Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(8):1786-1797. doi:10.7150/ijms.46811 This issue Cite
Research Paper
Efficacy and Safety of CAR-T Therapy for Relapse or Refractory Multiple Myeloma: A systematic review and meta-analysis
Qin Yang1, Xin Li1, Fangrong Zhang1, Qiaohui Yang4, Wen Zhou2,3 ![]() , Jing Liu1
, Jing Liu1 ![]()
1. Department of Hematology, The Third Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China.
2. Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education; Key Laboratory of Carcinogenesis, National Health and Family Planning Commission, Cancer Research Institute, School of Basic Medical Science, Central South University, Changsha, Hunan, P.R. China.
3. Cancer Research Institute, Central South University, Changsha, Hunan, P.R. China.
4. Institute of Reproductive and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, Hunan, P.R. China.
*These authors have contributed equally to this work.
Received 2020-4-8; Accepted 2021-1-23; Published 2021-2-18
Abstract

Background: Multiple myeloma (MM) is incurable in spite of recent treatment improvements, highlighting the development of new therapies. Chimeric antigen receptor (CAR) T-cell therapy has dramatically changed the therapeutic effectiveness in high-risk B-cell malignancies. For relapsed/refractory multiple myeloma (RRMM), preclinical evaluations of CAR-T therapy have shown promising efficacy, thus various active clinical trials are under way. Herein, we conducted this review to summarize efficacy and safety of CAR-T therapy and provide more evidence to guide clinical treatments.
Method: We systematically searched literature based on databases (PubMed, EMBASE, Cochrane Central Register of Controlled Trials), and conference abstracts reported from American Society of Hematology (ASH), European Hematology Association (EHA) and American Society of Clinical Oncology (ASCO), in addition to other sources (www.clinicaltrials.gov, article citations). Data assessed efficacy and safety of CAR-T therapy in patients with RRMM were extracted and evaluated, and then systematically analyzed by Comprehensive Meta-analysis 3.0 (CMA 3.0).
Results: A total of 23 studies including 350 participants from different countries, diagnosed as RRMM and treated with CAR-T therapy (containing 7 antigens targeted by CARs) were combined. In summary, we discovered the pooled overall response rate (77%), complete response rate (37%) and minimal residual disease (MRD) negativity rate within responders (78%). Furthermore, the pooled relapse rate of responders was 38% and median progression-free survival was 8 months. The pooled survival rate was 87% at last follow-up (median, 12 months). In addition, the pooled grade 3-4 rates of cytokine release syndrome (CRS) and neurologic toxicities (NT) were 14% and 13%, respectively.
Conclusion: Our study suggests that CAR-T therapy has demonstrated efficacy and safety in RRMM patients. BCMA-targeted CAR-T and anti-BCMA contained regimen have shown better efficacy.
Keywords: RRMM, CARs, CAR-T therapy, antigens, BCMA, co-stimulatory domain, systematic review, meta-analysis
Introduction
Multiple myeloma (MM) is a B-cell malignancy characterized by the aberrant expansion of clonal plasma cells and clinically manifested by hypercalcemia, renal dysfunction, anemia, and bone lesions. It accounts for 1-2% of all malignancies and ranks second among the hematological malignancies, just following non-Hodgkin lymphoma [1]. With the advent of proteasome inhibitors, immunomodulatory drugs and monoclonal antibodies, the overall survival of myeloma patients has been improved significantly over the last few years. In spite of recent development of therapeutic regimens, most patients will inevitably relapse and need salvage regimens, which are generally associated with a reduced duration of responses, highlighting the development of new therapies.
Chimeric antigen receptors (CARs) are engineered receptors that combine an antigen-recognition domain and T-cell signaling domains [2-4]. Expressing a CAR, T cells can specifically recognize a desired antigen, which was first described in the late 1980s [5, 6]. It was a highly effective form of adoptive cell therapy, and was approved by FDA in B cell acute lymphoblastic leukemia or large B cell lymphoma patients with the desirable remission rates [7, 8]. The impressive results provided rationales for developing CAR-T against MM. Preclinical evaluations of CAR-T cells showed promising efficacy for MM, especially for relapsed/refractory setting (RRMM). The first in-human trial on anti-BCMA CAR-T therapy was conducted by Brudno, which achieved a high response rate [9]. Subsequently, various ongoing clinical trials utilizing CAR-T technology have been performed to target myeloma antigens such as B cell maturation antigen (BCMA), CD19, CD138 and immunoglobulin light chains [10-13]. Up to now, the comprehensive analysis about this novel therapy on RRMM based on the current data has not been performed yet. And evaluation of the benefits of CAR-T therapy in patients with RRMM is necessary. Herein, we systematically review the current literature and report the results of a meta-analysis. It included a total of 23 studies incorporating 350 participants diagnosed as RRMM and treated with CAR-T therapy from 2016 to 2019, to study the efficacy and safety of CAR-T therapy and provide better understanding of this new treatment strategy.
Methods
Study design and search process
This systematic review and meta-analysis followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines in the Cochrane Handbook [14, 15]. Research articles that assessed the efficacy and safety of CAR-T therapy in patients with RRMM were the major objectives.
The search process was conducted based on two major resources as follows. Firstly, literature published on PubMed, EMBASE, Cochrane Central Register of Controlled Trials, and other sources (www.clinicaltrials.gov, article citations). Secondly, conference abstracts reported from American Society of Hematology (ASH), European Hematology Association (EHA) and American Society of Clinical Oncology (ASCO).
The following keywords were used to construct the search strategy: (chimeric antigen receptor OR car t therapy OR car t immunotherapy OR car t cell OR modified or engineered t cell) AND (multiple myeloma OR myeloma OR multiple myeloma relapse OR multiple myeloma refractory). Additionally, we boosted our literature search through a manual search of the reference lists of eligible articles.
Eligibility criteria
Two authors independently screened and judged the eligibility of identified articles, and disagreements were resolved by consensus. The included studies had to conform to the following criteria: (1) enrollment of subjects (age≥18 years) receiving CAR-T therapy, no matter what antigens CAR-T cells targeted; (2) evaluation of the efficacy and safety of CAR-T therapy; (3) one or more outcomes such as response, relapse or survival, and adverse events were reported.
Data extraction and quality assessment
The following data were extracted by 2 independent authors: the name of the first author, the year of publication, registration number, sample size, age and country of participants, lines of prior treatment, autologous stem cell transplantation (ASCT) before CAR-T, CAR construct, CAR antigens, T cell origin and subset, lymphodepletion, CAR-T doses, outcomes such as response, relapse or survival, and adverse events, duration of follow-up. The modified Institute of Health Economics (IHE) risk of bias tool [16] was used to perform the quality assessment of the included studies.
Statistical analysis
The following outcomes were to be measured, such as overall response, complete response, MRD negativity within responders, relapse, survival and adverse events. Data of each study were pooled to estimate the efficacy and safety of CAR-T therapy by using a random - effects models with the DerSimonian and Laird random-effects method (Comprehensive Meta-analysis 3.0, Englewood, USA). Heterogeneity was evaluated on Q-statistic and I2 statistics [17] and values of 25, 50 and 75% were used to represent low, medium and high quality respectively [18]. The source of heterogeneity would be explored if there was considerable heterogeneity. The evaluation of publication bias was performed by funnel plot and Egger test.
Results
Literature search results
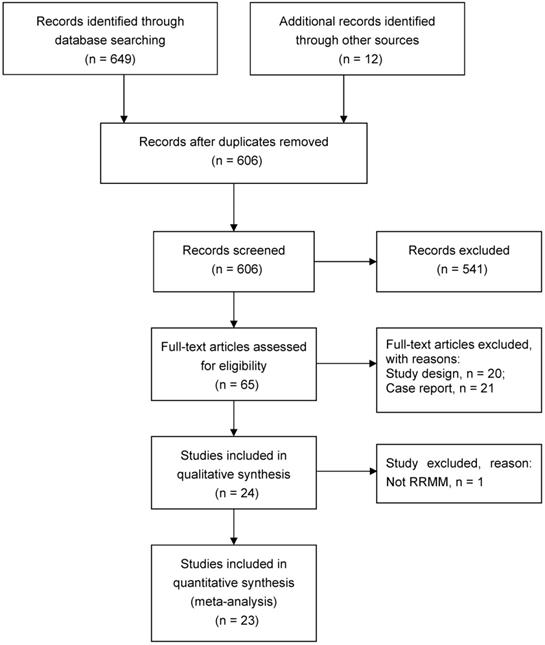
The database search identified 661 potentially eligible studies. After screening titles/abstracts and retrieving full-text articles, a total of 23 studies met the inclusion criteria, which included 350 participants from multiple centers diagnosed as RRMM and treated with CAR-T therapy from 2016 to 2019 [9, 11-13, 19-37] (Figure 1).
PRISMA flowchart of study search.
Quality assessment
The modified IHE tool included assessment of the study objective, design, study population, intervention and co-interventions, outcome measures (e.g., blinding, incomplete outcome data such as participants lost to follow-up, selective outcome reporting), statistical analysis, results, conclusions and conflicts of interest. Each item was scored as high risk, moderate risk or low risk of bias. The evaluation was made independently by two authors based on the criteria provided for the modified IHE risk of bias tool for interventional study designs. Disagreements were resolved by discussion. All the studies were assessed as low and moderate risk. Table S1 stated the details of the quality of the included studies.
Study characteristics
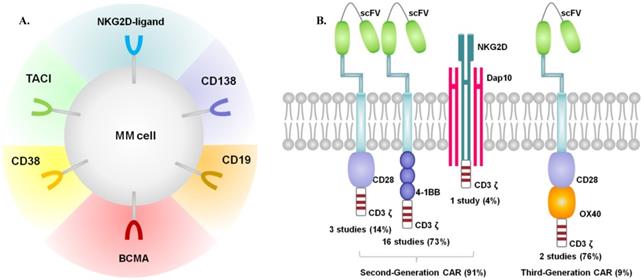
All of the 23 included studies, published between 2016 and 2019, were single-arm early phase studies. Most of the enrolled participants were middle-aged or elderly, with performance status 0-2 assessed by Eastern Cooperative Oncology Group (ECOG). All patients were diagnosed with RRMM and received multiple prior lines of treatments (proteasome inhibitors, immunomodulatory drugs, monoclonal antibodies or ASCT). Among the 23 studies, 2 studies had no statements on prior treatments [13, 26]. Most participants underwent ASCT as part of previous treatments. 1 study performed ASCT concurrently with CAR-T therapy [11], and data with ASCT were absent within 7 studies. Most studies performed lymphodepletion by fludarabine and cyclophosphamide. Melphalan was used in 1 study [11], and cyclophosphamide in 2 studies [20, 37].The lymphodepletion was not performed in 1 study [36], and the data were not available in 2 studies [12, 13]. T cells were genetically modified to express a CAR by using γ-retroviruses, lentiviruses, or transposon systems. The origin of T cells in most studies were autologous, while 1 study with both autologous and allologous T cells [35], and 5 studies with T cells of unknown origin. The characteristics of the included studies were shown in Table 1. Figure 2 presented the target antigens and CARs included in our review.
Characteristics of included studies
| Reference | Country | Sample size | Median age (range) | Sex (Male/Female) | Lines of Prior treatment | ASCT before CAR-T | LD | Antigen | CAR construct: vector/costimulatory molecule/scFv species | T cell origin | T cell subset | BCMA positivity requirement at enrollment | CAR-T dose | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brudno 2018 | Western | 26 | (18-70) | 13/13 | 10 (3-19) | 85% | Flu/Cy: 30mg/m2 /300mg/m2 daily on day -5 to -3 | BCMA | γ-retrovirus/CD28/murine | Autologous | CD4/CD8 | >50% | 0.3-9.0x106/Kg | Median 20 weeks |
| Shah 2018 | Western | 8 | 64 (54-70) | NA | 9 (4-17) | 88% | Flu/Cy: 30mg/m2 /300mg/m2 daily for 3 days | BCMA | Lentivirus/4-1BB/murine | Autologous | NA | Dose-escalation: >50% | 150x106 | Median 16 (2-27) weeks |
| Zhao 2018 | Eastern | 57 | 54 (27-72) | 34/23 | 3 (1-9) | 18% | Cy:300 mg/m2 on day -5 to -3 | BCMA | Lentivirus/4-1BB/Llama | Autologous | Unselected | Required (cutoff NR) | Median 0.5 (0.07-2.1)x106/Kg | Median 32 (2.8-82.8) weeks |
| Mailankody 2018 | Western | 44 | 53 (36-66) | NA | 10 (4-15) | 88% | Flu/Cy: 30mg/m2 /300mg/m2 on days -7 to -2 | BCMA | Lentivirus/4-1BB/human | NA | NA | NR | 50-150x106 | Median 5 (4-13) weeks |
| Jiang 2018 | Western | 16 | 55 (39-67) | NA | 4 (2-10) | 56% | Flu/Cy: 20-25mg/m2 /300- 500mg/m2 daily for 2-4 days | BCMA | NR/4-1BB/human | Autologous | NA | ≥50% | 0.5-1.8x108 | Median 8 (4-36) weeks |
| Mailankody 2018 | Western | 11 | NA | NA | 6 (4-14) | 100% | Flu/Cy: 30 mg/m2 /300 mg/m2 for 3 days | BCMA | Retrovirus/4-1BB/human | Autologous | NA | Required (cutoff NR) | 72-818x106 | >40 weeks |
| Gregory 2018 | Western | 12 | NA | NA | 3-9 | NA | Flu/Cy: 30 mg/m2/300mg/m2 for 3 days | BCMA | Transposon/4-1BB/human | Autologous | NA | Not required | 48-430x106 | >12 weeks |
| Green 2018 | Western | 7 | 63 (49-76) | NA | 8 (6-11) | 71% | Flu/Cy | BCMA | Lentivirus/4-1BB/human | Autologous | CD4/CD8 | ≥5% | 5-15x107 | Median 16 (2-26) weeks |
| Liu 2018 | Eastern | 17 | NA | NA | >2 | NA | Flu/Cy: 25 mg/m2 /300mg/m2 on days -5 to -3 | BCMA | γ-retrovirus/4-1BB/mouse | Autologous | NA | >5% | 9x106 | >60 weeks |
| Li 2018 | Eastern | 28 | NA | NA | NA | NA | Flu/Cy | BCMA | Lentivirus/CD28/murine | Autologous | NA | Required (cutoff NR) | 5.4-25.0x106 | Median 40 weeks |
| Raje 2019 | Western | 33 | 60 (37-75) | 21/12 | 7 (3-23) | 97% | Flu/Cy: 30mg/m2 /300mg/m2 daily on day -5 to -3 | BCMA | Lentivirus/4-1BB/murine | Autologous | CD4/ CD8 | Dose-escalation: ≥50%; dose-expansion: NR | 50-800x106 | Median 45 (24.8-91.2) weeks |
| Xu 2019 | Eastern | 17 | 55 (35-73) | 11/6 | 5 (3-11) | 47% | Flu/Cy: 25 mg/m2 daily for 3 days/250/300mg/m2 | BCMA | Lentivirus/4-1BB/Llama | NA | CD4/ CD8 | Required (cutoff NR) | 0.21-1.52x106/Kg | Median 60 (1.7-76.4) weeks |
| Li 2019 | Eastern | 9 | NA | NA | 4 (3-5) | NA | Flu/Cy | BCMA | Lentivirus/4-1BB/human | NA | NA | NR | 1.0-6.0x106/Kg | Median 9 weeks |
| Cohen 2019 | Western | 25 | 58 (44-75) | 17/8 | 7 (3-13) | 92% | Cy: 1.5g/m2 or no LD | BCMA | Lentivirus/4-1BB/human | Autologous | CD4/ CD8 | Not required | 1-50x107 | Median 54 weeks |
| Han 2019 | Eastern | 16 | NA | NA | Median 10 | NA | Flu/Cy: 30 mg/m2 on days -5 to -3/300-600 mg/m2 on days -5 to -4 | BCMA | Lentivirus/4-1BB/Llama | Autologous | NA | NR | 2-10x106 | Median 10 weeks |
| Garfall 2018 | Western | 10 | 61 (48-68) | 4/6 | 6 (2-10) | 100% | Melphalan: 140-200 mg/m2 | CD19 | Lentiviral/4-1BB | Autologous | Unselected | NR | 1-5x107 | >14 weeks |
| Guo 2016 | Eastern | 5 | 58 (48-68) | 1/4 | 11 (5-18) | 20% | NA | CD138 | Lentivirus/4-1BB/human | Autologous | CD8 | NR | Median 7.56 (4.4-15.1)x106 | Median 12 weeks |
| Ramos 2016 | Western | 8 | 56.5 (43-69) | 3/5 | NA | NA | NA | κ light chain | Retroviral/CD28/murine | Autologous | CD4/ CD8 | NR | 0.2-2x108/m2 | (6-96) weeks |
| Baumeister 2018 | Western | 5 | 70 (44-79) | 3/2 | ≥5 | 100% | No LD | NKG2D-ligands | γ-retroviral/ NKG2D-CAR | Autologous | CD4/ CD8 | NR | 1x106-3×107 | (14-107) weeks |
| Yan 2017 | Eastern | 8 | 57 (43-69) | 6/2 | 4 (2-7) | NA | Flu/Cy: 30 mg/m2/300 mg/m2 on days -5 to -3 | CD19 and BCMA | Lentivirus/CD28 + OX40 /murine | Auto/allo-logous | NA | Required (cutoff NR) | 25-82×106/kg | Median 5 (2-20 ) weeks |
| Yan 2019 | Eastern | 21 | 58 (49.5-61) | NA | 6 (5-8) | 14% | Flu/Cy: 30mg/m2for 3 days /750mg/m2 daily for 1 day | CD19 and BCMA | Lentiviral/4-1BB/human (anti-CD19) murine (anti-BCMA) | Autologous | Unselected | NR | CD19(1×106/Kg) BCMA (1×106/Kg) | Median25.6 (10.3-42.1) weeks |
| Li 2019 | Eastern | 12 | NA | NA | ≥2 | 17% | Flu/Cy: 25 mg/m2/250 mg/m2 on days -5 to -3 | CD38 and BCMA | NR | NA | NA | BCMA+ or CD38+ ≥ 50% | Median 2.17 (0.5-4.0)×106 | Median 22 (6-33) weeks |
| Popat 2019 | Western | 11 | 61 (45-69) | NA | 5 (3-6) | 73% | Flu/Cy: 30 mg/m2 daily/300mg/m2 daily for 3 days | TACI and BCMA | Retroviral/CD28 + OX40 | NA | NA | NR | 15-900×106 | NA |
Abbreviations: ASCT, Autologous stem cell transplantation; CAR-T, Chimeric antigen receptor-T; LD, Lymphodepletion; scFv, single-chain fragment variable; Flu, Fludarabine; Cy, Cyclophosphamide; BCMA, B cell maturation antigen; NA, not available; NR, not reported.
Summary of target antigens (A) and CARs (B) included in our review.
The pooled proportions of outcomes for RRMM with CAR-T
| Outcomes | No. of studies | Patients (n/N) | Pooled proportion (%) | 95% CI | Heterogeneity within study (I2, Q and p-value) | ||
|---|---|---|---|---|---|---|---|
| Q-value | I2 (%) | p-value | |||||
| Overall response | 23 | 274/350 | 77 | 68-85 | 51.714 | 57.458 | <0.01 |
| Complete response | 18 | 128/305 | 37 | 26-50 | 53.579 | 68.271 | <0.01 |
| MRD negativity within responders | 6 | 92/113 | 78 | 69-85 | 7.288 | 31.394 | <0.01 |
| Relapse* | 8 | 53/155 | 38 | 24-55 | 21.394 | 67.280 | <0.01 |
| Overall survival* | 8 | 160/188 | 87 | 71-95 | 26.824 | 73.904 | <0.01 |
| Grade 3-4 rates of CRS | 23 | 43/347 | 14 | 10-21 | 33.446 | 34.223 | >0.01 |
| Neurotoxicity | 19 | 37/277 | 13 | 8-22 | 36.330 | 50.454 | <0.01 |
*The time means the last follow-up; MRD: minimal residual disease; CRS: cytokine release syndrome.
Subgroup analysis of overall response and adverse events for RRMM with CAR-T
| Subgroups | Overall Response | Grade 3-4 rates of CRS | NT | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients (n/N) | Pooled proportion | 95% CI | p-interaction | Patients (n/N) | Pooled proportion | 95% CI | p-interaction | Patients (n/N) | Pooled proportion | 95% CI | p-interaction | |
| Country | <0.05 | >0.05 | <0.05 | |||||||||
| Eastern | 156/181 | 86 | 76-92 | 23/187 | 14 | 8-24 | 4/117 | 6 | 2-13 | |||
| Western | 118/169 | 69 | 56-79 | 20/160 | 14 | 8-23 | 33/160 | 23 | 15-34 | |||
| Trial site | >0.05 | >0.05 | >0.05 | |||||||||
| Single center | 113/151 | 75 | 60-86 | 22/157 | 13 | 7-23 | 11/104 | 10 | 4-23 | |||
| Multiple center | 161/199 | 80 | 66-88 | 21/190 | 15 | 9-25 | 26/173 | 15 | 7-29 | |||
| Trial status | >0.05 | >0.05 | >0.05 | |||||||||
| Completed | 107/141 | 72 | 51-86 | 18/131 | 17 | 9-30 | 9/114 | 10 | 4-25 | |||
| Ongoing | 167/209 | 80 | 68-88 | 25/216 | 14 | 8-21 | 28/163 | 15 | 8-27 | |||
| scFvs origin | >0.05 | >0.05 | >0.05 | |||||||||
| Human | 61/84 | 74 | 53-87 | 11/84 | 14 | 6-27 | 12/75 | 16 | 6-35 | |||
| Non-human | 190/228 | 83 | 72-90 | 28/225 | 14 | 8-23 | 25/169 | 14 | 6-28 | |||
| Co-stimulatory domain | >0.05 | >0.05 | >0.05 | |||||||||
| 4-1BB | 209/256 | 81 | 71-89 | 28/260 | 12 | 7-19 | 31/218 | 13 | 7-24 | |||
| CD28 | 45/61 | 72 | 45-90 | 10/51 | 21 | 8-44 | 6/23 | 25 | 5-66 | |||
| CARs | >0.05 | >0.05 | >0.05 | |||||||||
| 2nd generation | 254/322 | 78 | 68-86 | 38/316 | 14 | 9-20 | 37/246 | 15 | 9-25 | |||
| 3rd generation | 10/16 | 65 | 26-91 | 1/19 | 8 | 1-37 | 0/19 | 5 | 1-33 | |||
| Antigen target | <0.05 | >0.05 | >0.05 | |||||||||
| BCMA | 223/274 | 82 | 72-89 | 37/268 | 16 | 10-23 | 35/198 | 19 | 11-31 | |||
| Non-BCMA | 11/27 | 43 | 18-72 | 0/27 | 7 | 2-25 | 0/27 | 7 | 1-26 | |||
| CAR-T regimen | <0.05 | >0.05 | >0.05 | |||||||||
| Anti-BCMA contained | 266/323 | 81 | 73-88 | 43/320 | 15 | 10-22 | 37/250 | 15 | 9-25 | |||
| Anti-BCMA uncontained | 11/27 | 43 | 18-72 | 0/27 | 7 | 2-25 | 0/27 | 7 | 1-27 | |||
| CAR-T therapy mode | >0.05 | >0.05 | >0.05 | |||||||||
| Single-target | 234/301 | 77 | 66-85 | 37/295 | 14 | 9-21 | 35/225 | 16 | 10-27 | |||
| Dual-target | 40/49 | 80 | 55-93 | 6/52 | 14 | 5-34 | 2/52 | 6 | 2-21 | |||
Outcomes of meta-analysis
Basic pooled proportions of patients with CAR-T therapy
All the 23 studies reported the outcomes of overall response. 274 out of 350 patients in the 23 studies achieved overall response, with the pooled proportion being 77% (95% CI: 68-85; I2=57.458%; p< 0.01; Figure 3A). 128 out of 305 patients in 18 studies achieved complete response. The random-effects pooled proportion was 37% (95% CI: 26-50; I2=68.271%; p< 0.01; Figure 3B). In terms of MRD negativity within responders, 92 out of 113 participants in 6 studies obtained MRD negativity. The pooled proportion was 78% (95% CI: 69-85; I2=31.394; p< 0.01; Figure 3C). Our analysis revealed the overall response rate (77%) and MRD negativity rate within responders (78%) were impressive, although the number of patients with MRD data was not high. Our pooled complete response rate was 37%, and slightly lower than the best reported rate 43% [38].
The pooled proportions of efficacy and safety outcomes (A. Overall Response; B. Complete Response; C. MRD negativity within responders; D. Relapse at last follow-up; E. Overall Survival at last follow-up; F. Grade 3-4 rates of CRS; G. NT) for RRMM with CAR-T.
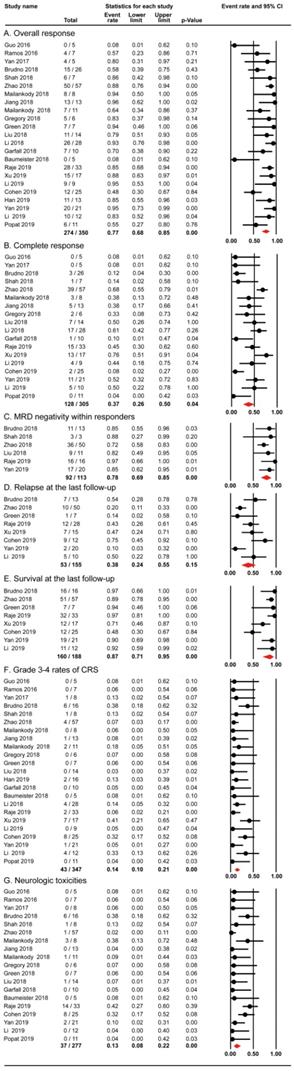
Furthermore, 8 studies evaluated relapse and survival outcomes at the last follow-up. The pooled proportion of relapse was 38% (95% CI: 24-55; I2=67.280; p< 0.01; Figure 3D) and the median progression-free survival was 8 months. The pooled proportion of survival (median follow-up, 12 months) was 87% (95% CI: 71-95; I2=73.904; p< 0.01; Figure 3E).
The important toxicities of cytokine release syndrome (CRS) and neurologic toxicities (NT) were also evaluated in this meta-analysis. 43 out of 347 patients in the 23 studies experienced grade 3-4 rates of CRS with the pooled proportion being 14% (95% CI: 10-21; I2=34.223; p> 0.01; Figure 3F). The pooled proportion of NT in 19 studies was 13% (95% CI: 8-22; I2=50.454; p< 0.01; Figure 3G). However, the criteria for CRS and NT grading varied previously and there has been a consensus now [39-41]. Our analysis showed the pooled rates of serious adverse events (CRS and NT) were relatively infrequent (14% and 13%).
Multiple factors associated with overall response rate and adverse events rates
The subgroup analysis of overall response and adverse events by country
13 studies were conducted in Western countries and 10 studies in Eastern countries. Further subgroup analysis revealed that the 86% (95%CI: 76-92) pooled overall response rate in Eastern countries were statistically different from the 69% (95%CI: 56-79) pooled overall response rate in Western countries. As to adverse events, there was no difference in grade 3-4 rates of CRS between Eastern and Western countries: 14% (95%CI: 8-24) versus 14% (95%CI: 8-23). A higher NT rate was observed in Western countries in comparison to Eastern countries: 23% (95%CI: 15-34) versus 6% (95%CI: 2-13). Based on the analysis above, it was presumable that the antimyeloma activity of CAR-T therapy in Eastern patients may be better than Western patients. The grade 3-4 rates of CRS showed no difference between them, while the NT was more prone to Western, indicating the possible effects of racial difference on the efficacy of CAR-T. However, the results should be interpreted with caution. Many factors including patient selection may contribute to the better response of Eastern patients. Additionally, the lower rate of NT events in Eastern patients may be related to reporting bias. Thus, more studies were needed to further understand the underlying mechanisms of the differences.
The subgroup analysis of overall response and adverse events by trial site
11 studies of 199 participants were performed in multiple centers, while 12 studies of 151 participants in single centers. We carried out a subgroup analysis by using trial site as a moderator. In conclusion, the pooled overall response rate of multiple centers was 80% (95%CI: 66-88) and showed no significant difference when compared with a 75% (95%CI: 60-86) overall response rate in single centers. There was also non-significant difference with regard to adverse events rates.
The subgroup analysis of overall response and adverse events by trial status
7 studies were completed and 16 studies are still ongoing. Our subgroup analysis of trial status categorized by completed and ongoing revealed non-significant difference in overall response rate: 72% (95%CI: 51-86) versus 80% (95%CI: 68-88), so did the adverse events rates.
The subgroup analysis of overall response and adverse events by scFvs origin
The single-chain variable fragments (scFvs) originated from murine (7 studies), llama (3 studies) and human (9 studies) in the included studies. Further subgroup analysis demonstrated that the pooled overall response rate of human scFvs origin was 74% (95%CI: 53-87) and showed no significant difference when compared with the 83% (95%CI: 72-90) overall response rate of non-human scFvs origin. The pooled CRS of grade 3-4 rates and NT rates showed no significant differences between human and non-human scFvs origin.
The subgroup analysis of overall response and adverse events by co-stimulatory domain
The main co-stimulatory molecules were CD28 and 4-1BB in the included studies. As defined, one co-stimulatory signaling domain is added in second-generation CARs, and two co-stimulatory signaling domains are added in third-generation type. Specially, NKG2D is categorized as second-generation type, which can naturally bind to the natively-encoded adaptor protein DAP10 and then provide a co-stimulatory signal upon ligand binding [19]. Based on the above classification, 20 studies used the second-generation CARs structure, and only 2 studies used the third-generation type.
We conducted subgroup analysis of co-stimulatory domains, which were categorized by 4-1BB- and CD28- contained second-generation CAR-T. The pooled overall response rate was 81% (95%CI: 71-89) and 72% (95%CI: 45-90) for 4-1BB- and CD28- contained second-generation CAR-T respectively. However, the difference wasn't significant statistically, as well as the adverse events rates. Therefore, we may get that 4-1BB- and CD28-contained second-generation CAR-T showed the same efficacy and safety.
Then we performed our subgroup analysis of second-generation and third-generation CARs. It demonstrated that the pooled overall response rate of second-generation CARs was 78% (95%CI: 68-86) and showed no significant difference when compared with a 65% (95%CI: 26-91) overall response rate of third-generation type. The adverse events rates were observed no difference as well. Theoretically, third-generation CARs were designed to enhance the efficacy with the advances in the field of CAR-T cell engineering. Nevertheless, some preclinical studies discovered that third-generation CARs had no difference or even worse function in comparison to second-generation CARs [42-45]. Our result showed the same activity between third-generation and second-generation CAR-T therapy, which deserved exploration for its reasons.
The subgroup analysis of overall response and adverse events by antigens targeted by CAR
Multiple antigen targets were included in our analysis. 19 studies used single-target CAR (15 BCMA, 1 CD19, 1 CD138, 1κ light chain, 1 NKG2D-ligands), and 4 studies used dual-target CAR (2 CD19 and BCMA, 1 BCMA and CD38, 1 BCMA and TACI).
We further conducted subgroup analysis by using the antigens as a moderator. The pooled overall response rate of BCMA-targeted CAR-T was 82% (95%CI: 72-89) when compared with non-BCMA-targeted one (43%, 95%CI: 18-72). The BCMA-targeted CAR-T had higher overall response rate than non-BCMA-targeted type (p< 0.05). Meanwhile, the adverse events rates revealed no significant difference. To sum up, BCMA-targeted CAR-T showed better efficacy than non-BCMA-targeted type, which may give a reasonable evidence to optimize the CAR-T therapy by choosing BCMA-targeted one.
Furthermore, we compared anti-BCMA contained regimen with anti-BCMA uncontained one. It turned out that the 81% (95%CI: 73-88) pooled overall response rate of anti-BCMA contained regimen were statistically different from the 43% (95%CI: 18-72) pooled overall response rate of anti-BCMA uncontained regimen. Meanwhile, the adverse events rates were observed no difference. By the subgroup analysis, we concluded that anti-BCMA contained regimen had higher overall response rate without bringing additional toxicities. Nevertheless, the number of non-BCMA targets in clinical trials was much smaller than BCMA, and non-BCMA targeted approaches were more of the time period during which they were done (early in CAR-T development). These may under- or over-estimate the pooled proportions.
Then we analyzed the pooled overall response rate of single-target CAR-T (77%, 95%CI: 66-85) when compared with dual-target CAR-T (80%, 95%CI: 55-93). The difference wasn't significant statistically, so did the adverse events rates. Theoretically, combining CAR-T cells with different targets in a cocktail infusion may combat antigen loss and resistance of CAR-T [46], and show better effectiveness than single-target one. But there was no difference between dual-target CAR-T therapy and single-target one, which need further investigation to testify this outcome and explain the reasons. All the subgroup analysis were demonstrated in the supplementary Figure 1-3.
Publication bias
Risk of publication bias was evaluated by funnel plot and Egger's tests. No evidence of potential publication bias was revealed for overall response, complete response, relapse and overall survival at last follow-up by visual inspection and Egger's tests. However, considerable publication bias was identified by visual inspection and Egger's tests regarding the outcomes of MRD negativity within responders, grade 3-4 rates of CRS and NT (Figure S4).
Discussion
Nowadays, cancer therapy has already transitioned from conventional chemotherapy to specific immune-based therapeutic strategies. As a state-of-the-art method, CAR-T therapy has achieved remarkable success in refractory hematological malignancies. Clinical trials of CD19-targeted CAR-T therapy were tested in acute and chronic leukemia with a 70%-90% response rate [47]. As for CAR-T therapy in MM, some previous studies have demonstrated the therapeutic potential. In this review, we assessed the efficacy and safety of CAR-T therapy based on 23 studies including 350 participants with RRMM globally. The analysis above revealed CAR-T therapy showed promising outcomes with tolerable toxicities in RRMM patients. Furthermore, BCMA-targeted CAR-T and anti-BCMA contained regimen contributed to better efficacy.
The design of co-stimulatory domains in CARs is a critical step to strengthen the function of T cells against antigens [48]. The co-stimulatory molecules are generally derived from either the CD28 receptor family (CD28, ICOS) or the tumor necrosis factor receptor family (4-1BB, OX40, CD27) [49]. And CD28 and 4-1BB have been most frequently used in clinical trials [3, 50]. The CAR-T therapy could produce more robust cytokines and enhance cytolytic capacity by adding CD28, 4-1BB, OX40, and other immune T-cell co-stimulators [3, 50-54]. The second-generation CARs have confirmed more supreme cytokine productivity and antitumor activity in mice in comparison to the first-generation ones [3]. It remains to be investigated whether other co-stimulatory molecules will exceed the well-established CD28 and 4-1BB domains. In addition, the profound understanding of influences on distinct domains is urgent so as to optimize CAR-T design and therapy.
MM is genetic and phenotypic heterogeneity. And this phenotypic heterogeneity includes differences in cell-surface antigen expressions [55]. Therefore, the choice of target antigens is crucial to determine antimyeloma activity and toxicity of CAR-T therapy. As observed in clinical trials involving anti-CD19 CAR-T therapy, hypogammaglobinemia was generated by destructing all B cells expressing CD19 [56, 57], regardless of normal or malignant one [48]. Thus, the targeted antigen should be absent on important normal tissues. Unfortunately, no plasma cells antigens were found to be strongly and uniformly expressed on all malignant plasma cells and not on normal cells. Further investigations should focus on this part to enhance the specificity of CAR-T therapy without increasing toxicities. In our review, we included multiple antigens targeted by CAR, most of which were BCMA. BCMA is known as CD269 and TNF receptor superfamily 17 (TNFRSF17) [58], and expressed in plasma cells and myeloma cells rather than in normal tissues and hematopoietic stem cells [46]. BCMA was recently reported to be uniformly expressed in most cases of MM by immunohistochemistry and flow cytometry [59], indicating that BCMA targeted CAR-T products may exert antimyeloma activity without serious adverse effects theoretically. In fact, plenty of BCMA targeted CAR-T products have been applied in active clinical trials. Several MM patients with infusion of anti-BCMA CAR-T have obtained objective responses, which is expected to be approved for clinical therapy in managing RRMM soon after. Other antigen targets, CD19, CD138, κ light chain and NKG2D-ligands, also have shown activities in RRMM. Further researches should aim to identify new targets for CAR-T therapy and optimize this strategy regarding tandem CAR-T or dual receptors within one T cell.
Toxicities such as CRS and NT restricted the widespread use of CAR-T therapy. CRS, the most important CAR-T toxicity, is an inflammatory syndrome caused by multiple cytokines produced by the CAR T cells themselves and other cells. It features hypotension, fevers, and tachycardia among many other abnormalities [48, 60]. NT are complex syndromes including encephalopathy, cognitive defects, dysphasias, seizures and cerebral edema, the pathogenesis of which remains poorly understood [60]. Multiple factors can result in CAR-T toxicity, including early and peak levels of certain cytokines, peak blood CAR-T cell levels, CAR-T cell doses, CAR design, patients' disease burden and so on. [60]. Toxicity management typically includes supportive care and immunosuppression with tocilizumab or corticosteroids [40, 61, 62]. In the future, further mechanistic understanding of these toxicities is an important area to improve the efficacy-to-toxicity ratio of CAR-T therapy.
In the section above, we have emphasized the therapeutic potential of CAR-T for MM by the promising early results, but this approach still has some limitations. Factors precluding durable remissions following CAR-T therapy include CAR-T cell manufacturing issues, limited CAR-T cell expansion and/or persistence, various resistance mechanisms and toxicities [63]. Firstly, this novel promising therapy requires a CAR T cell product to be successfully manufactured infused, activated and expanded, and then effectively mediates a cytotoxic response. Issues outlined above present barriers to effective efficacy and need to be improved. Secondly, poor persistent efficacy and resistance to CAR-T were observed in our analysis, which may be explained by immune escape due to target antigen loss or modulation [63]. Thirdly, selectivity of patients and cost-effectiveness should not be overlooked to expand patient access to CAR-T therapy. As for anti-BCMA CAR-T therapy, the possible reasons may include soluble BCMA blocking it from getting to the target and downregulation of the antigen's expression on malignant plasma cells over time. Based on these limitations, this novel treatment should be rationally applied in clinical practice.
In summary, our meta-analysis demonstrated impressive results of CAR-T cells, especially the anti-BCMA type, which can induce remarkable responses in highly pretreated RRMM patients. CAR-T therapy for MM is still at an early stage and more profound mechanisms are needed to be elucidated. In the future, phase 3 clinical trials should focus on investigating the efficacy and safety of CAR-T therapy so as to provide more evidence for more focused analysis of specific subgroups and make individualized treatments in subsequent studies. In addition, further researches should also optimize the design of CARs, explore different CAR-T products, and combine them with immunomodulatory drugs, checkpoint inhibitors, other CAR T cells, as well as gene-edited cellular products to enhance the safety and efficacy of this approach.
Strength and limitations
This systematic review and meta-analysis has the following strengths: we used a predefined search strategy and conducted data extraction and quality evaluation by two independent reviewers to minimize reviewer bias. Moreover, we systematically analyzed publication data about scFvs origin, co-stimulatory domains and antigens targeted by CAR, which was firstly evaluated in RRMM with CAR-T therapy. We conducted subgroup analysis by using country of enrolled participants, trial site and trial status as a moderator. These conclusions may contribute to making evidence-based decision and encouraging further research.
Nevertheless, our systematic review and meta-analysis has the following limitations. Firstly, quality of the included studies was assessed as considerable risk and statistical heterogeneity. Without the uniform criteria and baseline characteristics, data were heterogeneous. Moreover, several studies are still ongoing, and only publish preliminary outcomes, predicating the discrepancy of median follow-up. In a conclusion, a considerable risk of bias exists. Secondly, with a small number of articles, the estimate of subgroup analysis may under- or over-estimate the pooled proportions. Furthermore, we did not analyze the data on specific subgroups, including disease status, high-risk features, prior ASCT treatment, CAR-T doses, BCMA expression and CAR-T persistence due to lack of sufficient information. As the length of follow-up differs, the duration of response didn't be analyzed. Thirdly, no large-scale randomized controlled trials of CAR-T in RRMM have been reported. How much profit do this new strategy bring remains to be investigated.
Conclusion
Our study suggests that CAR-T therapy has demonstrated efficacy and safety in RRMM patients. BCMA-targeted CAR-T and anti-BCMA contained regimen have shown better efficacy. Our findings call for conducting large-scale randomized trials to obtain more evidence of CAR-T therapies in MM.
Abbreviations
CAR: Chimeric antigen receptor (CAR); RRMM: Relapsed/refractory multiple myeloma; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses; MRD: Minimal residual disease; CRS: Cytokine release syndrome; NT: Neurologic toxicities; BCMA: B cell maturation antigen; ASCT: Autologous stem cell transplantation; scFvs: single-chain variable fragments; SLAMF7: CS1 glycoprotein antigen; NKG2D: Natural killer group 2, member D.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Funds of China (No. 81870166), Hunan Provincial Innovation Foundation for Postgraduates (CX20200265), and Fundamental Research Funds for the Central Universities of Central South University (2020zzts768).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10:267-76
3. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388-98
4. Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015;36:494-502
5. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720-4
6. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024-8
7. US Food & Drug Administration: FDA approval brings first gene therapy to the United States. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm574058.htm.2017
8. US Food & Drug Administration: FDA approves tisagenlecleucel for adults with relapsed or refractory large B cell lymphoma. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm606540.htm
9. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N. et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol. 2018;36:2267-80
10. Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S. et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19:2048-60
11. Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M. et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight. 2019 4
12. Guo B, Chen M, Han Q, et a. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J Cell Immunother. 2016;2(1):28-35
13. Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O. et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J Clin Invest. 2016;126:2588-96
14. Higgins JPT Green S eds. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0: The Cochrane Collaboration, 2011. updated Mar 2011. https://www.handbook.cochrane.org
15. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097
16. Guo B, Moga C, Harstall C, Schopflocher D. A principal component analysis is conducted for a case series quality appraisal checklist. J Clin Epidemiol. 2016;69:199-207 e2
17. Borenstein M, Hedges LV, Higgins JP, Rothstein HR. A basic introduction to fixed-effect and random-effects models for meta-analysis. Res Synth Methods. 2010;1:97-111
18. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557-60
19. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK. et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol Res. 2019;7:100-12
20. Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E. et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129:2210-21
21. Green D, Pont M, Sather B, et a. Fully human Bcma targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood. 2018;132(Suppl 1):1011
22. Gregory T, Cohen A, Costello C, et a. Efficacy and safety of P-Bcma-101 CAR-T cells in patients with relapsed/refractory (r/r) multiple myeloma (MM). Blood. 2018;132(Suppl 1):1012
23. Han L, Gao Q, Zhou K, et a. The phase I clinical study of CART targeting BCMA with humanized alpaca-derived single-domain antibody as antigen recognition domain. J Clin Oncol. 2019;37(suppl):2535
24. Jiang S, Jin J, Hao S, et a. Low dose of human scFv-derived BCMA-targeted CAR-T cells achieved fast response and high complete remission in patients with relapsed/refractory multiple myeloma. Blood. 2018;132(Suppl 1):960
25. Li C, Mei H, Hu Y, et a. Improved efficacy and safety of a dual-target CAR-T cell therapy targeting BCMA and CD38 for relapsed/refractory multiple myeloma from a phase I study. EHA. 2019
26. Li C, Wang Q, Zhu H, et a. T cells expressing anti B-cell maturation antigen chimeric antigen receptors for plasma cell malignancies. Blood. 2018;132(Suppl 1):1013
27. Li C, Zhou J, Wang J, et a. Clinical responses and pharmacokinetics of fully human BCMA targeting CAR T-cell therapy in relapsed/refractory multiple myeloma. J Clin Oncol. 2019;37:8013
28. Liu Y, Chen Z, Fang H, et a. Durable Remission Achieved from Bcma-Directed CAR-T Therapy Against Relapsed or Refractory Multiple Myeloma. Blood. 2018;132(suppl 1):956
29. Mailankody S, Ghosh A, Staehr M, et a. Clinical Responses and Pharmacokinetics of MCARH171, a Human-Derived Bcma Targeted CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma: Final Results of a Phase I Clinical Trial. Blood. 2018;132(Suppl 1):959
30. Mailankody S, Htut M, Lee K, et a. JCARH125, anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: initial proof of concept results from a phase 1/2 multicenter study (EVOLVE). Blood. 2018;132(Suppl 1):957
31. Popat R, Zweegman S, et a. Phase 1 First-in-Human Study of AUTO2, the First Chimeric Antigen Receptor (CAR) T Cell Targeting APRIL for Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood. 2019;134(Suppl 1):3112
32. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D. et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2019;380:1726-37
33. Shah N, Alsina M, Siegel D, et a. Initial results from a phase 1 clinical study of bb21217, a next-generation anti Bcma CAR T therapy. Blood. 2018;132(Suppl 1):488
34. Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF. et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019;116:9543-51
35. Yan L, Shang J, Kang L, et a. Combined infusion of CD19 and Bcma-specific chimeric antigen receptor T cells for RRMM: initial safety and efficacy report from a clinical pilot study. Blood. 2017;130(Suppl 1):506
36. Yan Z, Cao J, Cheng H, Qiao J, Zhang H, Wang Y. et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol. 2019;6:e521-e9
37. Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y. et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11:141
38. Rajkumar SV, Kyle RA. Progress in Myeloma - A Monoclonal Breakthrough. N Engl J Med. 2016;375:1390-2
39. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN. et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25:625-38
40. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188-95
41. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW. et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139
42. Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z. et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther. 2014;25:1003-12
43. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH. et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165-74
44. Kunkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS. et al. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol Res. 2015;3:368-79
45. Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma. Mol Ther Methods Clin Dev. 2018;9:70-80
46. Lin Q, Zhao J, Song Y, Liu D. Recent updates on CAR T clinical trials for multiple myeloma. Mol Cancer. 2019;18:154
47. Xu D, Jin G, Chai D, Zhou X, Gu W, Chong Y. et al. The development of CAR design for tumor CAR-T cell therapy. Oncotarget. 2018;9:13991-4004
48. Mikkilineni L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. 2017;130:2594-602
49. Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells. 2019 8
50. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14:499-509
51. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161:2791-7
52. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL. et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676-84
53. Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol. 2015;33:9-15
54. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G. et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822-6
55. Paino T, Paiva B, Sayagues JM, Mota I, Carvalheiro T, Corchete LA. et al. Phenotypic identification of subclones in multiple myeloma with different chemoresistant, cytogenetic and clonogenic potential. Leukemia. 2015;29:1186-94
56. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA. et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099-102
57. Lokhorst HM, Schattenberg A, Cornelissen JJ, Thomas LL, Verdonck LF. Donor leukocyte infusions are effective in relapsed multiple myeloma after allogeneic bone marrow transplantation. Blood. 1997;90:4206-11
58. Berahovich R, Zhou H, Xu S, Wei Y, Guan J, Guan J. et al. CAR-T Cells Based on Novel BCMA Monoclonal Antibody Block Multiple Myeloma Cell Growth. Cancers (Basel). 2018 10
59. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ. et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688-700
60. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019;34:45-55
61. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011
62. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321-30
63. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16:372-85
Author contact
![]() Corresponding authors: Jing Liu, E-mail: jingliu0318com and Wen Zhou, E-mail: wenzhouedu.cn.
Corresponding authors: Jing Liu, E-mail: jingliu0318com and Wen Zhou, E-mail: wenzhouedu.cn.