCSCs, which was reversed by co-transfection with IHH. Collectively, these findings determine that the effect of miR-143 overexpression was neutralized by IHH overexpression.
In conclusion, our study demonstrated that miR-143 regulated growth and apoptosis in PCSCs. Moreover, the current study revealed that miR-143 directly targeted the IHH gene and inhibited its endogenous expression at both the mRNA and protein levels. These findings suggest that miR-143 may serve as a novel target for cartilage regeneration and repair.
Acknowledgements
This study was supported by Jiangsu province's youth medical talents project of China (No. 2017757), Suzhou science and technology support project of China (No. SYS2020155).
Authors' contributions
Fu-Yong Zhang, Yun-Fang Zhen, and Xiao-Dong Wang conceived and designed the study; Fu-Yong Zhang, Yun-Fang Zhen, Zhi-Xiong Guo, Jin Dai, Lun-Qing Zhu, Pei-Rong Liang, Guang-Hao Su, and Wen-Yan Zhang collected the samples, performed molecular analysis and wrote the manuscript; Yao Liu, Jian-Feng Fang, Quan-Wen Yuan, Feng Yao, Ya Liu,Yi Qiao,Ya Zhang, and Wan-Liang Guo made a critical review, adding improvements through comments and rewriting sentences; all authors read and approved the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Budd E, Waddell S, de Andrés MC. et al. The Potential of microRNAs for Stem Cell-based Therapy for Degenerative Skeletal Diseases. Current molecular biology reports. 2017;3:263-75
2. Fan MP, Si M, Li BJ. et al. Cell therapy of a knee osteoarthritis rat model using precartilaginous stem cells. European review for medical and pharmacological sciences. 2018;22:2119-25
3. Haraguchi R, Kitazawa R, Kohara Y. et al. Recent Insights into Long Bone Development: Central Role of Hedgehog Signaling Pathway in Regulating Growth Plate. Int J Mol Sci. 2019;20:5840
4. Ohba S. Hedgehog Signaling in Endochondral Ossification. J Dev Biol. 2016;4:20-5
5. Zou S, Chen T, Wang Y. et al. Mesenchymal stem cells overexpressing Ihh promote bone repair. J Orthop Surg Res. 2014;9:102
6. Chen L, Liu G, Li W. et al. Chondrogenic differentiation of bone marrow-derived mesenchymal stem cells following transfection with Indian hedgehog and sonic hedgehog using a rotary cell culture system. Cell Mol Biol Lett. 2019;24:16-24
7. Long F, Zhang XM. et al. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099-108
8. Correa D, Hesse E, Seriwatanachai D. et al. Zfp521 is a target gene and key effector of parathyroid hormone-related peptide signaling in growth plate chondrocytes. Dev Cell. 2010;19:533-46
9. Deng Q, Li P, Che M. et al. Activation of hedgehog signaling in mesenchymal stem cells induces cartilage and bone tumor formation via Wnt/β-Catenin. Elife. 2019;8:e50208
10. Amano K, Densmore M, Nishimura R. et al. Indian hedgehog signaling regulates transcription and expression of collagen type X via Runx2/Smads interactions. J Biol Chem. 2014;289:24898-910
11. Li N, Long B, Han W. et al. microRNAs: important regulators of stem cells. Stem cell research & therapy. 2017;8:110
12. Feng N, Xu B, Tao J. et al. A miR-125b binding site polymorphism in bone morphogenetic protein membrane receptor type IB gene and prostate cancer risk in China. Molecular biology reports. 2012;39:369-73
13. Su X, Liao L, Shuai Y. et al. MiR-26a functions oppositely in osteogenic differentiation of BMSCs and ADSCs depending on distinct activation and roles of Wnt and BMP signaling pathway. Cell death & disease. 2015;6:e1851
14. Mizuno Y, Tokuzawa Y, Ninomiya Y. et al. miR-210 promotes osteoblastic differentiation through inhibition of AcvR1b. FEBS letters. 2009;583:2263-8
15. Li E, Zhang J, Yuan T. et al. miR-143 suppresses osteogenic differentiation by targeting Osterix. Molecular and cellular biochemistry. 2014;390:69-74
16. Tian J, Rui YJ, Xu YJ. et al. MiR-143-3p regulates early cartilage differentiation of BMSCs and promotes cartilage damage repair through targeting BMPR2. Eur Rev Med Pharmacol Sci. 2018;22:8814-21
17. Zhang P, Yang W, Wang G. et al. miR-143 suppresses the osteogenic differentiation of dental pulp stem cells by inactivation of NF-κB signaling pathway via targeting TNF-α. Arch Oral Biol. 2018;87:172-9
18. Dong X, Lv B, Li Y. et al. MiR-143 regulates the proliferation and migration of osteosarcoma cells through targeting MAPK7. Arch Biochem Biophys. 2017;630:47-53
19. Hou Y, Feng H, Jiao J. et al. Mechanism of miR-143-3p inhibiting proliferation, migration and invasion of osteosarcoma cells by targeting MAPK7. Artif Cells Nanomed Biotechnol. 2019;47:2065-71
20. Sun X, Dai G, Yu L. et al. miR-143-3p inhibits the proliferation, migration and invasion in osteosarcoma by targeting FOSL2. Sci Rep. 2018;8:606
21. Gu Y, Ma L, Song L. et al. miR-155 Inhibits Mouse Osteoblast Differentiation by Suppressing SMAD5 Expression. BioMed research international. 2017;2017:1893520
22. Zhang Y, Wei QS, Ding WB. et al. Increased microRNA-93-5p inhibits osteogenic differentiation by targeting bone morphogenetic protein-2. PLoS One. 2017;12:e0182678
23. Xin M, Small EM, Sutherland LB. et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes & development. 2009;23:2166-78
24. Clapé C, Fritz V, Henriquet C. et al. miR-143 Interferes with ERK5 Signaling, and Abrogates Prostate Cancer Progression in Mice. PLoS One. 2009;4:e7542
25. Alenzi FQ. Links between apoptosis, proliferation and the cell cycle. Br J Biomed Sci. 2004;61:99-102
26. Fattahi S, Langroudi MP, Akhavan-Niaki H. Hedgehog signaling pathway: epigenetic regulation and role in disease and cancer development. J Cell Physiol. 2018;233:5726-35
27. Pan A, Chang L, Nguyen A. et al. A review of hedgehog signaling in cranial bone development. Frontiers in physiology. 2013;4:61
28. Mak KK, Kronenberg HM, Chuang PT. et al. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development. 2008;135:1947-56
29. Chung JH, Larsen AR, Chen E. et al. A IHH Homolog Transcriptionally Activated by p53 Suppresses Hedgehog Signaling. J Biol Chem. 2014;289:33020-31
30. Li Y, Zhang D, Chen C. et al. MicroRNA-212 displays tumor-promoting properties in non-small cell lung cancer cells and targets the hedgehog pathway receptor IHH. Mol Biol Cell. 2012;23:1423-34
Author contact
![]() Corresponding authors: Yao Liu, MM, Department of Orthopaedics, Children's Hospital of Soochow University, No. 92 Zhongnan Road, Suzhou 215000, China. E-mail: 191099718com; Xiao-Dong Wang, PhD, Department of Orthopaedics, Children's Hospital of Soochow University, No. 92 Zhongnan Road, Suzhou, 215000, China. E-mail: orthwxdcom.
Corresponding authors: Yao Liu, MM, Department of Orthopaedics, Children's Hospital of Soochow University, No. 92 Zhongnan Road, Suzhou 215000, China. E-mail: 191099718com; Xiao-Dong Wang, PhD, Department of Orthopaedics, Children's Hospital of Soochow University, No. 92 Zhongnan Road, Suzhou, 215000, China. E-mail: orthwxdcom.