Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(8):1810-1823. doi:10.7150/ijms.51654 This issue Cite
Research Paper
Hirsutella sinensis mycelium regulates autophagy of alveolar macrophages via TLR4/NF-κB signaling pathway
Juanhua Fu1*, Li Lu1*, Haining Wang1*, Yayi Hou1,2 ![]() , Huan Dou1,2
, Huan Dou1,2 ![]()
1. The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing 210093, China.
2. Jiangsu Key Laboratory of Molecular Medicine, Nanjing University, Nanjing 210093, China.
* These authors contributed equally to this work.
Received 2020-8-7; Accepted 2021-2-1; Published 2021-2-18
Abstract

Background: Hirsutella sinensis mycelium (HSM) has potent anti-pulmonary fibrotic activities and has been proposed as an effective treatment for idiopathic pulmonary fibrosis. Macrophages are the main innate immune cells in the lung tissue, playing key roles in pulmonary fibrosis repair and homeostasis. Excessive macrophage autophagy plays a vital role in pulmonary fibrosis. The protective effect of HSM on macrophages of bleomycin (BLM)-induced pulmonary fibrotic mice remain unclear.
Methods: In this study, we collected lung tissue and bronchoalveolar lavage fluid (BALF) samples from pulmonary fibrotic mice. Meanwhile, alveolar macrophages were isolated and murine macrophage RAW264.7 cell line was cultured for further study of HSM autophagy.
Results: First, we found that HSM decreased the number of autophagosomes, as well as the levels of LC3B and ATG5, and increased the protein level of P62 during the development of pulmonary fibrosis. Meanwhile, HSM reduced alveolar macrophages infiltration into the BALF and inhibited their accumulation in the fibrotic lung tissue. Flow cytometry analysis showed that HSM administration inhibited the autophagy marker LC3B expression in CD11bloCD11chi alveolar macrophages in BLM-induced lung fibrosis without affecting CD11bhiCD11clo interstitial macrophages. Transmission electron microscopy and JC-1 staining for mitochondrial membrane potential of alveolar macrophages also verified that the HSM significantly decreased autophagy in the alveolar macrophages of BLM-treated mice. In vitro, autophagosomes-lysosome fusion inhibitor chloroquine (CQ) was pre-incubated with RAW264.7 cells, and HSM reduced CQ-induced autophagosomes accumulation. TLR4 signaling inhibitor CLI095 reversed the above effects, suggesting HSM could reduce the cumulation of autophagosomes dependent on TLR4. Furthermore, lipopolysaccharide (LPS)-stimulated TLR4-related autophagy was significantly inhibited by HSM treatment. In addition, the protein expressions of TLR4 and phospho-NF-κB p65 were markedly inhibited in cells treated with HSM.
Conclusions: These results indicated that HSM could inhibit the autophagy of alveolar macrophages through TLR4/NF-κB signaling pathway to achieve anti-fibrotic effect.
Keywords: Hirsutella sinensis mycelium, pulmonary fibrosis, alveolar macrophage, autophagy, TLR4 signal pathway.
Introduction
Idiopathic pulmonary fibrosis (IPF), the archetypal fibrotic lung disease, is a serious disorder with unknown cause and increasing incidence [1-3]. The median survival after diagnosis is estimated to be 2-5 years [4, 5]. To treat IPF, Pirfenidone and Nintedanib have been included in an update of clinical treatment guidelines in 2015 [2]. However, these two anti-fibrotic medications are conditionally recommended, and have not been shown to reduce all-cause mortality in sufficiently powered studies [6, 7]. A recent conditionally recommended treatment for IPF is anti-acid therapy, proton pump inhibitors or histamine-2-receptor antagonists [8]. However, the scientific evidence of their potential beneficial effects on survival remains uncertain [9]. Thus, treatment strategies for the pathogenesis of IPF are needed.
In the studies on effective IPF treatments, Hirsutella sinensis mycelium (HSM), which is an asexual stage of Cordyceps sinensis, has exhibited potent anti-fibrotic activity [10-14]. Huang TT et al. reported that an ethanol extract of HSM inhibited transforming growth factor-β1 (TGF-β1)-induced differentiation of lung fibroblasts into myofibroblasts and reactive oxygen species production in lung epithelial cells [10]. Our laboratory also observed that HSM suppressed mTOR activation stimulated by recombinant TGF-β1 in A549 cells during fibroblast-myofibroblast trans-differentiation [11]. These in vitro studies were helpful to analyze the anti-fibrosis therapeutic mechanism of HSM, but the exact cause remains unknown. The bleomycin (BLM)-induced infiltration of inflammatory cells into bronchoalveolar lavage fluid (BALF) was found to be reduced by HSM ethanol extract treatment [10]. Interestingly, HSM relieved fibrotic damage, accompanied by a decrease in macrophage counts in our previous studies [11, 15]. Hence, it is important to examine whether the HSM effect on pulmonary fibrosis is related to macrophages.
Macrophages are the main innate immune cells in the lung tissue [16], and play key roles in the pulmonary fibrosis repair and homeostasis [17]. Collagen deposition can be regulated by macrophages, thereby participating in the processing of extracellular matrix [18]. Macrophages have been demonstrated to participate in regulating the survival of myofibroblasts [19]. Importantly, it has been demonstrated in a rodent BLM model that pulmonary macrophages could be the predominant source of TGF-β, a prominent fibrosis-inducing molecule [20-22]. Moreover, patients who developed an accelerated form of fibrotic lung disease were found to have dysregulated alveolar macrophages [23]. Some scientists have even suggested that targeting macrophages might have a resolution-promoting role during pulmonary fibrosis [24].
Autophagy is a conserved intracellular degradation pathway, and its disruption compromises homeostasis, which leads to pulmonary diseases [25-27]. Increased Light Chain 3 (LC3B) expression in alveolar macrophages was observed in IPF patients, suggesting that excessive macrophage autophagy worsened the disease [28]. Similarly, autophagy-related proteins, such as Beclin1 and P62, were expressed in macrophages from the fibrosis areas of paraquat-induced pulmonary fibrosis [29]. Autophagosomes accumulated in alveolar macrophages of human silicosis, and promoted macrophage activation to induce the development of fibrosis [30, 31]. Taken together, these data suggested that macrophage autophagy might play a vital role in pulmonary fibrosis.
The current study aimed to investigate the relationship between HSM alleviation of pulmonary fibrosis and macrophage autophagy, as well as the underlying molecular mechanism. The results showed that HSM could effectively inhibit excessive macrophage autophagy in BLM-induced pulmonary fibrosis mouse model, and the TLR4/NF-κB signaling pathway was required for HSM effect on macrophage autophagy.
Materials and Methods
Chemicals and reagents
HSM was obtained from Nanjing Zhongke Group (Nanjing, China), and the HSM solution was prepared as previously described [11]. BLM was purchased from Hisun Pharmaceutical Co., Ltd. (Zhejiang, China). Chloroquine (CQ), lipopolysaccharide (LPS), collagenase I and collagenase IV were obtained from Sigma (St. Louis, MO, USA). DNase I was purchased from Roche (Switzerland, UK). Dulbecco's Modified Eagle's Medium (DMEM) and Roswell Park Memorial Institute (RPMI) 1640 medium were obtained from Hyclone Laboratories (South Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Gibco (Grand Island, NY, USA). JC-1 was obtained from Fcmacs (Nanjing, China). TLR4 signaling inhibitor CLI095 was purchased from Invitrogen (Carlsbad, CA, USA). The antibodies used in this study were rabbit anti-GAPDH (Good HERE, Hefei, China), rabbit anti-LC3B (Cell Signaling Technology, MA, USA), rabbit anti-P62 (Cell Signaling Technology, MA, USA), rabbit anti-ATG5 (Cell Signaling Technology, MA, USA), rabbit anti-NF-κB p65 (Cell Signaling Technology, MA, USA), rabbit anti-phospho-NF-κB p65 (Cell Signaling Technology, MA, USA), rabbit anti-ERK (Cell Signaling Technology, MA, USA), rabbit anti-phospho-ERK (Cell Signaling Technology, MA, USA), rabbit anti-IRF 3 (Cell Signaling Technology, MA, USA), rabbit anti-phospho-IRF 3 (Cell Signaling Technology, MA, USA), PerCP anti-mouse CD45 (clone number: 30-F11) (Biolegend, CA, USA), APC anti-mouse CD11c (clone number: N418) (eBioscience, CA, USA), FITC rat anti-CD11b (clone number: M1/70) (BD Biosciences, CA, USA), PE anti-rabbit LC3B (clone number: D11) (Cell Signaling Technology, MA, USA), goat anti-rabbit IgG (H+L) (Fcmacs, Nanjing, China) and goat anti-rabbit IgG (Fcmacs, Nanjing, China).
Mouse model of pulmonary fibrosis
Specific pathogen-free (SPF) male C57BL/6 mice (aged 7-8- weeks, weighting 20 ± 2 g) were purchased from GemPharmatech [SCXK (SU) 2018-0008, Nanjing, China]. All animal experiments were strictly in accordance with the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committee of Nanjing University (Nanjing, China). All mice were housed in SPF condition with a 12:12 h light-dark cycle, and received water and food ad libitum. Mice were acclimated for at least a week before use.
The experimental process is shown in Figure 1A. A total of 40 male C57BL/6 mice were randomly divided into four groups: (i) CON group, (ii) HSM group, (iii) BLM group, (iv) BLM+HSM (200 mg/kg) group. The BLM stock solution was prepared by dissolving the compound in sterile saline and storing small aliquots at 4 °C. BLM (3 mg/kg) was intratracheally administered to induce pulmonary fibrosis on day 0. The CON and HSM groups were treated with the equivalent volume of vehicle (saline). From day 7 to 27 after the BLM administration, HSM diluted in 500μl saline or vehicle control (saline) was administered by oral gavage once daily. On day 28, all the mice were sacrificed.
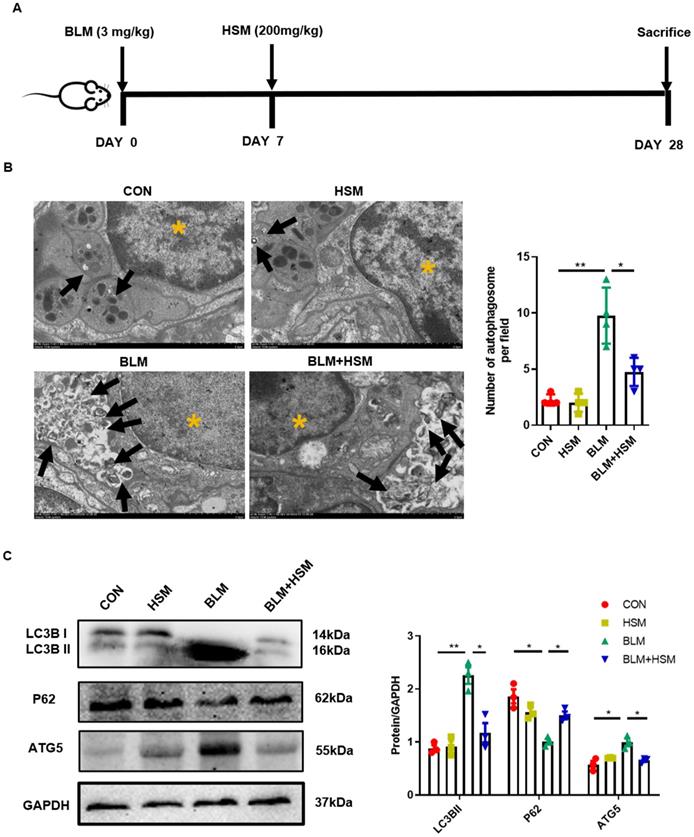
HSM down-regulates excessive autophagy in BLM-induced pulmonary fibrosis mice. (A) In vivo experimental design: Mice were intratracheally administered with BLM (3 mg/kg in 100 μl saline) on day 0, and HSM (200 mg/kg in 500 μl saline) was orally administered after the BLM administration from day 7 to day 27. On day 28, the mice were sacrificed and the corresponding samples were collected. (B) Transmission electron microscopy image of mouse lung tissue. Black arrow: autophagosome; yellow asterisk: cell nucleus; scale bar: 2 μm. The black bars represent the mean ± SEM values between the treated groups analyzed, n=4. (C) Analysis of the changes of LC3B, P62 and ATG5 proteins in mice of each group by Western blotting, n=3. Data are presented as means ± SEM of at least three separate experiments. *P < 0.05, **P < 0.01.
Sample collection
After the mice were sacrificed, the lungs and part of the trachea were harvested. The anterior lobe of the right lung was inflated, fixed in 4% paraformaldehyde for 24 h, and then paraffin embedded. The middle lobe of the right lung was collected for transmission electron microscopy (TEM) observation. After the ligation, 1 mL of PBS was injected into the lungs through the catheter, the lungs were washed five times and lavage fluids were collected for subsequent detection. After the lavage, lower lobe of left lung tissue was removed for flow cytometry analysis, gene and protein testing.
Isolation of alveolar macrophages and transmission electron microscopy observation
BALF was centrifuged (300 g, 5 min, 4 °C) to obtain a pellet, the pellet was lysed with erythrocyte lysate, then resuspended in RPMI-1640 medium (containing 10% FBS and 1% penicillin streptomycin). Cells (1×106 cells/well) were seeded in 6-well plates overnight. On the second day, the non-adherent cells were discarded, and 10 ml of EDTA-2Na was added to detach the adherent cells. After 30 min, the adherent cells were recovered, the supernatant was centrifuged, and washed three times with RPMI 1640 medium to obtain purified alveolar macrophages.
The purified alveolar macrophages and the lung tissue were fixed with phosphate buffer solution at pH=7.4 containing 2% paraformaldehyde and 2.5% glutaraldehyde. Alveolar macrophages were sliced at 70-80 nm with a diamond knife, and the slices were placed on a copper grid. Tissue samples were then fixed with 1% phosphate-buffered osmium tetroxide, embedded in Spurr's resin, and then sliced. After sectioning, the slices were stained with 1% uranyl acetate and 0.2% lead citrate. The slices were sent to Nanjing Google Bio Company (Nanjing, China) for imaging and observing the number of autophagosomes.
Cell culture and viability analysis
The murine monocyte/macrophage RAW264.7 cell line was cultured in a culture flask containing 10% FBS in DMEM high glucose medium and placed in an incubator at 37°C and 5% CO2 saturated humidity. In the in vitro experiment, RAW cells were pre-treated with CQ (20 μM) for 2h, followed by 16 μg/ml HSM treatment for 16 h. Similarly, LPS (0.1 μg/ml) and CLI095 (100 μg/ml) were used to treat cells for 2 h before using HSM (16 μg/ml and 160 μg/ml).
RAW264.7 cells (2 x 104 cells/well) were seeded in 96-well plates to assess cell viability. After overnight incubation, cells were incubated with various concentrations of HSM (6.25-1600 μg/ml) for 48 h. Subsequently, 10 μl of CCK-8 solution was added to each well and incubated for an additional 2 h. The absorbance at 450 nm was measured by a microplate reader (Bio Tek, Winooski, VT, USA). The effect of HSM on the viability of RAW264.7 cells was shown as ratios of the treated group to control group, and repeated for three times.
Western blotting
Lung tissues and cells were lysed in ice-cold RIPA lysis buffer containing protease and phosphatase inhibitors. The supernatants were collected by centrifugation and quantitated through BCA protein assay kit (Beyotime Institute of Biotechnology). An equivalent amount of 30 μg protein was separated by SDS-PAGE, transferred to a 0.22 μm PVDF membrane (Millipore Co, Bedford, MA, USA), and blocked in 5% BSA for 2 h at room temperature. The PVDF membrane was then incubated with the primary antibody overnight at 4 °C followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibodies for 2 h in the dark. An ECL kit (SupersignalTM West Pico PLUS, Thermo, USA) was used to detect the protein bands, and the grey level of proteins were calculated by the Image J software (National Institutes of Health, Bethesda, MD, USA). We diluted the antibodies as follows: rabbit anti-GAPDH (1:1000), rabbit anti-LC3B (1:1000), rabbit anti-P62 (1:1000), rabbit anti-ATG5 (1:1000), rabbit anti-NF-κB p65 (1:1000), rabbit anti-phospho-NF-κB p65 (1:1000), rabbit anti-ERK p65 (1:1000), rabbit anti-phospho-ERK (1:1000), rabbit anti-IRF 3 (1:1000), rabbit anti-phospho-IRF 3 (1:1000) and goat anti-rabbit IgG (1:4000). In the experiment, GAPDH was used as a protein loading control.
Extraction of total RNA and quantitative real-time PCR
Total RNA was isolated from RAW264.7 cells or lung tissues using the Trizol reagent (Invitrogen, USA). Thereafter, 1 mg RNA was reverse-transcribed into cDNA by HiScript® II Q PT SuperMix for qPCR kit (Vazyme Biotech, Nanjing, China). Next, qRT-PCR was performed on the BIOER Line Gene 9640 (Hangzhou, China). The relative expression of each gene was calculated by the 2-ΔΔCt method, repeated ≥3 times. The mRNA levels were normalized to that of GAPDH. The primers were synthesized by Springen (Nanjing, China). The 5′—3′ primer sequences were as follows:
m-LC3B: forward primer TGACTCACCTTGTGGTCCTAA, reverse primer CTTCCCAGAATCCAGTCTTTCC;
m-GAPDH: forward primer AGGTCGGTGTGAACGGATTTG, reverse primer TGTAGACCATGTAGTTGAGGTCA.
Flow cytometric analysis
ACK buffer was used to lyse the excess red blood cells in BALF. For the lung tissue, pulmonary lobes were mechanically disrupted in gentleMACSTM Dissociator (Miltenyi Biotec, Germany), digested in RPMI 1640 medium containing collagenase I (3 mg/ml), collagenase IV (3 mg/ml) and DNase I (2.5 μl) and filtered through a 70 μm cell strainer to remove major tissue fragments. Single-cell suspensions were then fixed with 4% paraformaldehyde and stained with Percp-conjugated CD45, APC-conjugated CD11c and FITC-conjugated CD11b antibodies for 30 min at 4 °C in the dark. After the cells were fixed and permeabilized, the intracellular antibody LC3B-PE was stained. Accuri C6 (BD Biosciences, CA, USA) was used to detect fluorescence, and FlowJo software (TreeStar, Ashland, OR, USA) was applied for data analysis.
Immunofluorescence
RAW264.7 cells were cultured on a glass coverslip. After various treatments, the cells were fixed with 4% paraformaldehyde at room temperature for 30 min, and 0.1% Triton X-100 at room temperature for 15 min to increase the permeability of the cell membrane. After blocking with 3% bovine serum albumin at room temperature for 60 min, the cells were incubated with rabbit anti-LC3B antibody at 4°C overnight followed by incubation with the goat anti-rabbit IgG (H+L) secondary antibody for 2 h at room temperature in the dark. The cells were stained with DAPI (Fcmacs, Nanjing, China), and finally the coverslip was fixed with neutral glue. Then the coverslip was observed under a FV10i confocal microscope (OLYMPUS, Japan).
Statistical analysis
All data were expressed as mean ± S.E.M. Statistical analysis was performed using Prism8 software, and one-way Analysis of Variance (ANOVA) was used for comparison between groups. A p-value <0.05 was considered statistically significant. The experimental results were repeated three more times, and each data point represents the mean of at least three samples.
Results
HSM down-regulates excessive autophagy in BLM-induced pulmonary fibrosis mice
As shown in Fig. 1A, we used BLM-induced pulmonary fibrosis model, and subsequently HSM was used for drug therapy (for details, please refer to the Materials and Methods section). In our previous studies, HSM was found to mitigate lung injury and fibrosis [11, 32], which prompted us to explore the underlying mechanism. Autophagy is involved in the pathological progression of IPF [33, 34]. Autophagosome formation and autophagic flux were reported to significantly increase in patients with lung fibrosis as well as BLM-induced pulmonary fibrosis animals [35, 36]. We first applied TEM to observe the autophagosome inside the lung tissue slices, as a golden standard [37]. We quantified the TEM sections and found that the number of autophagosomes were significantly increased in the BLM-induced fibrosis lung tissue, compared to uninjured saline control lung. After the HSM treatment, autophagosome formation per tissue area was significantly reduced compared with the BLM model group (Fig. 1B). LC3B hydrolyzes into cytosolic LC3B-I and lipid autophagosome membrane-bound LC3B-II. The conversion of LC3B-I to LC3B-II is an essential event for autophagosome formation. An increase in LC3B-II level is, therefore, a marker of activated autophagy [38-40]. As an LC3-interacting protein, P62 begins to degrade followed by the progression of autophagy [41]. ATG5 is another key autophagy protein required for conjugation of the ubiquitin-like protein LC3 to the phagophore [42]. Next, LC3B, P62 and ATG5 in lung tissue extracts were analyzed by western blot. LC3B-II level was remarkably increased at 28 days post BLM administration compared to saline control, while a decrease in P62 level was also observed (Fig. 1C). Hence, the autophagy level in the BLM model group was higher, consistent with a previous report [35]. Compared to tissue from the BLM group, significantly lower level of LC3B-II was observed in the BLM+HSM group, and the expression level of P62 was also up-regulated. Moreover, ATG5 expression showed a similar variation as LC3B-II between different groups. These results indicated that HSM regulates autophagic activity in the pathogenesis of pulmonary fibrosis.
HSM reduces the number of alveolar macrophages in the fibrotic lung
Since inflammatory cells in BALF interact with lung tissue cells to play an important role in pulmonary fibrosis, we attempted to identify whether these cells were associated with HSM regulation of autophagic activity in BLM-induced mice [43, 44]. Therefore, H&E staining was used to examine the changes in the numbers of various inflammatory cells in BALF. Based on the morphology of various cells, administration of BLM caused extensive infiltration of inflammatory cells, as shown by significant increases in total cells, macrophages, lymphocytes, eosinophils and neutrophils (Fig. 2A). As expected, HSM reversed the total cell and leukocyte accumulation in BALF of BLM-treated mice (Fig. 2A). Among these immune cells, macrophages attracted our attention because of their huge advantages in quantity. Next, the effect of HSM on macrophages was further analyzed by flow cytometry.
HSM reduces the number of alveolar macrophages in the fibrotic lung. (A) BALF was centrifuged and coated to count the changes of total cells, macrophages, lymphocytes, eosinophils and neutrophils. The CON, HSM, BLM and BLM+HSM mainly display representative morphological images of macrophages, lymphocytes, eosinophils and neutrophils. Scale: 10 μm, magnification: x200, n=4. (B) Gating strategy mainly used to identify macrophages in BALF and lung tissue of mice. The differential expression of CD11b and CD11c was used to distinguish pulmonary macrophages. (C) Effect of HSM on the number of total cells, CD45+ cells and alveolar macrophages in BALF, n=6. (D) Effect of HSM on the number of total cells, CD45+ cells, alveolar macrophages and interstitial macrophages in lung tissue, n=10. Data are presented as means ± SEM of at least three separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
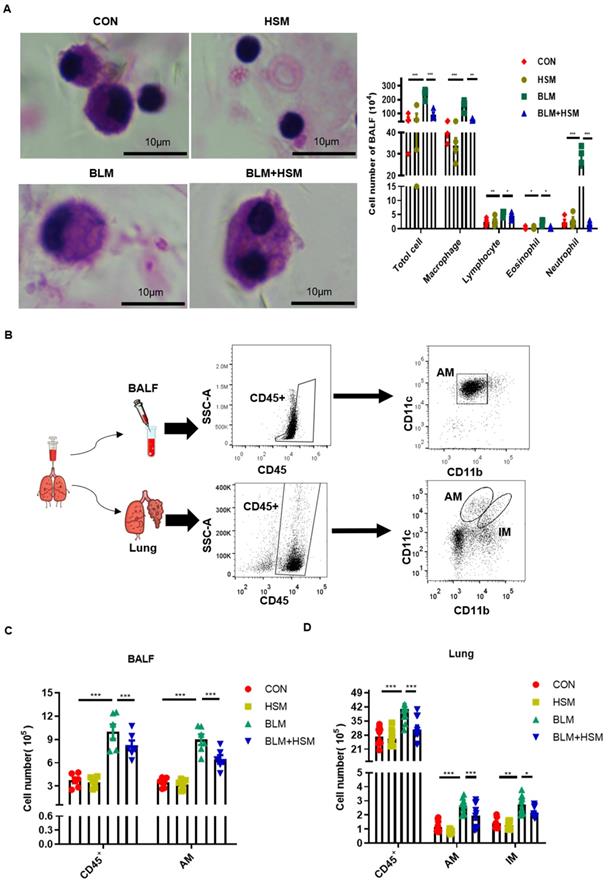
Pulmonary macrophages are mainly divided into two groups: alveolar macrophages (AMs) located in the airway space [45], and interstitial macrophages (IMs) residing in the lung parenchyma [45, 46]. AMs and IMs can be distinguished by the differential expression of their surface markers: integrin CD11b and CD11c. AMs express low levels of CD11b and high levels of CD11c, while IMs express high levels of CD11b and low levels of CD11c [47]. Therefore, flow cytometry was applied to detect cell surface proteins as previously described, and the specific experimental process is shown in Fig. 2B [48]. The number of total CD45+ leukocytes in the BALF of BLM-treated mice was higher than the control mice. In contrast, the treatment with HSM reduced the number of infiltrated CD45+ cells (Fig. 2C), which verified the results of cell smear. The AMs in BALF accounted for about 90% of CD45+ cells, and CD11bloCD11chi AMs increased on day 28 following BLM treatment, whereas HSM treatment reduced 35% of AM infiltration (Fig. 2C). In addition, macrophages were identified in the lung tissue. In contrast to BALF, CD11bloCD11chi AMs and CD11bhiCD11clo IMs had very small proportions of CD45+ cells, which were around 4%. However, the numbers of AMs and IMs showed that HSM considerably blocked the accumulation of these tissue macrophages (Fig. 2D). Based on these results, we considered that HSM reduces infiltration of AMs into the BALF and inhibits their accumulation in the lung tissue.
HSM inhibits autophagy of alveolar macrophages in fibrotic mice
Excessive macrophage autophagy was thought to be related to pulmonary fibrosis [30]. To evaluate whether the HSM effect on autophagy was associated with AMs, we monitored the LC3B protein level in leukocytes by flow cytometry. First, we observed that the total number of LC3B-expressing cells in lung tissue was significantly higher in the BLM model group compared with the saline-treated control group (Fig. 3A). In fibrotic lungs, BLM administration induced CD45+ leukocytes accounting for nearly 30% of the LC3B+ cells and these effects were significantly suppressed by HSM treatment (Fig. 3A). Furthermore, LC3B+ cells were elevated in AMs and IMs in the BLM group, which was reversed by HSM only in AMs, but not in IMs (Fig. 3A). Next, flow cytometry was used to evaluate the mean fluorescence intensity (MFI) of LC3B in lung tissue, MFI showed the same variation with the LC3B+ positive cell numbers (Fig. 3B). These data suggested that there was an inhibition effect of HSM on autophagy in AMs. Meanwhile, the change of LC3B expression was consistent with autophagy data of lung tissue in BALF, which was rich in over 90% AMs (Fig. 3C). Moreover, LC3B MFI in AMs from BALF samples in HSM-treatment group was further explained to be significantly downregulated compared with BLM model group (Fig. 3D). These results indicated that HSM inhibited LC3B expression in AMs in BLM-induced lung fibrosis, without affecting IMs.
HSM inhibits autophagy of alveolar macrophages in fibrotic mice. (A) The effect of HSM on the count of LC3B positive cells (total cells, CD45+ leukocytes, alveolar macrophages and interstitial macrophages) in lung tissue, n=10. (B) The effect of HSM on LC3B MFI of total cells, CD45+ leukocytes, alveolar macrophages and interstitial macrophages in lung tissue, n=10. (C) The effect of HSM on the number of LC3B+ total cells, CD45+ leukocytes and alveolar macrophages in BALF, n=6. (D) The effect of HSM on LC3B MFI of alveolar macrophages in BALF, n=6. (E) Primary alveolar macrophages were extracted from BALF of each group of mice, and the changes of autophagosomes were observed by transmission electron microscopy, n=4. Black arrow: autophagosome; yellow asterisk: cell nucleus; scale bar: 2 μm. Data are presented as means ± SEM of at least three separate experiments. **P < 0.01, ***P < 0.001.
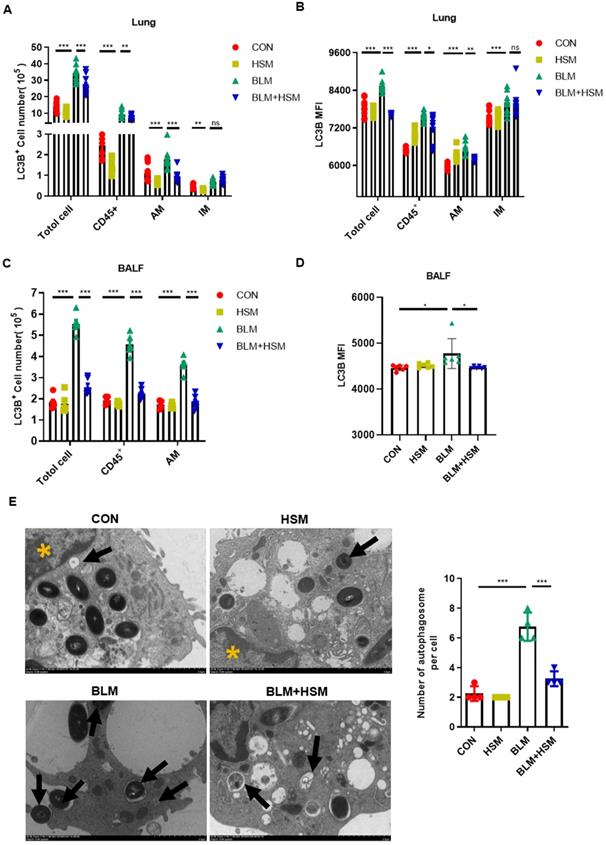
In order to verify the effect of HSM on autophagy of AMs, we separated AMs from BALF of different treatment groups and examined the changes of autophagosomes by TEM (Fig. 3E). The number of autophagosome in AMs from the BLM model group were increased, and this effect was attenuated by treatment with HSM (Fig. 3E). Besides, studies have reported autophagy can maintain mitochondrial homeostasis and promote cell survival under oxidative stress [49, 50]. JC-1 staining examined by flow cytometry was used to detect mitochondrial membrane potential (MMP) in AMs. The BLM model group had increased aggregated JC-1 in isolated AMs, HSM could promote more JC-1 monomer in AMs from fibrotic mice (Fig. S1A). These results suggested that HSM remarkably enhanced the collapse of the MMP. Bcl2 gene was identified to play a key role in cell survival and inhibition of apoptosis [51]. Bcl2 staining indicated that there were reduced F4/80+ apoptotic macrophages in the model group, HSM reversed the inhibition of apoptotic cells in the lungs (Fig. S1B). JC-1 aggregates are suggestive of a resistance to apoptosis, and double staining of Bcl2 and F4/80 did prove HSM render alveolar macrophages less resistant to apoptosis. These results together reminded us HSM significantly decreased autophagy in the AMs of BLM-treated mice.
HSM regulates autophagosome accumulation in macrophages dependent on TLR4 activity
After determining the effect of HSM on autophagy in AMs in vivo, RAW264.7 murine macrophage cells were used to study the autophagy regulatory effect of HSM in vitro. We first analyzed the cytotoxicity of HSM in RAW264.7 cells by CCK-8 assay. The results showed that HSM did not affect the cell viability at concentrations less than 400 μg/ml (Fig. 4A). Thus, we chose 16 μg/ml (HSM1) and 160 μg/ml (HSM2) as the final working concentration for the following experiments.
HSM regulates autophagosomes accumulation in macrophages dependent on TLR4 activity. (A) The effect of different concentrations HSM on the activity of RAW264.7 cells was analyzed by CCK-8 assay. (B) In vitro experiment to explore the effect of HSM on CQ-induced autophagy flux accumulation. RAW264.7 cells were pretreated with CQ (20 μM) for 2 h and then treated with or without HSM (16 μg/ml) for 16 h. Representative immunofluorescence image of LC3B in RAW264.7 cells treated with CQ and HSM. Scale bar: 40 μm. (C) The effect of HSM on the expression of LC3B mRNA was analyzed by q-PCR. (D) Western blotting to examine the effect of HSM on the expression of LC3B protein. (E) RAW264.7 cells were pretreated with CQ (20 μM) and CLI095 (100 μg/ml) for 2 h and then treated with or without HSM (16 μg/ml) for 16 h. Representative immunofluorescence image of LC3B in RAW264.7 cells treated with CLI095. Scale bar: 30 μm. Data are presented as means ± SEM of at least three separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
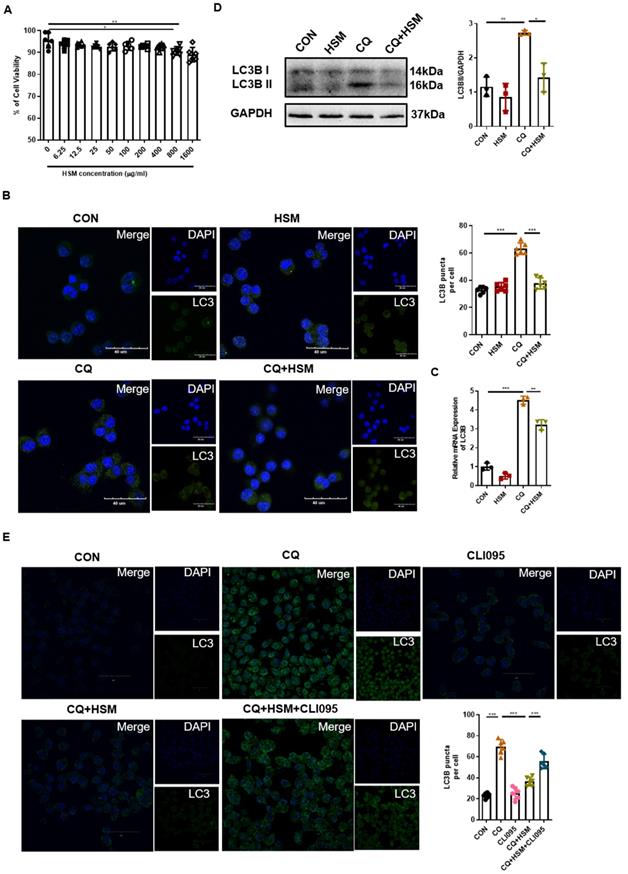
Abnormal autophagy activity as well as a block of autophagosome-lysosome can induce the accumulation of autophagosomes [52]. CQ inhibits the binding of autophagosomes to lysosomes by increasing the pH of lysosomes, thereby blocking the flux of autophagy, leading to the accumulation of LC3B protein and preventing autophagy from proceeding normally [53]. Next, we pre-treated RAW264.7 cells with CQ followed by HSM exposure, and immunofluorescence was used to detect the endogenous LC3B-II protein (Fig. 4B). CQ significantly induced LC3B-positive punctate structures in the macrophages, and HSM treatment resulted in a significant decrease in the number of LC3B-containing puncta (Fig. 4B). The LC3B gene expression and processed LC3B-II were measured by q-PCR and western blot, respectively. HSM showed similar reduction in CQ-induced autophagosome accumulation (Fig. 4C, 4D). Toll-like receptor 4 (TLR4) was reported to mediate autophagy in the pathological process of pulmonary fibrosis [54]. We checked whether the regulation of autophagy by HSM was related to TLR4 activity. CLI095, also known as TAK-242, selectively inhibits signaling mediated by the intracellular domain of TLR4 [55]. Interestingly, CLI095 could block the effect of HSM on the accumulation of autophagosomes in the RAW cells (Fig. 4E). Thus, the above results indicated that HSM can reduce the accumulation of autophagosomes dependent on TLR4 activity, which will help to restore autophagy.
Decreased TLR4/NF-κB signaling in macrophages with HSM incubation
LPS activates NF-κB translocation through TLR4 in AMs, and stimulates TLR4 in primary human macrophages and RAW264.7 cells to activate autophagy [56, 57]. To investigate the role of HSM on LPS-induced TLR4-associated autophagy in macrophages, we used LPS to activate TLR4 and then measured the expression of LC3B-related autophagosomes through immunofluorescence. In addition, we not only repeated the previous dose of HSM1 (16ug/ml), but also added the higher dose of HSM2 (160ug/ml) based on CCK-8 (Fig. 4A). Incubation of RAW cells with LPS led to the LC3B punctate staining, which is typical of autophagosomes (Fig. 5A). However, incubation of RAW264.7 cells with HSM1 after the LPS activation led to fewer autophagosomes, but HSM2 has no obvious effect (Fig. 5A). These data established the inhibition of abnormal autophagic response by HSM1 in murine macrophages after LPS stimulation.
Decreased TLR4/NF-κB signaling in macrophages with HSM incubation. (A) RAW264.7 cells were pretreated with LPS (0.1 μg/ml) for 2 h and then treated with or without HSM (HSM1:16 μg/ml and HSM2: 160 μg/ml) for 16 h. Representative immunofluorescence image of LC3B in RAW264.7 cells treated with LPS. Scale bar: 40 μm. (B) Effect of HSM on TLR4 protein expression by flow cytometry. (C) Effect of HSM on the expression of TLR4 downstream proteins NF-κB P65, ERK and IRF 3 was analyzed by western blotting. Data are presented as means ± SEM of at least three separate experiments. *P < 0.05, ***P < 0.001.
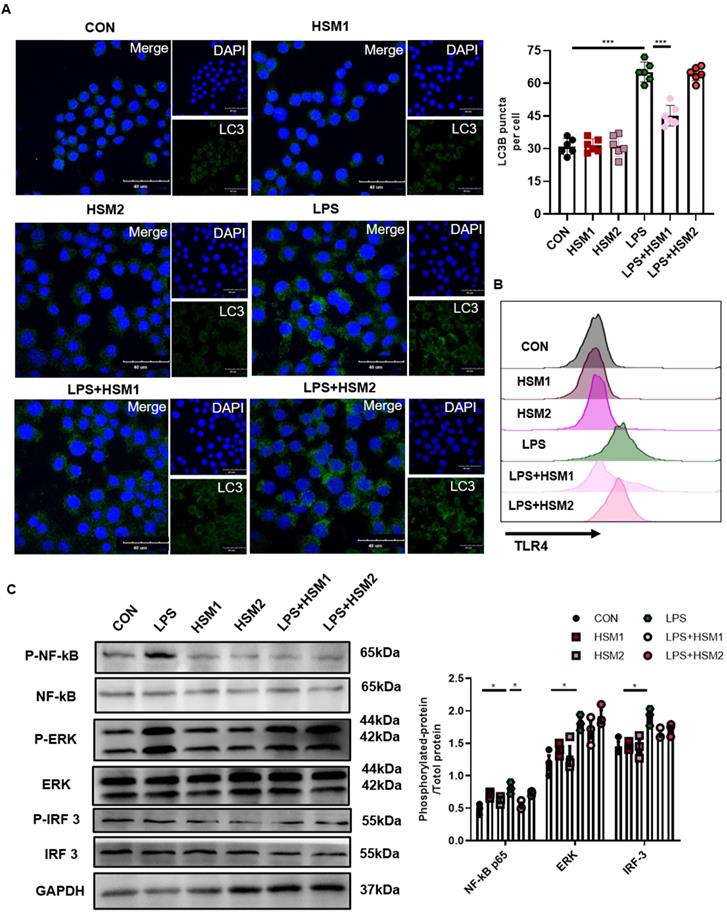
We hypothesized that the contribution of HSM to macrophage autophagy might be due to the regulation of TLR4 expression and its downstream signaling pathways. TLR4 protein expression was detected by flow cytometry. LPS-induced TLR4 expression was markedly inhibited in cells treated with HSM1 compared to cells induced with LPS only (Fig. 5B). Consistent with the effect on autophagy, HSM1 affected TLR4 expression rather than HSM2 (Fig. 5B). In addition, based on LPS-induced NF-κB, MAPK and IRF3 signaling pathways, HSM selectively abrogated the induction of phosphorylated NF-κB p65 (Fig. 5C). These results indicated that the effect of HSM1 on macrophage autophagy is mediated through the TLR4/NF-κB signaling pathway.
Discussion
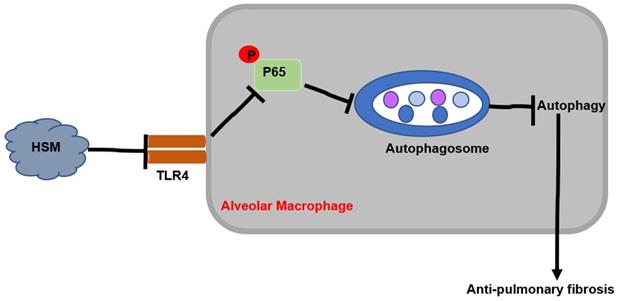
This study explored the potential protective effect of HSM on macrophages in attenuating BLM-induced pulmonary fibrosis. The results showed that HSM can inhibit excessive autophagic activity of AMs to achieve anti-fibrotic effect. In vitro studies showed that the effect of HSM on macrophage autophagy is mediated through the TLR4/NF-κB signaling pathway. These data suggested that AM autophagy might be a potentially novel and previously unrecognized target of HSM to limit pulmonary fibrosis, which had no effective therapies (Fig. 6).
Mechanism of HSM treatment of BLM-induced pulmonary fibrosis in mice.
Several studies have shown that pulmonary fibrosis is also accompanied by autophagy [35, 58-60], with an increase in the autophagic proteins. The level of microtubule associated protein LC3B, a unique molecular marker for autophagosomes, significantly increases in fibrotic lung tissues [35, 61]. Yang et al. demonstrated that the conversion of LC3B-I to LC3B-II is increased in the BLM-injured lung tissues [54]. A mouse autophagy RT-PCR array and protein level detection revealed that BLM-induced lung injury results in the up-regulation of ATG5 in lung tissue extracts [35]. This study showed that HSM reduced autophagy activation by increasing LC3B-II and ATG5, and decreasing P62 protein levels (Fig. 1C). Consistent with these results, a decrease in autophagosome formation was also revealed (Fig. 1B). Thus, we preliminarily considered that the anti-fibrotic effects of HSM were associated with the regulation of autophagic activity. Autophagy is a normal physiological process in the body to maintain homeostasis and survival. It is reported that autophagy plays different roles in the pathogenesis of pulmonary fibrosis. Atg4b-deficient mice displayed more extensive and severe fibrosis with increased collagen accumulation [35]. Impaired conventional ATG7-dependent macro-autophagy in lung endothelium increased susceptibility to BLM-induced fibrosis in mice [62]. However, some studies suggested that autophagy may cause harm and accelerate disease progression. In vivo, autophagy is activated in fibrotic patients [36]. There is widespread activation of autophagy in the lungs and pulmonary epithelial cells in the BLM-induced model [35]. This autophagy induction phenomenon also appears in the fibrosis model caused by amiodarone [59]. Additionally, the autophagy inhibitor 3-methyladenine reduced BLM-induced lung fibrosis [36].
Whether autophagy has a negative effect on human disease remains controversial, however, several studies have shown that macrophage autophagy may play a role in promoting fibrogenic diseases. IPF AMs show increased mitophagy and apoptosis resistance [28]. Autophagy of AMs has been reported to regulate silica-induced pulmonary fibrosis [30, 63, 64]. Our previous work and Fig. 2 demonstrated the anti-fibrotic effects of HSM associated with the regulation of AMs [11]. The current study showed that HSM inhibited autophagosome number and LC3B expression level in CD11bloCD11chi AMs without affecting IMs (Fig. 3A and B). And we verified this result by detecting the number of LC3B+ AM cells and the expression of LC3B MFI in BALF (Fig. 3C and D). This might indicate a role of AM autophagy in the anti-fibrosis mechanism of HSM.
Pulmonary macrophages may be used as targets for the treatment of pulmonary fibrosis due to their complexity and products in the process of fibrosis [65]. Depletion of pulmonary macrophages slows the regression of fibrosis during BLM-induced pulmonary fibrosis [24]. AMs are tissue-resident cells that play a primary role in maintaining immunological homoeostasis and host defense in lungs. Necrosis of monocyte-derived AMs can alleviate pulmonary fibrosis [66]. Moreover, AMs can also participate in the process of pulmonary fibrosis by producing TGF-β [28]. Engulfment of dysfunctional mitochondria by autophagosomes was reported in IPF AMs as well as apoptosis resistance [28]. The occurrence of autophagy is essential for maintaining the homeostasis of mitochondria. The autophagy inducer rapamycin can enhance the red fluorescence intensity under JC-1 staining and maintain the normal function of mitochondria [50]. Similarly, when HSM inhibited AM autophagy, MMP decreased and green fluorescence expression increased (Fig. S1A). Apoptosis and autophagy are two self-destructive processes in the body. Some research has indicated autophagy can regulate the occurrence of apoptosis. Autophagy is enhanced and conducive to cell adaptation and survival in various situations such as, the lack of growth factors and/or nutrition and hypoxia. Autophagy can inhibit apoptosis by removing pro-apoptotic proteins or adding caspase inhibitors [67, 68]. HSM promoted the apoptosis of F4/80+ macrophages in the lung under BLM treatment (Fig. S1B). These results further suggested the inhibitory effect of HSM on autophagy of alveolar macrophages.
In our study, we found low-dose of HSM (16ug/mL, HSM1) could decrease the LC3B punctate staining. Nevertheless, when the concentration of HSM rose to 160ug/mL (HSM2), the inhibitory effect on LC3B did not maintain (Fig. 5A). Similarly, HSM2 rather than HSM1 did not affect TLR4 expression on the macrophages and the activation of downstream signaling pathways (Fig. 5B and C). Such opposite effects between high and low doses of treatment have been previously reported. Welshons wrote in the journal PNAS that diethylstilbestrol doses of 0.02, 0.2, and 2.0 ng per g of body weight per day increased adult prostate weight, whereas a 200-ng-per-g dose decreased adult prostate weight in male offspring [69]. Tian et al. found that IL-2 high dose pretreatment ameliorated Con A-induced liver injury, while low dosage of IL-2 aggravated Con A-induced liver injury [70]. Similarly, effects of low and high doses of Abeta42 on electrical network and neuronal excitability in the rat prefrontal cortex were opposite [71]. In our study, such specific reasons underlying this phenomenon still need to be explored further. And the data strongly suggest that HSM may reduce the expression of LC3B and TLR4 signaling within a certain range of doses.
LPS-induced mTOR phosphorylation was down-regulated when TLR4 was knocked down, indicating that mTOR activation is caused by the TLR4 signaling pathway [72]. When the TLR4 signaling pathway is stimulated, MyD88 and TRIF can be recruited simultaneously to mediate the release of pro-inflammatory cytokines [73]. Our previous research found that HSM treatment of pulmonary fibrosis would cause the inhibition of mTOR activation and accompanied by inflammation suppression [11, 13]. These results suggest that TLR4 signaling may be related to the efficacy of HSM. Our results showed that HSM did exert its autophagy inhibition effect through TLR4 signaling (Fig. 4E). In addition, we also analyzed the changes in several other pathways downstream of TLR4. As downstream signaling pathways of TLR4, MAPK family, NF-κB and IRF3 have been reported to regulate cell growth and inflammation, leading to pulmonary fibrosis [74-76]. We found that HSM inhibited the activity of NF-κB but not the MAPK/ERK or IRF 3 pathway (Fig 5C). Consistent with this result, it has been reported that the inhibition of NF-κB signaling reduces the incidence of IPF in vivo, and reduces the proliferation of myofibroblasts and epithelial-mesenchymal transition in vitro [77-79]. These results indicated that HSM played an important role via the TLR4/NF-κB pathway.
In summary, the results of this study suggested that HSM exerts anti-fibrotic effects through the inhibition of autophagy in AMs, and the underlying mechanism is related to the inhibition of the TLR4/NF-κB p65 pathway. Hence, HSM may be a potential therapeutic candidate for the treatment of pulmonary fibrosis, and should be investigated in future studies.
Abbreviations
AM: alveolar macrophage; BALF: bronchoalveolar lavage fluid; BLM: bleomycin; CQ: Chloroquine; HSM: Hirsutella sinensis mycelium; IM: interstitial macrophage; IPF: Idiopathic pulmonary fibrosis; LC3B: Light Chain 3; LPS: lipopolysaccharide; MFI: mean fluorescence intensity; MMP: mitochondrial membrane potential; SPF: specific pathogen-free; TEM: transmission electron microscopy; TGF-β: transforming growth factor-β; TLR4: toll-like receptor 4.
Supplementary Material
Supplementary information.
Acknowledgements
The present work is supported by the Six Talent Peaks Project in Jiangsu Province (YY-021 and 2016-WSN-148), and Jiangsu Provincial Medical Youth Talent (QNRC2016005). We are also very grateful to Dr. Lizhi Xu (Medical School, Nanjing University) for her technical support.
Author contributions
DH and HYY designed the experiments. FJH and WHN performed the experiments. FJH, LL and WHN contributed to data analysis. FJH, LL and DH wrote the manuscript. All authors have reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Raghu G, Collard HR, Egan JJ. et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788-824
2. Raghu G, Rochwerg B, Zhang Y. et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015;192:e3-19
3. Samet JM, Coultas D, Raghu G. Idiopathic pulmonary fibrosis: tracking the true occurrence is challenging. Eur Respir J. 2015;46:604-6
4. du Bois RM. An earlier and more confident diagnosis of idiopathic pulmonary fibrosis. Eur Respir Rev. 2012;21:141-6
5. Navaratnam V, Fleming KM, West J. et al. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2011;66:462-7
6. King TE Jr, Bradford WZ, Castro-Bernardini S. et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083-92
7. Richeldi L, du Bois RM, Raghu G. et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071-82
8. Johannson KA, Strâmbu I, Ravaglia C. et al. Antacid therapy in idiopathic pulmonary fibrosis: more questions than answers? Lancet Respir Med. 2017;5:591-8
9. Tran T, Suissa S. The effect of anti-acid therapy on survival in idiopathic pulmonary fibrosis: a methodological review of observational studies. Eur Respir J. 2018 51
10. Huang T-T, Lai H-C, Ko Y-F. et al. Hirsutella sinensis mycelium attenuates bleomycin-induced pulmonary inflammation and fibrosis in vivo. Scientific Reports. 2015 5
11. Yue H, Zhao Y, Wang H. et al. Anti-fibrosis effect for Hirsutella sinensis mycelium based on inhibition of mTOR p70S6K phosphorylation. Innate Immun. 2017;23:615-24
12. Shou QY, Fu HY, Zhang LZ. et al. Study on treatment effect and mechanism of Hirsutella sinensis mycelium in idiopathic pulmonary fibrosis in rats. Zhongguo Zhong Yao Za Zhi. 2012;37:3618-23
13. Yue H, Liu F, Li L, Ma F, Shen S, Hou Y. Hirsutella sinensis mycelium relieves pulmonary fibrosis by inhibiting inflammatory factors and Treg cell function. Cellular & Molecular Immunology. 2016;32(03):354-357
14. Yue H, Liu F, Li L. et al. An Experimental Study of Hirsutella sinensis mycelium in Regulating Immunity and Improving Renal Fibrosis. Cellular & Molecular Immunology. 2016;32(10):1472-1476
15. Jacob CO, Hwang F, Lewis GD. et al. Tumor necrosis factor alpha in murine systemic lupus erythematosus disease models: implications for genetic predisposition and immune regulation. Cytokine. 1991;3:551-61
16. Arora S, Dev K, Agarwal B. et al. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223:383-96
17. Belchamber KBR, Donnelly LE. Targeting defective pulmonary innate immunity - A new therapeutic option? Pharmacol Ther. 2020;209:107500
18. Dancer RC, Wood AM, Thickett DR. Metalloproteinases in idiopathic pulmonary fibrosis. Eur Respir J. 2011;38:1461-7
19. Pradere JP, Kluwe J, De Minicis S. et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461-73
20. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245-57
21. Hicks J, Bullard DC. Review of autoimmune (lupus-like) glomerulonephritis in murine models. Ultrastruct Pathol. 2006;30:345-59
22. Tron F. Contribution of murine models of spontaneous lupus to the analysis of genetic factors in human disease. Ann Med Interne (Paris). 1990;141:217-21
23. Rouhani FN, Brantly ML, Markello TC. et al. Alveolar macrophage dysregulation in Hermansky-Pudlak syndrome type 1. Am J Respir Crit Care Med. 2009;180:1114-21
24. Gibbons MA, MacKinnon AC, Ramachandran P. et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184:569-81
25. Racanelli AC, Kikkers SA, Choi AMK. et al. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018;14:221-32
26. Zhao H, Wang Y, Qiu T. et al. Autophagy, an important therapeutic target for pulmonary fibrosis diseases. Clin Chim Acta. 2020;502:139-47
27. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-41
28. Larson-Casey JL, Deshane JS, Ryan AJ. et al. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity. 2016;44:582-96
29. Xu G, Wang X, Yu H. et al. Beclin 1, LC3, and p62 expression in paraquat-induced pulmonary fibrosis. Hum Exp Toxicol. 2019;38:794-802
30. Liu H, Fang S, Wang W. et al. Macrophage-derived MCPIP1 mediates silica-induced pulmonary fibrosis via autophagy. Part Fibre Toxicol. 2016;13:55
31. Chen S, Yuan J, Yao S. et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy. 2015;11:2346-57
32. Richardson B, Ray D, Yung R. Murine models of lupus induced by hypomethylated T cells. Methods Mol Med. 2004;102:285-94
33. Margaritopoulos GA, Tsitoura E, Tzanakis N. et al. Self-eating: friend or foe? The emerging role of autophagy in idiopathic pulmonary fibrosis. Biomed Res Int. 2013;2013:420497
34. Hosseinzadeh A, Javad-Moosavi SA, Reiter RJ. et al. Oxidative/nitrosative stress, autophagy and apoptosis as therapeutic targets of melatonin in idiopathic pulmonary fibrosis. Expert Opin Ther Targets. 2018;22:1049-61
35. Cabrera S, Maciel M, Herrera I. et al. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy. 2015;11:670-84
36. Meng Y, Pan M, Zheng B. et al. Autophagy Attenuates Angiotensin II-Induced Pulmonary Fibrosis by Inhibiting Redox Imbalance-Mediated NOD-Like Receptor Family Pyrin Domain Containing 3 Inflammasome Activation. Antioxid Redox Signal. 2019;30:520-41
37. Lucocq JM, Hacker C. Cutting a fine figure: On the use of thin sections in electron microscopy to quantify autophagy. Autophagy. 2013;9:1443-8
38. Kimura S, Fujita N, Noda T. et al. Chapter 1 Monitoring Autophagy in Mammalian Cultured Cells through the Dynamics of LC3. Autophagy in Mammalian Systems, Part B. 2009 p. 1-12
39. Ramkumar A, Murthy D, Raja DA. et al. Classical autophagy proteins LC3B and ATG4B facilitate melanosome movement on cytoskeletal tracks. Autophagy. 2017;13:1331-47
40. Feng Y, He D, Yao Z. et al. The machinery of macroautophagy. Cell Res. 2014;24:24-41
41. Pankiv S, Clausen TH, Lamark T. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45
42. Fussi N, Hollerhage M, Chakroun T. et al. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018;9:757
43. Baran CP, Opalek JM, McMaken S. et al. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:78-89
44. Kim TH, Lee YH, Kim KH. et al. Role of lung apolipoprotein A-I in idiopathic pulmonary fibrosis: antiinflammatory and antifibrotic effect on experimental lung injury and fibrosis. Am J Respir Crit Care Med. 2010;182:633-42
45. Guilliams M, De Kleer I, Henri S. et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977-92
46. Bedoret D, Wallemacq H, Marichal T. et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest. 2009;119:3723-38
47. Byrne AJ, Mathie SA, Gregory LG. et al. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70:1189-96
48. Misharin AV, Morales-Nebreda L, Mutlu GM. et al. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49:503-10
49. Chandramouleeswaran PM, Guha M, Shimonosono M. et al. Autophagy mitigates ethanol-induced mitochondrial dysfunction and oxidative stress in esophageal keratinocytes. PLoS One. 2020;15:e0239625
50. Cao W, Zhang J, Wang G. et al. Reducing-Autophagy Derived Mitochondrial Dysfunction during Resveratrol Promotes Fibroblast-Like Synovial Cell Apoptosis. Anat Rec (Hoboken). 2018;301:1179-88
51. Szegezdi E, Macdonald DC, T NC. et al. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941-53
52. Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010;221:117-24
53. Mauthe M, Orhon I, Rocchi C. et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435-55
54. Yang HZ, Wang JP, Mi S. et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. 2012;180:275-92
55. Kawamoto T, Ii M, Kitazaki T. et al. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40-8
56. Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269-390
57. Xu Y, Jagannath C, Liu XD. et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135-44
58. Mizumura K, Cloonan S, Choi ME. et al. Autophagy: Friend or foe in lung disease? 2016; 13 (Suppl 1): S40-7.
59. Mahavadi P, Knudsen L, Venkatesan S. et al. Regulation of macroautophagy in amiodarone-induced pulmonary fibrosis. J Pathol Clin Res. 2015;1:252-63
60. Cinacalcet for Cardiovascular Disease in Patients Undergoing Dialysis. New England Journal of Medicine. 2013; 368: 1842-5.
61. Mi S, Li Z, Yang HZ. et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. 2011;187:3003-14
62. Singh KK, Lovren F, Pan Y. et al. The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J Biol Chem. 2015;290:2547-59
63. Liu H, Cheng Y, Yang J. et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis. 2017;8:e2657
64. He X, Chen S, Li C. et al. Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages. Cells. 2020 9
65. Byrne AJ, Maher TM, Lloyd CM. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol Med. 2016;22:303-16
66. Misharin AV, Morales-Nebreda L, Reyfman PA. et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. 2017;214:2387-404
67. Shimizu S, Kanaseki T, Mizushima N. et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221-8
68. Zheng H, Jia L, Liu CC. et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J Neurosci. 2017;37:1772-84
69. vom Saal FS, Timms BG, Montano MM. et al. Prostate enlargement in mice due to fetal exposure to low doses of estradiol or diethylstilbestrol and opposite effects at high doses. Proc Natl Acad Sci U S A. 1997;94:2056-61
70. Zhang X, Wei HX, Rui S. et al. Opposite effects of high and low doses of interleukin-2 on T cell-mediated hepatitis in mice (interleukin-2 on hepatitis). Hepatol Int. 2010;4:641-8
71. Wang Y, Zhang G, Zhou H. et al. Opposite effects of low and high doses of Abeta42 on electrical network and neuronal excitability in the rat prefrontal cortex. PLoS One. 2009;4:e8366
72. Hu Y, Liu J, Wu YF. et al. mTOR and autophagy in regulation of acute lung injury: a review and perspective. Microbes Infect. 2014;16:727-34
73. Roy A, Srivastava M, Saqib U. et al. Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways. Int Immunopharmacol. 2016;40:79-89
74. Christman JW, Sadikot RT, Blackwell TS. The role of nuclear factor-kappa B in pulmonary diseases. Chest. 2000;117:1482-7
75. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911-2
76. Qiu H, Weng D, Chen T. et al. Stimulator of Interferon Genes Deficiency in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Front Immunol. 2017;8:1756
77. Chitra P, Saiprasad G, Manikandan R. et al. Berberine attenuates bleomycin induced pulmonary toxicity and fibrosis via suppressing NF-kappaB dependant TGF-beta activation: a biphasic experimental study. Toxicol Lett. 2013;219:178-93
78. Zhu T, Zhang W, Xiao M. et al. Protective role of andrographolide in bleomycin-induced pulmonary fibrosis in mice. Int J Mol Sci. 2013;14:23581-96
79. Wang YM, Ji R, Chen WW. et al. Paclitaxel alleviated sepsis-induced acute lung injury by activating MUC1 and suppressing TLR-4/NF-kappaB pathway. Drug Des Devel Ther. 2019;13:3391-404
Author contact
![]() Corresponding authors: Huan Dou, Email: douhuanedu.cn; Yayi Hou, Email: yayihouedu.cn.
Corresponding authors: Huan Dou, Email: douhuanedu.cn; Yayi Hou, Email: yayihouedu.cn.