Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The structure of PTX3
PTX3 in innate immunity
Role of PTX3 in related...
PTX3 in cancerous inflammation
PTX3 in retinal inflammation
PTX3 in metabolic inflammation
PTX3 in vascular inflammation
PTX3 in urinary tract...
PTX3 in airway inflammation
Manifestations of rheumatoid...
PTX3 in the pathogenesis of RA...
Pentraxin 3 with cardinal...
Conclusion and Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(8):1886-1898. doi:10.7150/ijms.54787 This issue Cite
Review
Perspectives on long pentraxin 3 and rheumatoid arthritis: several potential breakthrough points relying on study foundation of the past
Cheng Qiu1,2,3, Yichao Han1,2, Hanwen Zhang1,2, Tianyi Liu2, Haodong Hou1,2, Dan Luo4, Mingzhi Yu5, Kai Bian1,2, Yunpeng Zhao3, Xing Xiao1 ![]()
1. Department of Orthopedic Surgery, The First Affiliated Hospital of Shandong First Medical University, Jinan 250014, Shandong, P. R. China.
2. Cheeloo College of Medicine, Shandong University, Jinan 250012, Shandong, P. R. China.
3. Department of Orthopaedic Surgery, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan 250012, Shandong, P. R. China.
4. College of Stomatology, Qingdao University, Qingdao 266071, Shandong, P. R. China.
5. Key Laboratory of High Efficiency and Clean Manufacturing, School of Mechanical Engineering, Shandong University, Jinan 250061, Shandong, P. R. China.
Received 2020-10-20; Accepted 2021-1-24; Published 2021-3-3
Abstract

Rheumatoid arthritis (RA) is a systemic chronic autoimmune inflammatory disease which is mainly characterized by synovitis and results in a severe burden for both the individual and society. To date, the underlying mechanisms of RA are still poorly understood. Pentraxin 3 (PTX3) is a typical long pentraxin protein which has been highly conserved during evolution. Meanwhile, functions as well as properties of PTX3 have been extensively studied. Several studies identified that PTX3 plays a predominate role in infection, inflammation, immunity and tumor. Interestingly, PTX3 has also been verified to be closely associated with development of RA. We therefore accomplished an elaboration of the relationships between PTX3 and RA. Herein, we mainly focus on the associated cell types and cognate cytokines involved in RA, in combination with PTX3. This review infers the insight into the interaction of PTX3 in RA and aims to provide novel clues for potential therapeutic target of RA in clinic.
Keywords: Pentraxin 3, Rheumatoid arthritis, Synovitis, TNF-α, Inflammation
Introduction
Rheumatoid arthritis (RA) is a common autoimmune disease that afflicts 0.5-1% population all over the world [1]. Hitherto, the mechanism of RA is still poorly understood, while environmental factors and genetic causes may both contribute to the morbidity of RA [2]. Autoantigen should be considered as the crucial clue due to the nosogenesis in autoimmune diseases, including RA. Collagen type II, the chief contained protein of hyaline cartilage, has been recognized as a critical autoantigen [3]. Meanwhile, collagen induced and collagen-antibody induced autoimmune inflammatory arthritis mouse models have become an instrument to identify potential treatment against RA [4, 5]. RA is characterized as a chronic inflammation procedure, and the involved joints are infiltrated by inflammatory cells [6]. Therefore, the construction of inflammatory microenvironment in joints benefits from inducible lining of cell types sites. Provocative inflammation leads to gradual cartilage destruction and progressive bone erosion [7].
The classical long pentraxin, pentraxin 3 (PTX3), is expressed and released by various cell types in response to inflammatory cytokine stimulation and microbial invasion [8-10]. PTX3 is affiliated with the soluble pattern recognition receptors with high conservative structure both in mice and human during evolution [11]. Currently, persistent studies have acknowledged that PTX3 plays an indispensable role in immunity, inflammation, infection, tissue remodeling and fertility [12].
Herein, we mainly sum up the roles of PTX3 in RA mingling with our own insights. By virtue of accumulative evidence, we recognize that PTX3 may play a crucial role in RA progression. On this stage, we expect that future studies may focus on these clues provided by us to hopefully elucidate more about the mechanisms of RA.
The structure of PTX3
Pentraxins are recognized as a category of ancient protein superfamily with a highly conservative structure in evolution [13]. The pentraxins are manifested as their carboxy-terminal with an identical 203 amino acid pentraxin domain [14]. According to the primary structure of the subunit, they can be classified as two groups: long pentraxins and short pentraxins [14]. The classical short pentraxins, C-reactive protein (CRP) and serum amyloid P (SAP) component, are acute-phase proteins major produced by liver in response to inflammation, whereas PTX3 is the typical prototype of long pentraxin proteins (Figure 1) [15-17].
The gene and protein structure of PTX3, CRP and SAP. What is known is that all three molecules are secreted protein. There are 3 exons located on PTX3 gene which is different from 2 exons on the genes of CRP and SAP. Preliminary peptides of PTX3, CRP and SAP are endowed with signal peptide located on their N-terminal domain. Long PTX3, along with CRP and SAP, is characterized by pentraxin signature which is the pentraxin family sign.
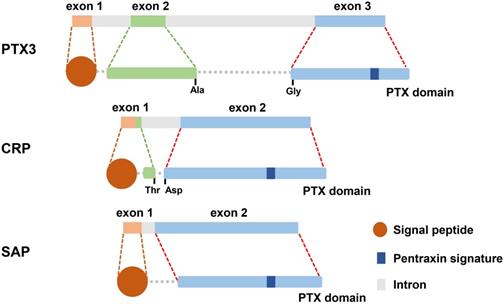
PTX3 contains 381 amino acids, including a 17-amino acid signal peptide, which is characterized by a sheet-like secondary structure and a spiral arrangement of domain [18]. However, SAP consists of calcium-dependent ligand binding and a unique flat β-film structure similar to leguminous lectins [19]. And CRP is composed of five non-covalently bound precursors arranged symmetrically around the central hole. The CRP promoter has 206 amino acids and is folded into two anti-parallel slices with a flat jelly topology [20].
The primary sequence of PTX3 is highly conserved during evolution. PTX3 protomer is assembled into an N-terminal region and a 203 amino acids long C-terminal domain, and then formed multimers are stabilized by disulfide bonds [21]. Two cysteines are responsible for determining the secondary structure of the domain at amino acid positions 210 and 271 to form the disulfide bridge [22]. The C-terminal domain (203 amino acids) of PTX3 has homology with the classic short pentraxins (8 amino acids in length) and is conserved among all members of the pentraxin family [15, 21]. Currently, the functions of the N-terminal domain (178 amino acids) of PTX3 are still unidentified [23].
PTX3 in innate immunity
PTX3, a soluble pattern recognition molecule (PRM), is a key factor in humoral innate immune response to microorganisms [24]. Pentraxins and complement system can opsonize apoptotic cells, promptly facilitating the uptake of cellular debris to control the maintenance of peripheral immune tolerance [25]. Meanwhile, PTX3 interacts with a variety of ligands including growth factors, extracellular matrix components and selected pathogens [26]. The complement C1q is the first characteristic ligand described by PTX3 [14]. The plastic-immobilized C1q binds to PTX3, which also interacts with the C1q spherical head, especially the charged residues located at the top of the molecule [13, 27]. PTX3 directly regulates the activation of the classical complement cascade by combining with C1q, in addition to activating indirect complement mediated immune response [14]. Intriguingly, PTX3 also blocks the relevant site of action by binding to C1q in the liquid phase to inhibit complement activation [13]. Moreover, PTX3 also interacts with the factor H (FH) and C4 binding protein (C4BP) to adjust the complement cascade [24]. Clearance of apoptotic cells is related to the interaction of PTX3 and C1q [16]. The glycosylation of PTX3 may abrogate the interaction between PTX3 and C1q, and subsequent PTX3-mediated complement activation is then blocked [21].
Toll-like receptor (TLR) participates in the production of C1q and PTX3 by immature dendritic cells (imDCs) [28]. During acute phase immune response, imDCs, which produce phagocytosed antigens, execute antigen presentation regulated by PTX3. Antimicrobial resistance-related functions of PTX3 are related to the interaction of conserved ancestral domains in innate immunity [17], as well as tissue repair [24]. PTX3 has been proven as the opsonin for bacteria and fungi, promoting medullary cell recognition and phagocytosis in a complement- and FcγR-dependent manner, as well as neutralizing viral infections [15]. For instance, the neutrophil derived protein PTX3 is essential for controlling fungal growth [14].
To date, there are numerous findings on the role of PTX3 in innate immune system. In conclusion, PTX3 facilitates the recognition of microbial moieties and the activation of complements. Additionally, PTX3 plays a role in the activation of some adaptive immune responses, including macrophage polarization and DC maturation.
Role of PTX3 in related inflammation
The extraordinary role of PTX3 in inflammation has been identified by various studies. Herein, we mainly summarize the classic types of inflammatory responses in which PTX3 involved, comprising cancerous inflammation, retinal inflammation, metabolic inflammation, vascular inflammation, urinary tract inflammation and airway inflammation below (Table 1).
The role of PTX3 in related typical inflammations
| Type of inflammation | Role of PTX3 | Reference |
|---|---|---|
| Cancerous inflammation | The hypermethylation of PTX3 promoter with subsequent diminished PTX3 expression emerges in human esophageal squamous cell carcinoma, selected mesenchymal and epithelial tumors. Ptx3-/- mice are susceptible to mesenchymal tumor and dermal cancer, accompanied by increased macrophages infiltration, secreted inflammatory cytokines, neovascularization, increased C3 deposition and C5a levels, and lower FH localization. Ptx3-/- mice with tumors are mostly characterized by trp53 mutations. The inhibitory role of PTX3 on several tumor cells is prone to rely on FGF2. PTX3 deficiency also promotes migration and invasion of tumor cells in gastric cancer. | [29-34] |
| Retinal inflammation | The expression of PTX3 is increased in cultured RPEC when stimulated with inflammatory cytokines or lipid peroxides in vitro. The plasma level of PTX3 and its gene expression in RPEC and choroid are both independent of AMD status. PTX3 regulates the retinal inflammation mainly by binding to FHL-1. In an AMD model, mice with enhanced C3a and IL-1β levels in RPEC and macrophage accumulation in the choroid via complement activation show a deficiency of PTX3. | [35-38] |
| Metabolic inflammation | In terms of obesity, PTX3 controls angiogenesis by binding to FGF2. PTX3-deficiency induces polarization of M2-like macrophages to inhibit lipid accumulation and metabolic inflammation in obesity, accompanied by increased CD31 and VEGF in adipose tissue. | [30, 39-41] |
| Vascular inflammation | The absence of PTX3 promotes neointimal hyperplasia after vascular injury and macrophage infiltration. PTX3 regulates vascular inflammation by interplaying with adhesion molecules on the vessel wall, associated cytokines and chemokines. Elevated levels of PTX3 in a cardiovascular disease might be associated with the severity of the disease. | [42-46] |
| Urinary tract inflammation | Rapid expression of PTX3 to recruit neutrophils to fight against bacterial infection is induced by pathogenic E. coli stimulation via inhibition of TLR4-MyD88 pathway. PTX3-deficiency is highly associated with the increased severity of infection and the increased sensitivity to pathological inflammation. | [47-50] |
| Airway inflammation | Th17-dominant CD4+ T cells mediate airway inflammatory response in mice with PTX3-deficiency when exposed to OVA. The depletion condition of PTX3 is identified to emerge the phenotype of dysregulated IL-2 and reprograming CD4+ T cells. PTX3 is churned out in airway smooth muscle bundles and epithelial cell layers by inflammatory cell infiltration. Higher expression of PTX3 induces ASMC migration and promotes airway remodeling. | [51-56] |
PTX3 in cancerous inflammation
Based on uniform concept of various insights, the occurrence of tumor is highly associated with the co-function of multi-factors. The infiltrated acclimation of immune cells in intricate tumor microenvironment is a typical characteristic for neoplasm progression. Chronic tumorous inflammation largely encourages cancer cells to survive and proliferate, and adjacent molecules including selectins and chemokines with their receptors, are co-opted by cancer cells for migration or metastasis [29].
Pentraxins are associated with the regulation of chronic tumorous inflammation and play a governing role in tumor development [29, 30]. Among the entire pentraxins family members, PTX3 is implied to achieve reduced expression in several malignant cancers' progressions. Human esophageal squamous cell carcinoma, selected mesenchymal and epithelial tumors were reported to be related with the hypermethylation of PTX3 promoter, with subsequent diminished production [31]. Meanwhile, cancer cells succumbing to inhibition were observed using a methylation inhibitor, 5-Aza-2'-deoxycytidine, which could sustain PTX3 expression, implying that PTX3 may serve as a potential therapeutic target [32]. Furthermore, 12-Dimethylbenzo pyrene/retinoic acid treatment is associated with mesenchymal tumor and dermal cancer, and the susceptibility of Ptx3-/- mice to those is greatly increased. In-depth analysis of two carcinogenic models suggested that PTX3 deficiency was involved in more pronounced inflammation, infiltrating macrophages, pro-inflammatory cytokines, pronounced neovascularization and increased C3 deposition and C5a levels. Furthermore, tumors that occur to Ptx3-/- mice are mostly distinguished by trp53 mutations and oxidative DNA damage [30]. The shortage of PTX3 also decreases FH localization, but increases C3 deposition and tissue damage. Lack of C3 or inhibition of CCL2, followed by a decrease in tumor infiltrating macrophages, is sufficient to restore the phenotype observed in Ptx3-/- mice [30, 33]. Therefore, PTX3 may be assumed to play a protective role for carcinogenesis in inflammatory tumor microenvironment relying on regulation of complement activation.
Recently, a novel therapeutic strategy for cancers is raised by virtue of overexpression of PTX3 which inhibits fibroblast growth factor-2 (FGF2) dependent tumor growth [34]. PTX3 induced inflammatory compartment production may be similarly involved in these processes.
PTX3 in retinal inflammation
Retinal inflammation plays a key role in the development of age-related macular degeneration (AMD), which deposits the complement system in the acute phase of the macula, leading to inflammatory disorders. Complement protein FH binds to CRP, as well as PTX3 that down-regulates inflammatory response. The truncated form of the unknown FH, which is also called factor F-like protein 1 (FHL-1), may have independent immunoregulatory functions in the human retinal environment [35]. PTX3 is present in the macula of human eyes, and the polymorphism of complement factor H Y402 (Y402H) related to AMD alters the binding of FHL-1 to PTX3 [36]. In acute stage, pentameric protein CRP deposites in the macula, which activates the complement system and leads to dysregulation of inflammation. Taking into consideration of the interplay between PTX3 and FH which partly is caused by the binding site of CCP7, similarly integrated with CRP. As expected, the solid phase binding experiments were verified by showing the interaction of full length FH with immobilized PTX3 in relevant studies, and it was also proved that FHL-1 also interacts with PTX3 under the same conditions. In human donor ocular tissues, PTX3 binds to FH on CCP7, CCP19 and CCP20, which is capable of regulating the classical pathway of complement and the lectin pathway, and local expression at the site of inflammation [37]. It is worthy of note that PTX3 enhances the interaction of FH with apoptotic cells, resulting in a regulatory enhancement of iB3b [36]. However, plasma levels of PTX3 and its gene expression in retinal pigment epithelial cell (RPEC) and choroid are both independent of AMD status. Interestingly, PTX3 expression was increased in cultured RPEC which was stimulated with inflammatory cytokines or lipid peroxides. Furthermore, the genetic defect of PTX3 was amplified in an animal model of AMD that increased both C3a and IL-1β levels in RPEC and macrophage accumulation in the choroid via complement activation [38]. Therefore, it could be inferred that PTX3 regulates the retinal inflammatory response possibly by binding to FHL-1.
PTX3 in metabolic inflammation
Obesity is usually the result of an imbalance of excess nutrients and metabolic homeostasis, leading to a chronic inflammatory state and an increase in systemic proinflammatory mediators and adipose tissue-retaining immune cells phenotype from tolerant to pro-inflamed. Once the immune cells in adipose tissue transform into proinflammatory phenotype, they will produce adipokines and inflammatory cytokines [39]. These factors promote the recruitment of immune cells, allowing the persistence of local and systemic inflammation. Progressive accumulation of fat may result from excessive nutrition, elevated free fatty acids, inflammation and activation in adipose tissue. This further restrains inflammation of adipose tissue and insulin resistance [39]. The absence of PTX3 shows the phenotypic polarization of M2 macrophages, which retains the macrophages of adipose tissue, thereby preventing the accumulation of lipid and inflammation in obesity caused by high-fat diet [40]. Related experiments also demonstrated that PTX3 controls angiogenesis by binding to FGF2. These results also showed a direct correlation between expression of vascular endothelial growth factor (VEGF), adipose tissue angiogenesis and M2 macrophage activation [30]. Similarly, PTX3 deficiency is associated with increased expression of CD31 and VEGF in adipose tissue, suggesting an improvement in vascular network, which may also contribute to reduced accumulation of lipid in Ptx3-/- mice [30, 41].
PTX3 in vascular inflammation
Vascular inflammation plays an important role in vascular homeostasis and restricted immunity of organs and tissues in the whole body, reflecting the dynamic interaction of circulating cells, blood molecules and vascular structure [42]. PTX3 is mainly produced in the inflammatory sites as a crucial orchestrator for regulation of inflammation. Neutrophils release PTX3 at an early stage, while the synthesis of endothelial cells and macrophages is to maintain the production of PTX3 [43]. PTX3 inhibits the phagocytosis of neutrophils in late apoptosis and promotes their accumulation in the vascular wall in vascular stenosis, such as small vasculitis. The inadequacy of PTX3 is associated with increased accumulation of macrophages and exaggerated monocytes in atherosclerotic lesions. In addition, Ptx3-/- mice showed adhesion molecules on the vessel wall, and increased expression of cytokines and chemokines, thus indicating that PTX3 regulates vascular-related inflammatory responses [44]. Preliminary findings indicated that PTX3 with the potential pathogenic capacity in cardiovascular disease, was described as an early marker of major local activation of innate immunity and inflammation of vascular wall [45]. In addition, ascending PTX3 in cardiovascular disease may show protective physiological response, which might be related to the severity of the disease [46].
PTX3 in urinary tract inflammation
The pathogenicity of urinary tract infection (UTI) is an influential causation of urinary tract inflammation, and host immunity emerging in the urinary tract is an indispensable pathophysiological feature in urinary tract infections [47, 48]. PTX3 is a humoral PRM for urinary tract infection caused by pathogenic Escherichia coli (E. coli), which is one of the major reasons for human UTI [48]. PTX3 produced by urothelial cells and renal cells plays an important role in controlling urinary tract inflammation, and the deficiency of PTX3 may lead to excessive inflammation [48]. Local and rapid expression of PTX3 stimulated by pathogenic E. coli is inhibition of TLR4-MyD88 pathway during UTI, aiming to enhance the recruitment of neutrophils to fight against the bacteria [49]. In related studies, PTX3-deficiency is highly associated with increased both severity of infection and sensitivity to pathological inflammation. Notably, Ptx3-/- mice expressed higher levels of bacterial load in bladder and kidney, neutrophil infiltration, chemokine levels and tissue damage [50]. Therefore, PTX3 may play a positive role during urinary tract inflammation.
PTX3 in airway inflammation
Airway inflammation is characterized by infiltration of inflammatory cells and airway hyperresponsiveness, and then chronic inflammation mediates subsequent airway remodeling [51]. Recent study revealed that PTX3 is highly related to allergic inflammation from asthmatic patients and ovalbumin-induced mice model. Furthermore, augmented airway response mediated by Th17-dominant CD4+ T cells secreting IL-6 and IL-23, which were sustained by DC cells, was observed in Ptx3-/- mice when exposed to ovalbumin (OVA) stimulation. The depletion of PTX3 promotes the phenotype of deregulated IL-2 and reprograming CD4+ T cells [52]. Several studies indicated that PTX3 produced by epithelial cells has a regulatory effect on airway smooth muscle cell (ASMC) and is churned out in airway smooth muscle bundles and epithelial cell layers, in parallel to inflammatory cell infiltration [53]. Primary human ASMC produces higher levels of PTX3 at baseline with TNF stimulation [54], while higher expression of PTX3 induces ASMC migration and promotes airway remodeling [55]. Recent reports indicated that PTX3 inhibits FGF2-induced airway smooth muscle migration [56]. It is possible that PTX3 may exert potential dual influence by enhancing local airway chemokine expression in human ASMC, while counteracting FGF2-induced human ASMC migration to down-regulate airway remodeling.
Manifestations of rheumatoid arthritis
RA is an autoimmune chronic inflammatory disease that has the characteristics of joint swelling, pain, progressive cartilage damage, destruction of bone and systemic complications, even leading to severe disability [57]. The main pathological aberrance of RA in joint may attribute to hyperplastic synovium, brutal bone erosion and cartilage destruction. At present, antibodies against cyclocitrulline peptide (ACPA) is a more relative biomarker, compared to rheumatoid factor (RF), that predicts invasive disease. Effective treatment can reduce the concentration of RF and ACPA [58]. In RA, several studies demonstrated that inflammatory cytokines can trigger the transition from systemic immunity to arthritis and sustain a detectable proinflammatory environment, and in some cases cytokine interactions prevent inflammation from subsiding [59]. Pro-inflammatory cytokines such as interleukin-1beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) released from the inflamed and damaged tissue of the local inflammatory response, and subsequent secretions including histamine, serotonin, bradykinin, ATP, prostaglandin E2 (PGE2) and protons (H+) play a predominant role in RA. The focus of treatment is shifted to early intervention in the process of disease development to prevent inflammatory joint damage [60].
Currently, to further interpret the underlying mechanisms of RA is fundamental for both intervention and treatment of RA patients. Overall it still remains to be devoted to exploring drugs or cure treatments with significant effects for RA and patients will benefit from amelioration of this malady in terms of current situation.
PTX3 in the pathogenesis of RA with main cell types
Synoviocyte
Synovitis is the major manifestation in RA. PTX3 expressed by numerous cell types after stimulation with cytokines (e.g. IL-1β and TNF-α), TLR agonists or microbes in inflammation is found to be increased in rheumatoid arthritis synovial fluid. Additionally, it is known that PTX3 is also expressed in synoviocyte in vitro [61]. PTX3 is mainly expressed in type B synoviocytes of arthritic patients with the stimulation of TNF-α, but not IL-1β [62]. On the other hand, PTX3 expressed highly by synoviocytes from RA is not affected by anti-TNF antibodies, IL-1 receptor antagonists or a combination of both [61]. However, synoviocytes from RA patients were implied to produce higher levels of PTX3 in vitro while inhibited by transforming growth factor-beta (TGF-β) and interferon-gamma (IFN-γ) [63]. Yokota et al found that simvastatin significantly impairs RA process, and they further identified that simvastatin down-regulates the expression of monocyte chemoattractant protein-1 (MCP-1) and PTX3 in fibroblast-like synoviocyte derived from RA patients [64]. Furthermore, Satomura et al also discovered that the acute-phase reactant, serum amyloid A (SAA), had the efficacy of induction of PTX3 in synoviocyte [65]. In synoviocyte, FGF-2 is reported to play a dramatic role in enhancement of alkaline phosphatase (ALP) activity, the expression of chondrogenic genes (Sox9, Col2α1 and Aggrecan), osteogenic genes (Foxc2, osteocalcin and Col1α1) and VEGF-A, while suppressed by PTX3 addition [66]. Recently, Zhao et al demonstrated that PTX3 suppresses the progression of RA by inhibiting the autocrine and paracrine stimulation of FGF-2 on synovial fibroblast [67].
Chondrocyte
The original ingredient of cartilage is exclusively generated by chondrocyte. Chondrocytes involved in inflammatory microenvironment are prone to death mostly presented as apoptosis [68], bringing about the homeostasis disruption in cartilage. The detectable level of PTX3 is implied in chondrocyte. Furthermore, Haglund et al reported the activation of TLR4 was induced by lipopolysaccharide (LPS) that significantly promotes the production of PTX3 and other inflammatory cytokines in articular chondrocytes [69]. Additionally, Barksby et al also indicated the genes of PTX3, IL-8 and several matrix metalloproteinases (MMPs) were up-regulated with stimulation of IL-1α along with oncostatin M in chondrocyte [70]. The direct interaction with inducible expression of PTX3 in chondrocyte may mainly rely on the proinflammatory cytokine stimulation. In addition, it is well known that FGF-2 is a stimulator for chondrocyte proliferation upon cartilage wounding [71]. However, whether PTX3 potentially serves as an antagonist for FGF-2 to inhibit chondrocyte proliferation is unknown.
Macrophage
The notable function of PTX3 is possible to antagonize macrophage infiltration directly during inflammatory process. Ptx3 knockout mice are used to improve the susceptibility of macrophage (CD68+) infiltration in 3-methylcholanthrene injection site [30]. Also, the expression of PTX3 was evaluated to overexpress in fibrosarcoma, whereas the recruitment of infiltrated macrophage was decreased, as well as neovascularization [72]. And, other evidence demonstrated that PTX3 acted as a potent chemotactic agent through interaction with neuropeptide Y to enhance the migration of macrophage indirectly [73]. The powerful phagocytosis of macrophages is highly embodied in gouty arthritis, and PTX3 contributes to promoting the uptake of monosodium crystals for macrophages via increasing IL-1β and chemokine CXCL1 production [74]. Previous assessment identified that distinct phenotypic macrophages are emerged in both synovium lining and cartilage tissue, and the polarization of macrophage also exists in RA joints with significant production of TNF-α and IL-1β [75]. The role of PTX3 is in line with the macrophage skewing which results in increased M1 macrophage polarization and diminished M2 macrophage, whereas the opposite of PTX3 deficiency [39]. At the same time, up-regulated PTX3 also functioned in promoting the macropinocytosis of M1 macrophages for low-density lipoprotein (LDL) [76]. IL-10 is a common marker of M2 macrophages, assuming that the regulatory bridge may appear in the confrontation between M1 and M2 macrophages, and the role of IL-10 has been elucidated to stimulate the production of PTX3 [26]. The production of PTX3 and IL-10 is elevated in macrophages in response to PGE2 [77].
Dendritic cell
Dendritic cell (DC) is a typical professional APC with the function of immune tolerance and T cell activation in the pathogenesis of RA. Both C1q and PTX3 are produced by imDC in response to TLR engagement [78]. Moreover, PTX3 plays a critical role in recognition of diverse pathogens and modulation of complement activation via connecting to C1q and DCs' recognition of pathogens [79]. Moreover, during the inflammatory conditions of RA, exaggerated apoptosis of cells is a common feature and subsequent necrosis debris release is emerged once apoptotic cells cannot be cleared promptly [80]. PTX3 hereof binds to apoptotic cells, inhibiting its recognition by DC instead of macrophage, and then embarks on normal binding late in the apoptotic process the same as binding to apoptotic cells better than necrotic ones in vitro [17]. Furthermore, PTX3 serves as an opsonin by enhancing the phagocytosis of DCs. It inhibits the release of both TNF-α and IL-10 by LPS-challenged DCs, but the inhibitory effect is blocked in the presence of apoptotic cells [81]. PTX3 suppresses the phagocytosis of DCs and the expression of immunosuppressive cytokines in order to control cross-presentation of self-epitopes expressed by apoptotic cells, promoting innate immune response to pathogens. Taken together, PTX3 presumably influences the production of such chemokines and pro-inflammatory cytokines in the synovium of RA patients by targeting on DCs.
Osteoblast and osteoclast
Similarly, PTX3 increases the potential of osteoblast precursors to differentiate to osteoclasts by affecting the receptor activator of nuclear factor-κ B ligand (RANKL) mediated production of precursor osteoblasts [82]. Furthermore, PTX3 enhances the expression of RANKL, leading to upregulated precursor osteoblasts, which causes the excessive formation of osteoclasts. In the inflammatory bone microenvironment, PTX3 influences TNF-α by inducing precursor osteoblasts to produce RANKL, which promotes the formation of osteoclasts and subsequent bone erosion [83]. PTX3 may mainly manipulate the RANKL/osteoprotegerin (OPG) ratio on osteoblast precursor, instead of affecting osteoclastogenesis directly.
Neutrophil
PTX3 is positively expressed in neutrophils progenitor cells (promyelocytes, myelocytes and metamyelocytes) during neutrophils maturation [84]. Immune signals from cytokines caused neutrophils to rapidly release performed PTX3 from secondary particles. On the other hand, PTX3 prevents excessive recruitment of neutrophils to inflammatory sites [85]. Incremental overexpression of PTX3 and reactive oxygen species (ROS) in neutrophils is associated with impaired endothelial function, and it may be a marker of inflammation in hemodialysis patients [86]. Furthermore, neutrophils with PTX3 deficiency are prone to the detriment of fungi and they may increase the risk of invasive aspergillosis in patients when treated with hematopoietic stem cell transplantation [87]. Notably, neutrophils migrate to the joint cavity early and play a role in inflammation in RA.
Lymphocyte
It is known that PTX3 plays an indispensable role in the regulation of CD4+ T lymphocyte-mediated inflammation [52]. The lack of PTX3 promotes a Th17-dominated CD4+ T cell response in the lung and enhances the survival of CD4+ T cell [52]. Furthermore, it is suggested that PTX3 inhibits the cross-expression of antigens derived from apoptotic cells on CD8+ T cells [17]. Given the ability to recognize limited sets of microbial molecular patterns, pentraxins are considered to be ancestors of antibodies produced by follicular B cells [11]. Subsets of effector T cells, such as CD4+ T cells (TH17) that produce IL-17, play an important role in the pathology of RA, and the role of Treg in peripheral immune tolerance in RA patients remains in dispute [88]. To date, a pathogenic role of these cells may contribute to the development and activity of RA and the therapy for autoimmune diseases is still largely dependent on nonspecific immunosuppression.
Pentraxin 3 with cardinal RA-involved molecule ensemble
TNF-α
The classical cytokine, TNF-α, is identified to exhibit a pleiotropic effect on immunity and inflammation. The majority of TNF-α originates from monocytes, macrophages, DC cells, activated B lymphocytes and T lymphocytes, which can bind to two receptors (TNFR1 and TNFR2) [89]. TNFR1 is universally expressed on nearly all cell types that recruit several molecules after combining with ligand TNF-α, and then activates downstream signaling, nuclear factor kappa-B (NF-κB) in particular, mostly to activate and exacerbate inflammation. However, TNFR2 is mainly verified to regulate the inflammation reaction level and inhibit cell apoptosis, which could account for why TNFR2 is expressed exclusively on immune cells [90]. Bidirectional function of TNF-α is validated to amplify in autoimmune diseases, especially the deleterious effect. During the treatment of autoimmune diseases, particularly exemplified by RA, application of available anti-TNF biologics (monoclonal antibodies, e.g., certolizumab, adalimumab, golimumab and infliximab or soluble TNF receptors, e.g., etanercept) achieves seminal breakthrough since the new start of millennium [91]. However, except financial burden and tolerance limitation for few patients, anti-TNF therapy is also associated with multiple, although relatively rare, side effects: increased risk of intracellular infection, particularly reactivation of Mycobacterium tuberculosis, and increased risk of secondary autoimmune manifestations and lymphoma [92].
It is well established that TNF-α plays a critical role in the development of RA and several signaling transduction pathways (NF-κB, JAK/STAT, Akt, p38/MAPK, ERK and Wnt/β-catenin) are identified to be involved in it. To date, in RA, the signaling of NF-κB is pivotal to TNF-α in macrophage, synoviocyte and chondrocyte [93]. As an inflammatory biomarker, previous study already indicated the TNF-α-induced expression of PTX3 in synoviocyte of RA patients [61]. Currently, the up-regulation of PTX3 by TNF-α in dental pulp cells was also confirmed via the activation of NF-κB signaling [94]. Similarly, with the activation of NF-κB signaling in intestinal ischemia and reperfusion injury, PTX3 was demonstrated to be a sensor that promotes the production of inflammatory mediators such as TNF-α and chemokine CXCL1 [95]. However, the presence of PTX3 was reported to down-regulate IL-1β, TNF-α and MCP-1 expression through repressing the NF-κB signaling activation in macrophages, whilst the detectable concentrations of transforming growth factor-β (TGF-β) was examined [45].
IL-1β
IL-1β plays a critical role in the degradation of articular cartilage by stimulating both synovial fibroblasts and chondrocytes to secrete MMPs, cathepsins, and mast cell proteinases. Furthermore, in rheumatoid synovium, the upregulation of IL-1β promotes the expression of MMPs, which thereby exacerbates synovial inflammation, increasing joint destruction and bone resorption [96]. Interestingly, several studies demonstrated that IL-1β mediated the expression of PTX3 in RA. However, in macrophages, the production of IL-1β, TNF-α and MCP-1 are reduced significantly in the presence of PTX3. Thus, PTX3 may reflect the degree of inflammation indirectly [97].
IL-6
Among abundant cytokines in circulation during inflammation, IL-6 exerts a powerful systemic effect, which results in common complications in RA patients, including changes in cholesterol metabolism, atherosclerosis and even mood disorders [98]. Neiman et al had shown that IL-6 may serve as a possible marker for predicting structural damage [99]. IL-6, associated with disease progression and joint destruction, is greatly increased in the synovial fluid of RA patients [100]. Dessein et al indicated that IL-6 is closely related to the biomarkers of endothelial dysfunction due to inflammatory cytokines released from inflamed joints in RA [101]. Relevant reports also identified that the concentration of IL-6 can be used as an assessment of the degree of cardiovascular disease in RA [102]. However, PTX3 is indirectly affected by the expression of IL-6 [103]. Therefore, PTX3 and IL-6 are not directly interrelated, but IL-6 may indirectly induce the expression of PTX3 to promote the formation of inflammatory microenvironment of RA [104].
TGF-β
The immunosuppressive effects of TGF-β have long been emphasized. Meanwhile, RA, an inflammatory disease targetting joints, is caused by abnormal responses of T and B cells. It has been demonstrated that the levels of IL-6, IL-17 and TGF-β in RA patients' peripheral blood serums are significantly elevated [105]. The development of TGF-β in RA seems to be determined by the anatomic background of cytokine signaling. Also, the modulation of TGF-β activity could be a potential therapy for Th17-mediated RA [106]. Luchetti et al reported that TGF-β down-regulated the expression of PTX3 mRNA in RA synoviocytes [61]. PTX3 has a potential relationship with TGF-β in RA, which might shed light on the treatment of RA.
P-selectin
P-selectin is responsible for the interaction of leukocytes and endothelial cells, and promotes tissue damage in antigen-induced arthritis [107]. The expression of PTX3 is strongly attributed to the adhesion of monocytes to activated endothelial cells stimulated by TNF-α, which may rely on the interaction between PTX3 and P-selectin [108]. Simultaneously, the interaction between P-selectin and PTX3 was identified as the negative feedback loop for recruitment of leukocytes, and P-selectin knockout mice were refractive to PTX3-evoked endothelial dysfunction [109]. Meanwhile, Livija Deban et al reported the identification of interaction between P-selectin and PTX3, and further elucidated the mechanisms that PTX3 bound to P-selectin via its N-linked glycosidic moiety encoded by the third exon [110]. The results may display the role of PTX3 to suppress leukocyte recruitment and inflammation in a P-selectin-dependent way. Herein, it could be speculated that PTX3 may perform an essential role during the onset of RA through acting on endothelial cells and Treg with P-selectin.
Conclusion and Perspectives
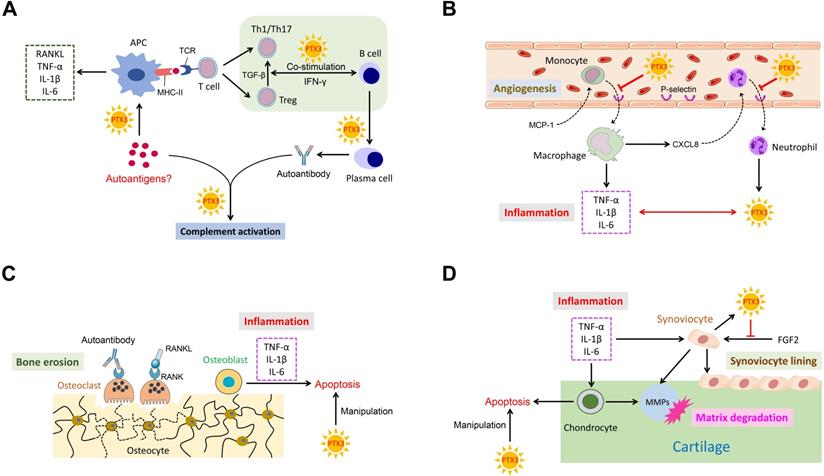
PTX3 is known to play an indispensable role in several pathological conditions. Conspicuously, as a messenger, the elevated expression of PTX3 is necessary for occurrence and development of inflammation. Importantly, RA is an intricate systemic disorder that typically characterized by the co-function of multiple related cell types in affected synovial joints. Cardinal cell types implicated in RA mainly comprise synoviocyte, chondrocyte, macrophage, dendritic cell, osteoblast, osteoclast, neutrophil and lymphocyte. Relevant cells in RA pathogenesis also function in conjunction with secreted cytokines. Consequently, we depict a schematic representation to summarize these potential interactions with PTX3 involved in RA (Figure 2).
The ensemble of PTX3 with related cell types and cytokines in RA. (A) PTX3 promotes autoantigens recognition by APCs, mediates T cells differentiation and activation, regulates the co-stimulation of B cells with T cells, and activates complement system by interacting with the antigen-antibody complex. (B) PTX3 inhibits the recruitment of inflammatory cells by targeting on adhesion molecules and interacts with inflammatory cytokines. (C) PTX3 manipulates apoptotic osteoblasts. (D) PTX3 manipulates apoptotic chondrocytes and antagonizes the function of FGF2 for synoviocyte cell lining.
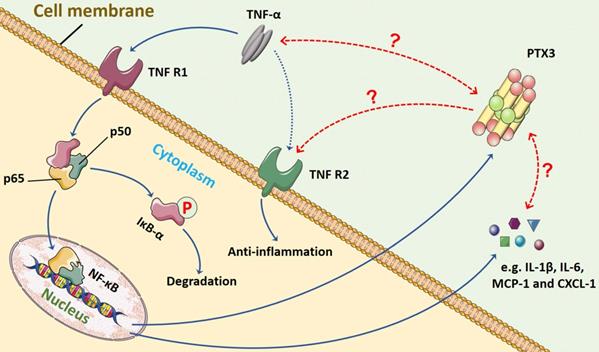
In terms of the relationship between TNF-α and PTX3, we presume that a feedback loop might exist between them, and the crosstalk may commonly manipulate inflammation progression through NF-κB signaling pathway (Figure 3). The TNF-α/TNFR/NF-κB/PTX3 axis may contribute to a novel therapeutic target for RA.
The possible feedback loop between PTX3 and TNF-α via NF-κB signaling pathway in inflammation. In the progression of inflammation, including RA, the pro-inflammatory cytokine TNF-α mainly binds to TNFR1 that activates NF-κB signaling. The major step is the translocation of the dimer (p50 and p65) into nucleus once IκB-α is inactivated and degraded. The transduction of downstream genes mediated by TNF-α involves abundant inflammatory cytokines (e.g. IL-1β and IL-6) and chemokines (e.g. MCP-1 and CXCL-1). Also, the production of biomarker PTX3 is stimulated by TNF-α. However, whether the interaction between PTX3 and TNF-α exists remains unveiled. The TNF-α/TNFR/NF-κB/PTX3 axis may contribute to a feedback loop co-regulating inflammation.
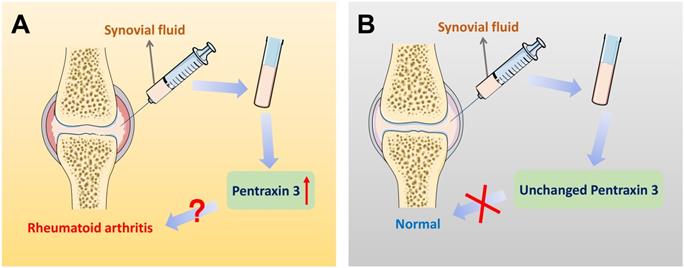
On account of the current non-specific markers for differential diagnosis of RA, further interpretations of precise markers are required. Therefore, inflammatory molecule PTX3 draws attention. Different expression of PTX3 in synovial fluid between RA patients and the control group is significantly available. Correlations could be identified between different stages of RA progression and the elevated expression of PTX3. The expression of PTX3 might be used as a marker for the evaluation of the activity of RA disease from which the clinical differential diagnosis will benefit. Moreover, compared to the conventional diagnostic marker CRP for RA patients, plausible correlation analysis is also given between PTX3 and RA both in serum and synovial fluid (Table 2). In accord with CRP, locally produced PTX3 in synovial fluid is also endowed with the capacity to imply RA inflammation. Additionally, the concentrations of PTX3 in patients with RA that dynamically lined with the disease, compared to the osteoarthritis (OA) normal. In view of the specific presence of PTX3 in synovial fluid, it may be preferable to choose synovial fluid as a determination of RA diagnosis (Figure 4). Notwithstanding, it may also raise the issue of investigating the cause of churned out PTX3 in synovial fluid of RA patients.
The comparison between PTX3 and CRP
| PTX3 | CRP | |
|---|---|---|
| Category of Pentraxins | Long | Short |
| Gene localization in human chromosome | 3q25 | 1q23 |
| Gene structure | 3 exons separated by 2 introns | 2 exons |
| Source | Dendritic cell, Monocyte, Macrophage, Synoviocyte, Neutrophil, Adipocyte and Fibroblast | Major hepatocyte |
| Stimulus | TNF-α and IL-1β | IL-6 |
| Major biological process | Inflammation, Innate immunity, Infection and Tumor | Inflammation |
| Expression in serum of RA patients | Up-regulation | Up-regulation |
| Expression in synovial fluid of RA patients | Up-regulation | Up-regulation |
| Association with the severity of RA | Correlation* | Correlation |
| Expression in serum after RA treatment | Unchanged | Decreased |
*The concentrations of PTX3 in both serum and synovial fluid are related with the severity of Stage II-Ⅳ of RA by Sharma et al. A positive correlation of PTX3 in synovial fluid with disease severity is shown by Mustafa Serdar et al. There is no correlation of PTX3 in blood with RA according to Lieh-bang Liou et al and Gia Deyab et al. The definition of correlation is provisional on account of the limited current status.
Pentraxin 3 in synovial fluid might be regarded as a biomarker for the specific differential diagnosis of rheumatoid arthritis. (A) In the patients of rheumatoid arthritis, there exists a significantly higher level of PTX3 in joint synovial fluid, compared to OA controls. Also, when the expression of PTX3 is abnormally up-regulated in an examinee's synovial fluid, the diagnosis of the disease is likely to be rheumatoid arthritis. (B) In contrast, a lower level of PTX3 is observed in normal people. However, in synovial fluid, when the production of PTX3 remains unchanged, there is no evidence indicating health but at least rheumatoid arthritis may be ruled out.
Abbreviations
RA: rheumatoid arthritis; PTX3: pentraxin 3; CRP: C-reactive protein; SAP: serum amyloid P; PRM: pattern recognition molecule; FH: factor H; TLR: Toll-like receptor; imDC: immature dendritic cell; FGF2: fibroblast growth factor-2; AMD: age-related macular degeneration; C4BP: C4 binding protein; FHL-1: factor F-like protein 1; Y402H: factor H Y402; RPEC: retinal pigment epithelial cell; VEGF: vascular endothelial growth factor; UTI: urinary tract infection; E. coli: Escherichia coli; OVA: ovalbumin; ASMC: airway smooth muscle cell; ACPA: against cyclocitrulline peptide; RF: rheumatoid factor; IL-1β: interleukin-1beta; TNF-α: tumor necrosis factor-alpha; PGE2: prostaglandin E2; H+: protons; SAA: serum amyloid A; LPS: lipopolysaccharide; LDL: low-density lipoprotein; DC: dendritic cell; RANKL: receptor activator of nuclear factor kappa-B ligand; ALP: alkaline phosphatase; MCP-1: monocyte chemoattractant protein-1; ROS: reactive oxygen species; OPG: osteoprotegerin; NF-κB: nuclear factor kappa-B; OA: osteoarthritis.
Acknowledgements
This work was supported by the College Student Science and Technology Innovation Fund of Shandong University (Grant No. 2019369), the Cross-disciplinary Fund of Shandong University (Grant No. 2018JC007) and the National Natural Science Foundation of China (Grant No. 81501880).
Author Contributions
Cheng Qiu and Xing Xiao performed the literature search and were major contributors in the writing of the manuscript. Yichao Han, Hanwen Zhang, Tianyi Liu, Haodong Hou, Dan Luo, Mingzhi Yu and Kai Bian performed the literature search, provided suggestions and co-wrote the manuscript. Yunpeng Zhao provided suggestions and revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388:2023-38
2. Glant TT, Mikecz K, Rauch TA. Epigenetics in the pathogenesis of rheumatoid arthritis. BMC Med. 2014;12:35
3. Trentham DE, Dynesius-Trentham RA, Orav EJ, Combitchi D, Lorenzo C, Sewell KL. et al. Effects of oral administration of type II collagen on rheumatoid arthritis. Science. 1993;261:1727-30
4. Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. 2007;2:1269-75
5. Khachigian LM. Collagen antibody-induced arthritis. Nat Protoc. 2006;1:2512-6
6. Arandjelovic S, Perry JSA, Lucas CD, Penberthy KK, Kim TH, Zhou M. et al. A noncanonical role for the engulfment gene ELMO1 in neutrophils that promotes inflammatory arthritis. Nat Immunol. 2019;20:141-51
7. Nanjundaiah SM, Venkatesha SH, Yu H, Tong L, Stains JP, Moudgil KD. Celastrus and its bioactive celastrol protect against bone damage in autoimmune arthritis by modulating osteoimmune cross-talk. J Biol Chem. 2012;287:22216-26
8. Alles VV, Bottazzi B, Peri G, Golay J, Introna M, Mantovani A. Inducible expression of PTX3, a new member of the pentraxin family, in human mononuclear phagocytes. Blood. 1994;84:3483-93
9. Breviario F, d'Aniello EM, Golay J, Peri G, Bottazzi B, Bairoch A. et al. Interleukin-1-inducible genes in endothelial cells. Cloning of a new gene related to C-reactive protein and serum amyloid P component. J Biol Chem. 1992;267:22190-7
10. Lee GW, Lee TH, Vilcek J. TSG-14, a tumor necrosis factor- and IL-1-inducible protein, is a novel member of the pentaxin family of acute phase proteins. J Immunol. 1993;150:1804-12
11. Chorny A, Casas-Recasens S, Sintes J, Shan M, Polentarutti N, Garcia-Escudero R. et al. The soluble pattern recognition receptor PTX3 links humoral innate and adaptive immune responses by helping marginal zone B cells. J Exp Med. 2016;213:2167-85
12. Garlanda C, Bottazzi B, Bastone A, Mantovani A. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol. 2005;23:337-66
13. Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ. et al. Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur J Immunol. 2003;33:465-73
14. Mantovani A, Garlanda C, Doni A, Bottazzi B. Pentraxins in innate immunity: from C-reactive protein to the long pentraxin PTX3. J Clin Immunol. 2008;28:1-13
15. Bottazzi B, Inforzato A, Messa M, Barbagallo M, Magrini E, Garlanda C. et al. The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. J Hepatol. 2016;64:1416-27
16. Gershov D, Kim S, Brot N, Elkon KB. C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med. 2000;192:1353-64
17. Baruah P, Propato A, Dumitriu IE, Rovere-Querini P, Russo V, Fontana R. et al. The pattern recognition receptor PTX3 is recruited at the synapse between dying and dendritic cells, and edits the cross-presentation of self, viral, and tumor antigens. Blood. 2006;107:151-8
18. Doni A, Stravalaci M, Inforzato A, Magrini E, Mantovani A, Garlanda C. et al. The Long Pentraxin PTX3 as a Link Between Innate Immunity, Tissue Remodeling, and Cancer. Front Immunol. 2019;10:712
19. Xi D, Luo T, Xiong H, Liu J, Lu H, Li M. et al. SAP: structure, function, and its roles in immune-related diseases. Int J Cardiol. 2015;187:20-6
20. Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol. 2001;38:189-97
21. Inforzato A, Peri G, Doni A, Garlanda C, Mantovani A, Bastone A. et al. Structure and function of the long pentraxin PTX3 glycosidic moiety: fine-tuning of the interaction with C1q and complement activation. Biochemistry. 2006;45:11540-51
22. Deban L, Bottazzi B, Garlanda C, de la Torre YM, Mantovani A. Pentraxins: multifunctional proteins at the interface of innate immunity and inflammation. BioFactors. 2009;35:138-45
23. Inforzato A, Baldock C, Jowitt TA, Holmes DF, Lindstedt R, Marcellini M. et al. The angiogenic inhibitor long pentraxin PTX3 forms an asymmetric octamer with two binding sites for FGF2. J Biol Chem. 2010;285:17681-92
24. Doni A, Garlanda C, Mantovani A. Innate immunity, hemostasis and matrix remodeling: PTX3 as a link. Semin Immunol. 2016;28:570-7
25. Nauta AJ, Daha MR, van Kooten C, Roos A. Recognition and clearance of apoptotic cells: a role for complement and pentraxins. Trends Immunol. 2003;24:148-54
26. Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol. 2010;28:157-83
27. Roumenina LT, Ruseva MM, Zlatarova A, Ghai R, Kolev M, Olova N. et al. Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry. 2006;45:4093-104
28. Doni A, Peri G, Chieppa M, Allavena P, Pasqualini F, Vago L. et al. Production of the soluble pattern recognition receptor PTX3 by myeloid, but not plasmacytoid, dendritic cells. Eur J Immunol. 2003;33:2886-93
29. Garlanda C, Bottazzi B, Magrini E, Inforzato A, Mantovani A. PTX3, a Humoral Pattern Recognition Molecule, in Innate Immunity, Tissue Repair, and Cancer. Physiol Rev. 2018;98:623-39
30. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P. et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. 2015;160:700-14
31. Rodrigues PF, Matarazzo S, Maccarinelli F, Foglio E, Giacomini A, Silva Nunes JP. et al. Long Pentraxin 3-Mediated Fibroblast Growth Factor Trapping Impairs Fibrosarcoma Growth. Front Oncol. 2018;8:472
32. Wang JX, He YL, Zhu ST, Yang S, Zhang ST. Aberrant methylation of the 3q25 tumor suppressor gene PTX3 in human esophageal squamous cell carcinoma. World J Gastroenterol. 2011;17:4225-30
33. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C. et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67:9417-24
34. Ronca R, Giacomini A, Di Salle E, Coltrini D, Pagano K, Ragona L. et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell. 2015;28:225-39
35. Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. Identification of factor H-like protein 1 as the predominant complement regulator in Bruch's membrane: implications for age-related macular degeneration. J Immunol. 2014;193:4962-70
36. Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A. et al. Binding of the long pentraxin PTX3 to factor H: interacting domains and function in the regulation of complement activation. J Immunol. 2008;181:8433-40
37. Daigo K, Inforzato A, Barajon I, Garlanda C, Bottazzi B, Meri S. et al. Pentraxins in the activation and regulation of innate immunity. Immunol Rev. 2016;274:202-17
38. Woo JM, Kwon MY, Shin DY, Kang YH, Hwang N, Chung SW. Human retinal pigment epithelial cells express the long pentraxin PTX3. Mol Vis. 2013;19:303-10
39. Bonacina F, Moregola A, Porte R, Baragetti A, Bonavita E, Salatin A. et al. Pentraxin 3 deficiency protects from the metabolic inflammation associated to diet-induced obesity. Cardiovasc Res. 2019;115:1861-72
40. Elias I, Franckhauser S, Ferre T, Vila L, Tafuro S, Munoz S. et al. Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes. 2012;61:1801-13
41. Noma H, Mimura T, Yasuda K, Shimura M. Role of inflammation in diabetic macular edema. Ophthalmologica. 2014;232:127-35
42. Ramirez GA, Rovere-Querini P, Blasi M, Sartorelli S, Di Chio MC, Baldini M. et al. PTX3 Intercepts Vascular Inflammation in Systemic Immune-Mediated Diseases. Frontiers in Immunol. 2019;10:1135
43. Carrizzo A, Procaccini C, Lenzi P, Fusco C, Villa F, Migliarino S. et al. PTX3: an inflammatory protein modulating ultrastructure and bioenergetics of human endothelial cells. Immun Ageing. 2019;16:4
44. Napoleone E, Di Santo A, Bastone A, Peri G, Mantovani A, de Gaetano G. et al. Long pentraxin PTX3 upregulates tissue factor expression in human endothelial cells: a novel link between vascular inflammation and clotting activation. Arterioscler Thromb Vasc Biol. 2002;22:782-7
45. Shiraki A, Kotooka N, Komoda H, Hirase T, Oyama JI, Node K. Pentraxin-3 regulates the inflammatory activity of macrophages. Biochem Biophys Rep. 2016;5:290-5
46. Norata GD, Marchesi P, Pulakazhi Venu VK, Pasqualini F, Anselmo A, Moalli F. et al. Deficiency of the long pentraxin PTX3 promotes vascular inflammation and atherosclerosis. Circulation. 2009;120:699-708
47. Thurman JM, Nemenoff RA. Never make assumptions: the complicated role of complement in urinary tract infections. Kidney Int. 2016;90:469-71
48. Jaillon S, Moalli F, Ragnarsdottir B, Bonavita E, Puthia M, Riva F. et al. The humoral pattern recognition molecule PTX3 is a key component of innate immunity against urinary tract infection. Immunity. 2014;40:621-32
49. Yadav M, Zhang J, Fischer H, Huang W, Lutay N, Cirl C. et al. Inhibition of TIR domain signaling by TcpC: MyD88-dependent and independent effects on Escherichia coli virulence. PLoS Pathog. 2010;6:e1001120
50. Chromek M, Slamova Z, Bergman P, Kovacs L, Podracka L, Ehren I. et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. 2006;12:636-41
51. Patel DF, Peiro T, Shoemark A, Akthar S, Walker SA, Grabiec AM. et al. An extracellular matrix fragment drives epithelial remodeling and airway hyperresponsiveness. Sci Transl Med. 2018 10
52. Balhara J, Shan L, Zhang J, Muhuri A, Halayko AJ, Almiski MS. et al. Pentraxin 3 deletion aggravates allergic inflammation through a TH17-dominant phenotype and enhanced CD4 T-cell survival. J Allergy Clin Immunol. 2017;139:950-63.e9
53. Zhang J, Shan L, Koussih L, Redhu NS, Halayko AJ, Chakir J. et al. Pentraxin 3 (PTX3) expression in allergic asthmatic airways: role in airway smooth muscle migration and chemokine production. PLoS One. 2012;7:e34965
54. Zhang J, Koussih L, Shan L, Halayko AJ, Chen BK, Gounni AS. TNF up-regulates Pentraxin3 expression in human airway smooth muscle cells via JNK and ERK1/2 MAPK pathways. Allergy Asthma Clin Immunol. 2015;11:37
55. Cappuzzello C, Doni A, Dander E, Pasqualini F, Nebuloni M, Bottazzi B. et al. Mesenchymal Stromal Cell-Derived PTX3 Promotes Wound Healing via Fibrin Remodeling. J Invest Dermatol. 2016;136:293-300
56. Camozzi M, Zacchigna S, Rusnati M, Coltrini D, Ramirez-Correa G, Bottazzi B. et al. Pentraxin 3 inhibits fibroblast growth factor 2-dependent activation of smooth muscle cells in vitro and neointima formation in vivo. Arterioscler Thromb Vasc Biol. 2005;25:1837-42
57. Alam J, Jantan I, Bukhari SNA. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed Pharmacother. 2017;92:615-33
58. Bohler C, Radner H, Smolen JS, Aletaha D. Serological changes in the course of traditional and biological disease modifying therapy of rheumatoid arthritis. Ann Rheum Dis. 2013;72:241-4
59. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis - shaping the immunological landscape. Nat Rev Rheumatol. 2016;12:63-8
60. Kay J, Upchurch KS. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology. 2012;51(Suppl 6):vi5-9
61. Luchetti MM, Piccinini G, Mantovani A, Peri G, Matteucci C, Pomponio G. et al. Expression and production of the long pentraxin PTX3 in rheumatoid arthritis (RA). Clin Exp Immunol. 2000;119:196-202
62. Cieslik P, Hrycek A. Long pentraxin 3 (PTX3) in the light of its structure, mechanism of action and clinical implications. Autoimmunity. 2012;45:119-28
63. Presta M, Camozzi M, Salvatori G, Rusnati M. Role of the soluble pattern recognition receptor PTX3 in vascular biology. J Cell Mol Med. 2007;11:723-38
64. Yokota K, Miyoshi F, Sato K, Asanuma Y, Akiyama Y, Mimura T. Geranylgeranyl-pyrophosphate regulates secretion of pentraxin 3 and monocyte chemoattractant protein-1 from rheumatoid fibroblast-like synoviocytes in distinct manners. Clin Exp Rheumatol. 2011;29:43-9
65. Satomura K, Torigoshi T, Koga T, Maeda Y, Izumi Y, Jiuchi Y. et al. Serum amyloid A (SAA) induces pentraxin 3 (PTX3) production in rheumatoid synoviocytes. Mod Rheumatol. 2013;23:28-35
66. Li Y, Cai H, Fang W, Meng Q, Li J, Deng M. et al. Fibroblast growth factor 2 involved in the pathogenesis of synovial chondromatosis of temporomandibular joint. J Oral Pathol Med. 2014;43:388-94
67. Zhao S, Wang Y, Hou L, Wang Y, Xu N, Zhang N. Pentraxin 3 inhibits fibroblast growth factor 2 induced osteoclastogenesis in rheumatoid arthritis. Biomed Pharmacother. 2020;131:110628
68. Adan N, Guzman-Morales J, Ledesma-Colunga MG, Perales-Canales SI, Quintanar-Stephano A, Lopez-Barrera F. et al. Prolactin promotes cartilage survival and attenuates inflammation in inflammatory arthritis. J Clin Invest. 2013;123:3902-13
69. Haglund L, Bernier SM, Onnerfjord P, Recklies AD. Proteomic analysis of the LPS-induced stress response in rat chondrocytes reveals induction of innate immune response components in articular cartilage. Matrix Biol. 2008;27:107-18
70. Barksby HE, Hui W, Wappler I, Peters HH, Milner JM, Richards CD. et al. Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: implications for cartilage destruction and repair. Arthritis Rheum. 2006;54:540-50
71. Khan IM, Palmer EA, Archer CW. Fibroblast growth factor-2 induced chondrocyte cluster formation in experimentally wounded articular cartilage is blocked by soluble Jagged-1. Osteoarthritis Cartilage. 2010;18:208-19
72. Annese T, Ronca R, Tamma R, Giacomini A, Ruggieri S, Grillo E. et al. PTX3 Modulates Neovascularization and Immune Inflammatory Infiltrate in a Murine Model of Fibrosarcoma. Int J Mol Sci. 2019;20:4599
73. Choi B, Shin MK, Kim EY, Park JE, Lee H, Kim SW. et al. Elevated Neuropeptide Y in Endothelial Dysfunction Promotes Macrophage Infiltration and Smooth Muscle Foam Cell Formation. Front Immunol. 2019;10:1701
74. Batista NV, Barbagallo M, Oliveira VLS, Castro-Gomes T, Oliveira RDR, Louzada-Junior P. et al. The Long Pentraxin 3 Contributes to Joint Inflammation in Gout by Facilitating the Phagocytosis of Monosodium Urate Crystals. J Immunol. 2019;202:1807-14
75. Ambarus CA, Noordenbos T, de Hair MJ, Tak PP, Baeten DL. Intimal lining layer macrophages but not synovial sublining macrophages display an IL-10 polarized-like phenotype in chronic synovitis. Arthritis Res Ther. 2012;14:R74
76. Lai HY, Hsu LW, Tsai HH, Lo YC, Yang SH, Liu PY. et al. CCAAT/enhancer-binding protein delta promotes intracellular lipid accumulation in M1 macrophages of vascular lesions. Cardiovasc Res. 2017;113:1376-88
77. Hsiao YW, Li CF, Chi JY, Tseng JT, Chang Y, Hsu LJ. et al. CCAAT/enhancer binding protein delta in macrophages contributes to immunosuppression and inhibits phagocytosis in nasopharyngeal carcinoma. Sci Signal. 2013;6:ra59
78. Baruah P, Dumitriu IE, Peri G, Russo V, Mantovani A, Manfredi AA. et al. The tissue pentraxin PTX3 limits C1q-mediated complement activation and phagocytosis of apoptotic cells by dendritic cells. J Leukoc Biol. 2006;80:87-95
79. Doni A, Garlanda C, Bottazzi B, Meri S, Garred P, Mantovani A. Interactions of the humoral pattern recognition molecule PTX3 with the complement system. Immunobiology. 2012;217:1122-8
80. Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634-5
81. Rodewohl A, Scholbach J, Leichsenring A, Koberle M, Lange F. Age-dependent cellular reactions of the human immune system of humanized NOD scid gamma mice on LPS stimulus. Innate Immun. 2017;23:258-75
82. Lee EJ, Song DH, Kim YJ, Choi B, Chung YH, Kim SM. et al. PTX3 stimulates osteoclastogenesis by increasing osteoblast RANKL production. J Cell Physiol. 2014;229:1744-52
83. Teitelbaum SL. Osteoclasts, integrins, and osteoporosis. J Bone Miner Metab. 2000;18:344-9
84. Razvina O, Jiang S, Matsubara K, Ohashi R, Hasegawa G, Aoyama T. et al. Differential expression of pentraxin 3 in neutrophils. Exp Mol Pathol. 2015;98:33-40
85. Folwaczny M, Karnesi E, Berger T, Paschos E. Clinical association between chronic periodontitis and the leukocyte extravasation inhibitors developmental endothelial locus-1 and pentraxin-3. Eur J Oral Sci. 2017;125:258-64
86. dell'Oglio MP, Simone S, Ciccone M, Corciulo R, Gesualdo M, Zito A. et al. Neutrophil-dependent pentraxin-3 and reactive oxygen species production modulate endothelial dysfunction in haemodialysis patients. Nephrol Dial Transplant. 2017;32:1540-9
87. Cunha C, Aversa F, Lacerda JF, Busca A, Kurzai O, Grube M. et al. Genetic PTX3 deficiency and aspergillosis in stem-cell transplantation. N Engl J Med. 2014;370:421-32
88. van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM. et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum. 2011;63:73-83
89. Turner M, Feldmann M. Comparison of patterns of expression of tumour necrosis factor, lymphotoxin and interleukin-6 mRNA. Biochem Biophys Res Commun. 1988;153:1144-51
90. Yang S, Wang J, Brand DD, Zheng SG. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front Immunol. 2018;9:784
91. Detert J, Bastian H, Listing J, Weiß A, Wassenberg S, Liebhaber A. et al. Induction therapy with adalimumab plus methotrexate for 24 weeks followed by methotrexate monotherapy up to week 48 versus methotrexate therapy alone for DMARD-naive patients with early rheumatoid arthritis: HIT HARD, an investigator-initiated study. Ann Rheum Dis. 2013;72:844-50
92. Kvamme MK, Lie E, Uhlig T, Moger TA, Kvien TK, Kristiansen IS. Cost-effectiveness of TNF inhibitors vs synthetic disease-modifying antirheumatic drugs in patients with rheumatoid arthritis: a Markov model study based on two longitudinal observational studies. Rheumatology. 2015;54:1226-35
93. Loh C, Park SH, Lee A, Yuan R, Ivashkiv LB, Kalliolias GD. TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: transcriptomic and epigenomic analysis. Ann Rheum Dis. 2019;78:1205-14
94. Kim Y, Park JS, Park HJ, Kim MK, Kim YI, Bae SK. et al. Pentraxin 3 Modulates the Inflammatory Response in Human Dental Pulp Cells. J Endod. 2018;44:1826-31
95. Souza DG, Amaral FA, Fagundes CT, Coelho FM, Arantes RM, Sousa LP. et al. The long pentraxin PTX3 is crucial for tissue inflammation after intestinal ischemia and reperfusion in mice. Am J Pathol. 2009;174:1309-18
96. Ruscitti P, Cipriani P, Carubbi F, Liakouli V, Zazzeroni F, Di Benedetto P. et al. The role of IL-1beta in the bone loss during rheumatic diseases. Mediators Inflamm. 2015;2015:782382
97. Schett G, Dayer JM, Manger B. Interleukin-1 function and role in rheumatic disease. Nat Rev Rheumatol. 2016;12:14-24
98. Ridgley LA, Anderson AE, Pratt AG. What are the dominant cytokines in early rheumatoid arthritis? Curr Opin Rheumatol. 2018;30:207-14
99. Neiman JA, Raman R, Chan V, Rhoads MG, Raredon MS, Velazquez JJ. et al. Photopatterning of hydrogel scaffolds coupled to filter materials using stereolithography for perfused 3D culture of hepatocytes. Biotechnol Bioeng. 2015;112:777-87
100. Fonseca JE, Santos MJ, Canhao H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8:538-42
101. Dessein PH, Joffe BI, Singh S. Biomarkers of endothelial dysfunction, cardiovascular risk factors and atherosclerosis in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R634-43
102. Qu D, Liu J, Lau CW, Huang Y. IL-6 in diabetes and cardiovascular complications. Br J Pharmacol. 2014;171:3595-603
103. Tekeoglu I, Harman H, Sag S, Altindis M, Kamanli A, Nas K. Levels of serum pentraxin 3, IL-6, fetuin A and insulin in patients with rheumatoid arthritis. Cytokine. 2016;83:171-5
104. Sag MS, Sag S, Tekeoglu I, Kamanli A, Nas K, Harman H. The Relationship Between Serum Pentraxine 3 Levels and Hematological Markers in Patients With Rheumatoid Arthritis. Arch Rheumatol. 2018;33:39-44
105. Pan F, Xiang H, Yan J, Hong L, Zhang L, Liu Y. et al. Dendritic Cells from Rheumatoid Arthritis Patient Peripheral Blood Induce Th17 Cell Differentiation via miR-363/Integrin alphav/TGF-beta Axis. Scand J Immunol. 2017;85:441-9
106. Xu X, Zheng L, Bian Q, Xie L, Liu W, Zhen G. et al. Aberrant Activation of TGF-beta in Subchondral Bone at the Onset of Rheumatoid Arthritis Joint Destruction. J Bone Miner Res. 2015;30:2033-43
107. Schmitt-Sody M, Metz P, Gottschalk O, Birkenmaier C, Zysk S, Veihelmann A. et al. Platelet P-selectin is significantly involved in leukocyte-endothelial cell interaction in murine antigen-induced arthritis. Platelets. 2007;18:365-72
108. Fang J, Chen Q, He B, Cai J, Yao Y, Cai Y. et al. Tanshinone IIA attenuates TNF-alpha induced PTX3 expression and monocyte adhesion to endothelial cells through the p38/NF-kappaB pathway. Food Chem Toxicol. 2018;121:622-30
109. Carrizzo A, Lenzi P, Procaccini C, Damato A, Biagioni F, Ambrosio M. et al. Pentraxin 3 Induces Vascular Endothelial Dysfunction Through a P-selectin/Matrix Metalloproteinase-1 Pathway. Circulation. 2015;131:1495-505 discussion 505
110. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V. et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol. 2010;11:328-34
Author contact
![]() Corresponding author: Xing Xiao, Department of Orthopedic Surgery, The First Affiliated Hospital of Shandong First Medical University, Jinan 250014, Shandong, China. E-mail: 1883com.cn.
Corresponding author: Xing Xiao, Department of Orthopedic Surgery, The First Affiliated Hospital of Shandong First Medical University, Jinan 250014, Shandong, China. E-mail: 1883com.cn.