Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(5):1225-1239. doi:10.7150/ijms.51743 This issue Cite
Research Paper
Identifying daily changes in circRNAs and circRNA-associated-ceRNA networks in the rat pineal gland
Yi Zheng#, Hao Jiang#, Hao-Qi Wang, Hai-Xiang Guo, Dong-Xu Han, Yi-Jie Huang, Yan Gao, Bao Yuan ![]() , Jia-Bao Zhang
, Jia-Bao Zhang ![]()
Department of Laboratory Animals, Jilin Provincial Key Laboratory of Animal Model, Jilin University, Changchun 130062, Jilin, P.R. China.
#These authors contributed equally to this work.
Received 2020-8-9; Accepted 2020-12-22; Published 2021-1-14
Abstract

Circular RNAs (circRNAs) are a new class of covalently closed circular RNA molecules that are involved in many biological processes. However, information about circRNAs in the pineal gland, particularly that of rats, is limited. To establish resources for the study of the rat pineal gland, we performed transcriptome analysis of the pineal glands during the day and night.
In this study, 1413 circRNAs and 1989 miRNAs were identified in the pineal gland of rats during the night and day using the Illumina platform. Forty differentially expressed circRNAs and 93 differentially expressed miRNAs were obtained, among which 20 circRNAs and 37 miRNAs were significantly upregulated during the day and 20 circRNAs and 56 miRNAs were significantly upregulated during the night.
As circRNAs have been reported to work as miRNA sponges, we predicted 15940 interactions among 40 circRNAs, 93 miRNAs and 400 mRNAs with differential diurnal expression using miRanda and TargetScan to build a ceRNA regulatory network in the rat pineal gland.
The diurnal expression profile of circRNAs in the rat pineal gland may provide additional information about the role of circRNAs in regulating changes in melatonin circadian rhythms. The analyzed data reported in this study will be an important resource for future studies to elucidate the altered physiology of circRNAs in diurnal rhythms.
Keywords: Pineal gland, RNA sequencing, microRNAs, circular RNAs
Introduction
The pineal gland is one of the central organs that regulates the circadian system. It converts external light signals into endogenous signals by secreting melatonin, the hormone of the night. In mammals, the nerve endings of sympathetic axons in the superior cervical ganglia (SCG) release norepinephrine (NE) and control melatonin synthesis [1]. NE stimulates the alpha and beta adrenergic receptor. The alpha adrenergic receptor stimulation increases intracellular Ca2+ concentration and the beta adrenergic receptor activates the adenylate cyclase (AC)/cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathway, which converts the basic leucine zipper (bZIP) TF cAMP-responsive element binding protein (CREB) into its phosphorylated form (pCREB) [2, 3]. At night, the cAMP/PKA/pCREB pathway [4] and other NE-stimulated signaling mechanisms [5] play a role in increasing the expression and activity of aralkylamine N-acetyltransferase (AANAT) and acetylserotonin O-methyltransferase (ASMT) [6-8], which are the last two enzymes in melatonin synthesis. Then cAMP-dependent phosphorylation triggers the binding of AANAT to 14-3-3 proteins (30 and 33 kDa), a family of highly conserved isoforms that exist as dimers. The pAANAT/14-3-3 complex shields AANAT from metabolic processes that lead to proteolysis. And the AANAT enzyme activity are more than 100-fold increased. Fianlly this mechanism significantly increase melatonin secretion. Whereas the regulation of melatonin secretion in the pineal glands of fish, amphibians, reptiles, and birds is different from that in mammals [9], the rat is one of the preferred experimental species to study the role of the pineal gland in melatonin secretion and some melatonin-deficient diseases in humans, such as adolescent idiopathic scoliosis [10], mammary tumorigenesis [11], Alzheimer's disease [12] and hypoxic-ischemic brain damage [13].
In addition to mRNAs, relevant experiments have shown that microRNAs (miRNAs) are also involved in melatonin secretion from the pineal gland. In the rat pineal gland, miR-483 was the first miRNA found to inhibit melatonin secretion [14]. And the four most abundant miRNAs are miR-182, miR-183, miR-127 and miR-96 and account for nearly half of the total miRNA population in the pineal gland. Then, miR-182 and miR-325-3p were found to regulate rat pineal function with HIBD [13, 15]. LncRNAs were also found to act as miRNA sponges to activate the CLOCK gene in the rat pineal gland [16]. However, studies on circular RNAs (circRNAs) in the rat pineal gland are still very rare.
CircRNAs have recently been identified as a new type of ncRNA [17]. CircRNAs are closed RNA molecules that are mainly produced from premRNA through variable shear processing in the eukaryotic transcriptome [18]. The circRNA ring structure makes it less sensitive to RNA exonuclease (RNase R) than linear RNA [19]. CircRNAs are conserved in different species and are abundantly expressed in the brain [20]. As shown in previous studies, circRNAs play important regulatory roles at the transcriptional and posttranscriptional levels [21]. CircRNAs also serve as competitive endogenous RNAs (ceRNAs) by binding intracellular miRNAs and blocking the inhibitory effects of miRNAs on target genes [22]; for example, circRNA-ZFR functions as a miR-101-3p sponge to increase Cullin 4B expression [23]. Relevant studies have also found that very few circRNAs are directly translated [24, 25]; for example, one such circRNA, circPPP1R12A, encodes a novel protein that promotes colon cancer. Although circRNAs have important biological functions, the roles of circRNAs in the pineal gland remain to be explored. Therefore, the expression and functions of circRNAs in the pineal gland must be identified.
This study explored the diurnal expression of circRNAs, miRNAs and mRNAs in the rat pineal gland using RNA-seq. The sequencing results were used to analyze, predict and construct possible ceRNA interaction networks. These results may identify new functions and roles of circRNAs in the pineal gland and provide ideas for further research into the mechanisms of ncRNAs in this tissue.
Materials and methods
Ethics statement
All animal procedures were performed according to the guidelines of the Guide for the Care and Use of Laboratory Animals of Jilin University, and the experimental protocols were approved by the Institutional Animal Care and Use Committee of Jilin University, Changchun, China. The permit number was SY201901011.
Animals
Sprague-Dawley (SD) rats were obtained from Liaoning Changsheng Biotechnology Co., Ltd. The pineal gland and other tissues were extracted from eight-week-old male SD rats. The rats were acclimated at room temperature (21±2 °C) on a 12:12 h light/dark cycle for at least 1 week with free access to food and water. All rats used in this study were euthanized with a CO2 anesthesia machine. CO2 levels were increased to 99.9% at a rate of 30% per minute and maintained at this level for 10 minutes to ensure that all rats were euthanized. Some rats were anesthetized and sacrificed in bright sunlight at Zeitgeber time (ZT) 7 and were considered the light group. Three of these rats were named C01, C02 and C03 (or S01, S02, and S03). Other rats were anesthetized and sacrificed under dark conditions (under red light) at ZT 19 and were considered the dark group. Three of these rats were named C04, C05 and C06 (or S04, S05, and S06). The rats named C01~06 were used for RNA-seq, and the other rats were used for other experiments.
RNA isolation
Total RNA was isolated using TRIzol reagent (Invitrogen). RNA degradation and contamination, particularly DNA contamination, were monitored on 1.5% agarose gels. RNA concentrations and purity were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). RNA integrity was assessed using the RNA Nano 6000 Assay Kit and the Agilent Bioanalyzer 2100 System (Agilent Technologies, CA, USA).
Library preparation for sequencing
For circRNAs and mRNA, the Ribo-Zero rRNA Removal Kit (Epicenter, Madison, WI, USA) was used to remove rRNA. Sequencing libraries were generated using the NEBNext® UltraTM Directional RNA Library Prep Kit for Illumina® (NEB, USA) according to the manufacturer's recommendations, and index codes were added to attribute sequences to each sample. After adding fragmentation buffer to break the rRNA-depleted RNA at random, first-strand cDNAs were synthesized using random hexamer primers and reverse transcriptase. Then, second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. The remaining overhangs were converted into blunt ends. After adenylation of the 3' ends of DNA fragments, the NEBNext adaptor with a hairpin loop structure was ligated to prepare for hybridization. AMPure XP Beads (Beckman Coulter, Beverly, USA) were used to preferentially select insert fragments with a length of 150-200 bp. Then, PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index(X) Primer.
For miRNAs, a total amount of 2.5 ng of RNA per sample was used as input material for RNA sample preparation. Sequencing libraries were generated using NEBNext® Small RNA Library Prep Set for Illumina (NEB, USA) according to the manufacturer's recommendations, and index codes were added to attribute sequences to each sample. Briefly, first, the 3' SR adaptor was ligated. The SR RT primer hybridizes to the excess of the 3' SR adaptor and transforms the single-stranded DNA adaptor into a double-stranded DNA molecule to prevent the formation of adaptor dimers. Second, the 5' SR adaptor was ligated. Then, reverse transcription was performed to synthesize the first chain. Finally, PCR amplification and size selection were conducted. A PAGE gel was used to electrophoretically screen fragments, and gel extraction was performed on the fragments to obtain small RNA libraries.
The PCR products were subsequently purified (AMPure XP system), and the library quality was assessed using the Agilent Bioanalyzer 2100 system.
Clustering and sequencing
The clustering of the index-coded samples was performed on the cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (for circRNA and mRNA) and v4-cBot-HS (for miRNA) (Illumina) according to the manufacturer's instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq X Ten platform, and 2*150-bp paired-end reads (for circRNA and mRNA) and 50-nt single-end reads (for miRNA) were generated. The construction of the libraries and the RNA-Seq were performed by the Biomarker Biotechnology Corporation (Beijing, China). The sequencing data were uploaded in GSE146193.
Quality control for libraries
Raw reads in the fastq format were initially processed using in-house Perl scripts. In this step, clean reads were obtained by removing reads containing adapters and poly-N sequences and low-quality reads from the raw data.
For miRNAs, reads were trimmed and cleaned by removing sequences smaller than 15 nt or longer than 35 nt. At the same time, the Q-score and GC content of the clean data were calculated [26]. Q-score = -10*log10 P (in the formula, P is the probability of error in base recognition). All downstream analyses were based on high-quality clean reads. At the same time, Pearson's correlation coefficient was calculated to evaluate correlations between biological replicates of the sequencing samples [27].
After clean reads were obtained, they were aligned with the rat genome to obtain the location in the rat genome (Rnor_6.0) and the unique sequence characteristics of the sequencing sample. The reads that mapped to the specific site in the rat genome are called mapped reads, and the corresponding data are called mapped data.
Identification of circRNAs, mRNAs and miRNAs
We used CIRI (CircRNA Identifier) tools and find_circ software to identify circRNAs [28, 29]. CIRI software uses the reads to compare with the reference gene sequence, generates SAM files, analyzes the CIGAR values in SAM files, and scans PCC signals (paired chiastic clipping signals) from SAM files. The CIGAR value in the junction read is characterized by xS/HyM or xMyS/H, where X and y represent the number of bases, M is on mapping, S is soft clipping, and H is hard clipping. For double-end reads, the CIRI algorithm considers a pair of reads, one of which can be mapped to circRNA, and the other one also needs to be mapped to circRNA's sequence interval. For single exons forming rings, or the circular structure formed by “long exon 1-short exon - long exon 2”, CIGAR values should be xS/HyMzS/H and (x+y)S/HzM or xM(y+z)S/H. CIRI software can separate these two cases very well. For splicing signals GT/AG, CIRI will also consider other weak splicing information, such as AT/AC, and the algorithm will extract exon boundary positions from the GTF/GFF file and filter false positives with known boundaries. Since the ring-forming splicing sites of the circRNA reads could not be directly aligned to the genome, the find_circ software first took 20 bp as anchor points at both ends of reads that could not be aligned with the genome and then took anchor points as independent reads to the genome for alignment and searched for the unique matching sites. If the alignment position of two anchor points is reversed in the linear direction, the reads of anchor points are extended until the junction position of circRNA is found. At this time, the sequences of both sides are GT/AG splicing signals, and the circRNA reads are judged to be circRNAs.
For mRNA, the transcriptome was assembled using StringTie based on the reads mapped to the reference genome. The assembled transcripts were annotated using the gffcompare program. Three computational approaches, including CPC/CNCI/Pfam/CPAT, were combined to sort nonprotein coding RNA candidates from putative protein-coding RNAs in the unknown transcripts. Putative protein-coding RNAs were filtered out using a minimum length and exon number threshold. Transcripts with lengths greater than 200 nt and more than two exons were selected as lncRNA candidates and further screened using CPC/CNCI/Pfam/CPAT, which has the power to distinguish protein-coding genes from noncoding genes.
The intersection of the results obtained using the two methods was the final prediction. In terms of the identification of known miRNAs, we compared the read sequences with the reference genome using the mature miRNA column in miRBase [30]. Read sequences that were identical to known miRNAs were considered known miRNAs. For sequences that have not been identified as known miRNAs, miRDeep2 software was used to predict new miRNAs [31].
Analysis of differentially expressed RNAs
For circRNA, miRNA and mRNA, the expression levels were determined from the read counts and were normalized using the transcripts per million (TPM) algorithm [32]. The differential expression analysis was performed for the two groups (C01, C02, C03 and C04, C05, C06) using the DESeq R package. CircRNAs with an adjusted P value <0.05 and an absolute value of a log2 (fold change) >2 and miRNAs with an adjusted FDR <0.01 and an absolute value of a log2 (fold change) >0.5 identified using DESeq were considered differentially expressed.
Prediction of miRNA target genes
The miRanda (animal), RNAhybrid and TargetScan databases were used to predict the miRNAs targeting mRNAs and circRNAs. The input files were miRNA, mRNA and circRNA base sequence files that we obtained from the RNA-seq. Then, the circRNAs, mRNAs and miRNAs with different expression were selected to construct the ceRNA network.
Functional annotation of circRNA host genes and miRNA target genes
Some circRNA sequences can be aligned to a gene sequence, which is defined as the circRNA host gene. The functions of circRNA host genes and miRNA target genes were annotated using the NR database [33] (a nonredundant protein sequence database), the Swiss-Prot database [34] (a manually annotated, nonredundant protein sequence database), the Gene Ontology (GO) database [35], the Clusters of Orthologous Groups of proteins (COG) database [36], the euKaryotic Orthologous Groups of proteins (KOG) database [37], and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [38].
Reverse transcription and qPCR
First-strand cDNAs were generated using a FastKing RT Kit (with gDNase) (Tiangen) with random primers according to the manufacturer's protocol. Primers for the differentially expressed circRNAs were obtained from RiboBio Biotechnology Co., Ltd (Support Table S13). qPCR was performed using a Mastercycler ep Realplex2 system (Eppendorf, Germany). The products were quantified using Super Real PreMix Plus (SYBR Green, Tiangen, China) according to the manufacturer's instructions. The relative expression levels of circRNAs were normalized to GAPDH, and the dark group was considered the control group.
Statistical analysis
All data are presented as the means ± standard deviations of three independent experiments. Significant differences between multiple groups were determined using one-way ANOVA with SPSS 19.0 for Windows. P < 0.05 was considered a statistically significant difference.
Results
Sequencing quality control
The original sequences obtained from sequencing contain joint sequences or low-quality sequences. Quality control of the original data was conducted to obtain high-quality sequences (i.e., clean reads) and to ensure the accuracy of the subsequent analysis. After sequencing quality control, a total of 127.99 Gb clean data were obtained for circRNA samples. Per sample has an average of 71.3 M reads, and the Q-score (30) percentage of each sample was not less than 95.05% (Support Table S1). For the miRNAs, the number of clean reads in each miRNA sample was greater than 17.75 M after quality control, and the Q-score (30) percentage of each sample was not less than 98.32% (Support Table S2).
Sequence alignment and subsequent analysis were performed using the rat genome as a reference. Clean reads were sequentially aligned with the rat genome to obtain the location in the rat genome or gene, as well as the characteristic information about the sequence that was unique to the sample.
According to the comparison of circRNA sequencing results, the ratio of reads to the rat genome reached 99.99% (Support Table S3). Clean reads obtained from miRNA sequencing were sequentially compared with the Silva database, GtRNAdb database, Rfam database and Repbase database using Bowtie software. The reads were categorized and annotated as scRNA, rRNA, snRNA, snoRNA, tRNA and unannotated reads containing miRNA sequences. The results showed that an average of 67% of the unannotated reads were identified as miRNAs (Support Table S4). Data utilization was normal, and the selected reference genome was sufficient for subsequent analysis.
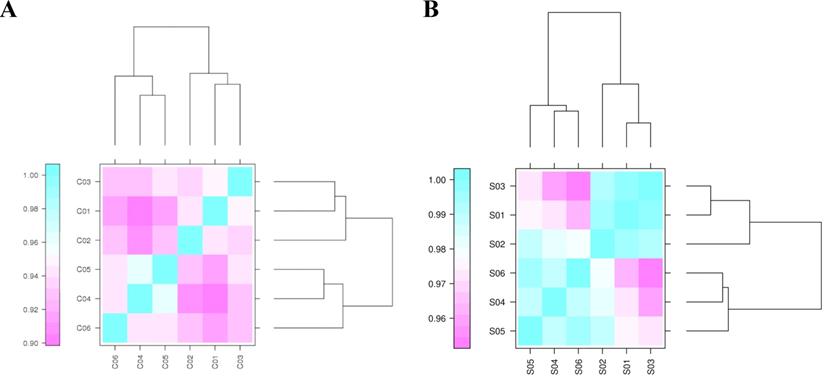
An assessment of the correlation of biological replicates is important for analyzing transcriptome sequencing data. It tests the repeatability of biological experiments, evaluates the reliability of differentially expressed genes, and assists with screening abnormal samples. Pearson's correlation coefficient (r) was calculated to evaluate the correlations of biological replicates. The closer r2 is to 1, the stronger the correlation between the two repeated samples. The r2 statistics of any pair of circRNA and miRNA samples are shown in Figure 1, Support Table S5 and S6. Most circRNAs and miRNAs in the two groups showed similar expression trends and a high replicate correlation (all r2 values were above 0.90).
Correlation coefficients for circRNA and miRNA samples. “C” represents the sample for circRNA-seq. C01, C02, and C03 indicate the samples harvested under light conditions. C04, C05, and C06 represent the samples collected under dark conditions. “S” indicates the sample for miRNA-seq. S01, S02, and S03 are the samples collected under light conditions. S04, S05, and S06 represent the samples collected under dark conditions.
Deep sequencing analysis of circRNAs and miRNAs in rats
We performed RNA-seq to detect circRNA and miRNA expression in rat pineal glands between day and night. We found that a total of 1413 circRNAs were expressed in the pineal gland (Support Table S7). The circRNAs were named according to the positions on the genome that the software recognizes on both ends of their base sequences. The lengths of the circRNAs were distributed from 200-3000+ nt. Most circRNAs were exonic circRNAs, but circRNAs with lengths greater than 3000 nt were mainly derived from intergenic regions (Figure 2A). Although the circRNAs were distributed among all 22 chromosomes, they were mainly distributed on chromosomes 3, 5 and 6 (Figure 2B). Then, we mapped the measured circRNAs to the genome and drew a circos diagram (Figure 2C).
The circRNAs exhibiting diurnal expression patterns in the rat pineal gland. (A) Length distribution of all circRNAs. (B) Distributions of all circRNAs on chromosomes. (C) Statistical analysis of the circRNA distribution in the genome and a circos plot of the expression levels in each sample. The histogram shows the number of circRNAs at each position, and the color represents the expression distribution of each sample: 0-1: blue, 1-10: green, 10-100: orange, and 100+: red. The heat map from the outside to the inside indicates C01, C02, C03, etc. (D) Diagram of circRNAs that are expressed in all three replicates of a group (light or dark). (E) Numbers of the three types of circRNAs coexpressed in the light group and dark group.
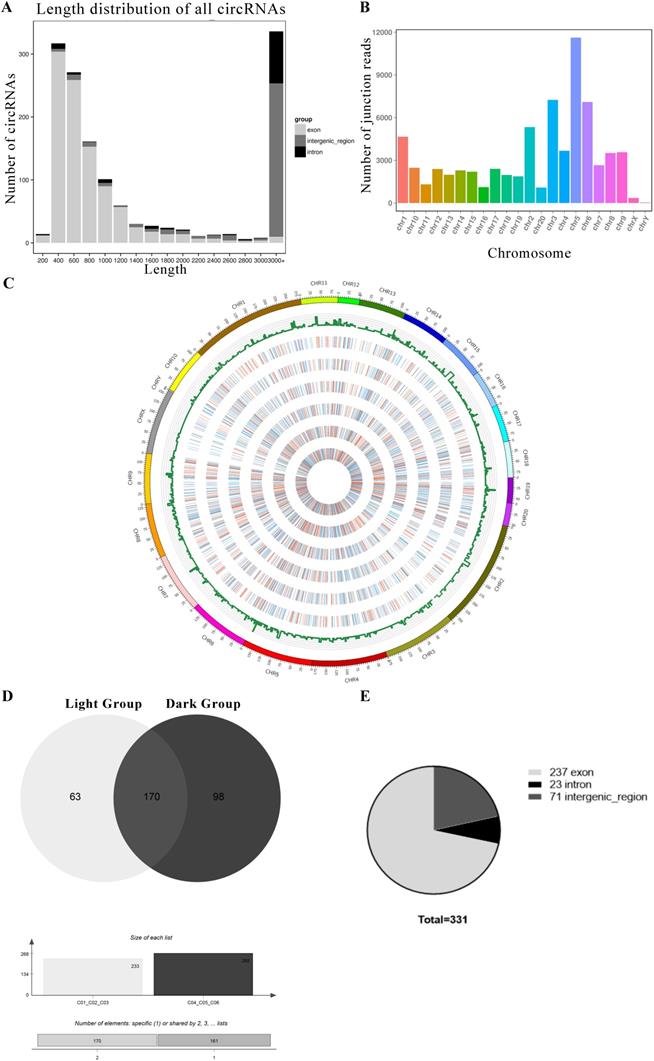
However, not all the circRNAs were both expressed in the light group and dark group, and we selected the circRNAs expressed in all three replicates of the light group or the dark group to be used in the next analysis. Sixty-three circRNAs were mainly expressed under light conditions. They were expressed in all light samples and not expressed in at least one dark sample. Ninety-eight circRNAs were mainly expressed under dark conditions. In addition, 170 circRNAs were coexpressed in all samples (Figure 2D). The selected circRNA sources were statistically analyzed, and 72% of circRNAs were derived from exons, 7% were derived from introns, and 21% were derived from intergenic regions (Figure 2E). The relevant mRNA expression information is provided in Support File 1.
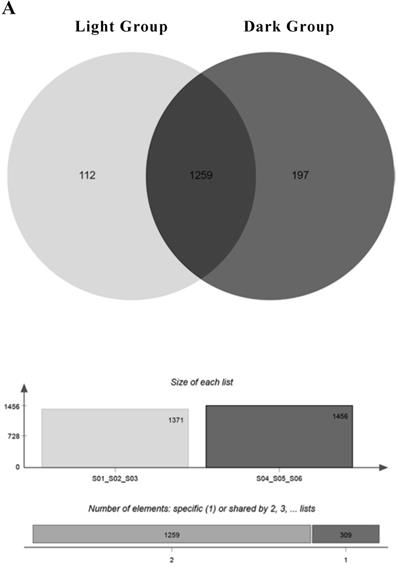
A total of 1989 miRNAs were detected using miRDeep2, including 638 known miRNAs and 1351 newly predicted miRNAs (Table 1). Since miRNAs are highly conserved among species, we classified the detected known miRNAs and new miRNAs into miRNA families based on sequence similarity (Support Table S9). One hundred twelve miRNAs were mainly expressed under light conditions. They were expressed in all light groups and not expressed in at least one dark sample. One hundred ninety-seven miRNAs were mainly expressed under dark conditions, and 1259 miRNAs were expressed in all groups (Figure 3A). The relevant information of miRNA sequencing is explained in Support File 2.
The number of miRNAs expressed in the sample
| Sample-ID | Known-miRNAs | Novel-miRNAs | Total |
|---|---|---|---|
| S01 | 568 | 1065 | 1633 |
| S02 | 579 | 1128 | 1707 |
| S03 | 565 | 1058 | 1623 |
| S04 | 586 | 1161 | 1747 |
| S05 | 572 | 1072 | 1644 |
| S06 | 579 | 1116 | 1695 |
| Total | 638 | 1351 | 1989 |
The miRNAs that are expressed in all three replicates of a group (light or dark).
Enrichment of the differentially expressed circRNAs and miRNAs
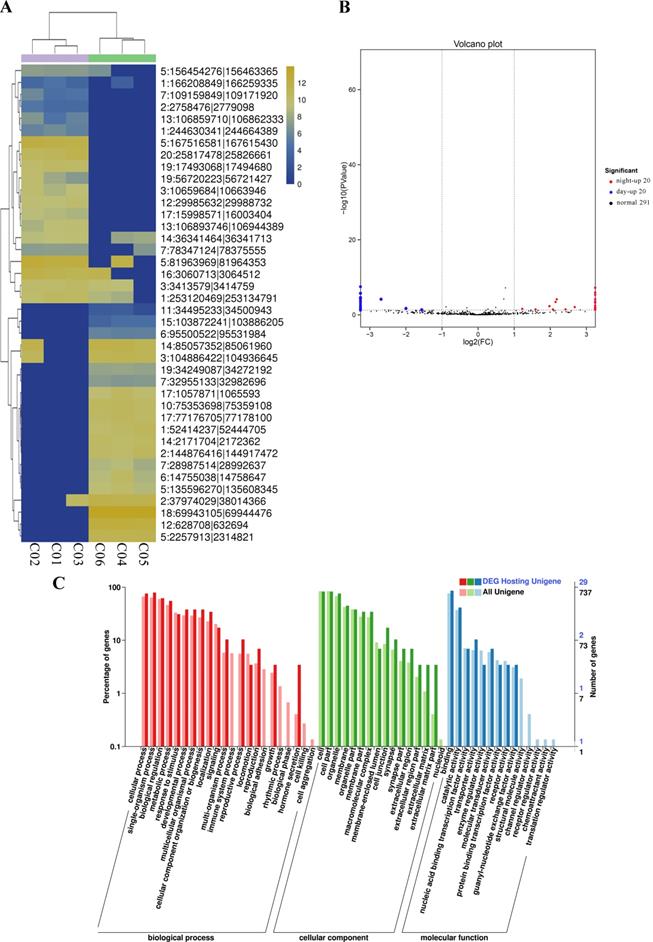
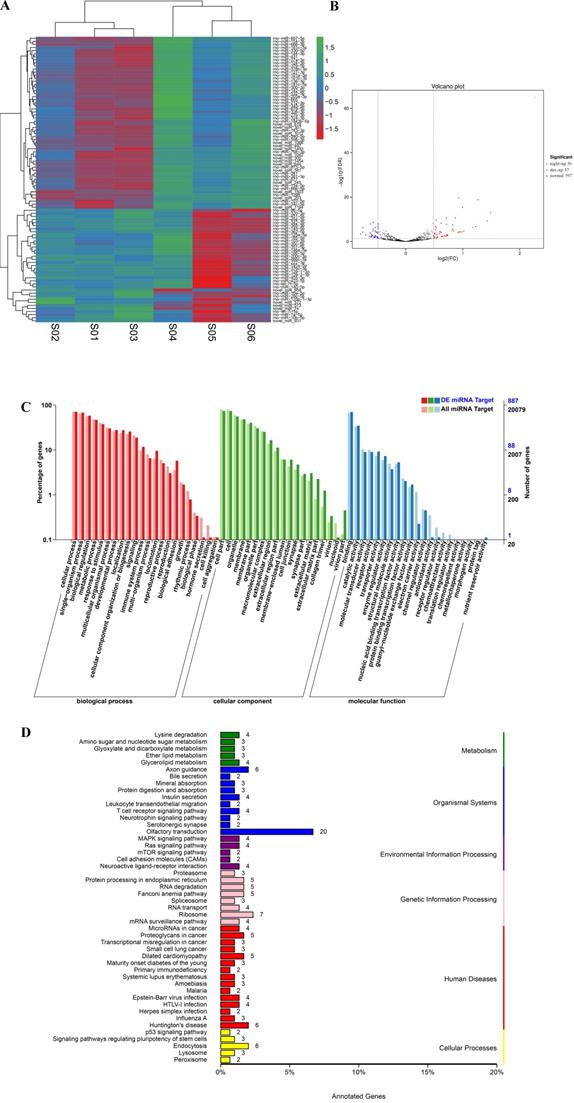
After the expression of all circRNAs and miRNAs was statistically analyzed, the RNA expressed only in individual samples was excluded. Then, DEseq was used to analyze the diurnal differential expression of circRNAs and miRNAs. For comparisons of the processed data between the light and dark groups, a fold change > 2 and P value < 0.05 were selected as the criteria for circRNAs with significant differential expression (Figure 4A and Support Table S8). Ultimately, 40 differentially expressed circRNAs were obtained, among which 20 circRNAs were significantly upregulated during the day and 20 circRNAs were significantly upregulated during the night (Figure 4B and Table 2). We performed GO and KEGG enrichment analyses for all the circRNA host genes and for the host genes of the differentially expressed circRNAs. The GO enrichment results are presented in Figure 4C and Support Table S7. In addition, a topGO-directed acyclic graph was constructed with the enriched terms. Ultimately, 29 circRNA host genes were annotated with GO terms, including “protein serine/threonine kinase activity” (GO: 0004674) and “ion channel activity” (GO: 0005216). The results of the KEGG pathway enrichment analysis of the differentially expressed circRNA host genes are shown in Support Table S7. Twenty of the genes were enriched in the KEGG pathways bacterial invasion of epithelial cells, bile secretion and CoA biosynthesis. All data for differentially expressed circRNAs are provided in Support Tables S7-8.
The top 10 circRNAs with the most significant differentially expressed
| #ID | circRNA length | P-Value | log2FC | regulated |
|---|---|---|---|---|
| 17:77176705|77178100 | 380 | 0.000 | -Inf | day-up |
| 6:95500522|95531984 | 31463 | 0.000 | -2.176 | day-up |
| 1:52414237|52444705 | 329 | 0.000 | -Inf | day-up |
| 19:34249087|34272192 | 23106 | 0.000 | -Inf | day-up |
| 12:628708|632694 | 313 | 0.000 | -2.142 | day-up |
| 7:32955133|32982696 | 27564 | 0.002 | -Inf | day-up |
| 7:28987514|28992637 | 568 | 0.004 | -Inf | day-up |
| 14:2171704|2172362 | 359 | 0.004 | -Inf | day-up |
| 17:1057871|1065593 | 7723 | 0.005 | -Inf | day-up |
| 3:104886422|104936645 | 50224 | 0.006 | -1.951 | day-up |
| 1:244630341|244664389 | 1749 | 0.000 | Inf | night-up |
| 13:106859710|106862333 | 327 | 0.000 | Inf | night-up |
| 13:106893746|106944389 | 1800 | 0.000 | Inf | night-up |
| 5:167516581|167615430 | 762 | 0.000 | Inf | night-up |
| 20:25817478|25826661 | 9184 | 0.000 | Inf | night-up |
| 17:15998571|16003404 | 673 | 0.000 | Inf | night-up |
| 16:3060713|3064512 | 566 | 0.000 | 2.687 | night-up |
| 3:10659684|10663946 | 399 | 0.001 | Inf | night-up |
| 12:29985632|29988732 | 353 | 0.001 | Inf | night-up |
| 7:109159849|109171920 | 12072 | 0.002 | Inf | night-up |
Differential expression of circRNAs between day and night. (A) Cluster analysis of the differentially expressed circRNAs. The different columns represent different samples, and the different rows represent different circRNAs. The color represents the level of circRNA expression in the sample (log2TPM+1). (B) Volcano plot of circRNA expression between the two groups. The abscissa represents the logarithm of the difference in circRNA expression between two groups. The ordinate represents the negative logarithm of the P value. The blue dots represent circRNAs with day-up expression, the red dots represent circRNAs with night-up expression, and the black dots represent circRNAs with no significant differences in expression. (C) GO map of all the circRNA host genes and the host genes of the differentially expressed circRNAs. The abscissa shows the GO classification, the left ordinate shows the percentage of genes, and the right ordinate shows the number of genes. All Unigene: the host genes of all circRNAs. DEG Hosting Unigene: the host genes of the differentially expressed circRNAs.
Similarly, for comparisons of the processed data between the day and night groups, a fold change > 0.5 and FDR<0.01 were selected as the criteria for miRNAs with significant differential expression (Figure 5A and Support Table S10). Ultimately, 93 differentially expressed miRNAs were obtained, among which 37 miRNAs were significantly upregulated during the day and 56 miRNAs were significantly upregulated during the night (Figure 5B and Table 3). We performed GO and KEGG enrichment analyses of all the miRNA target genes and the target mRNAs of the differentially expressed miRNAs. The GO enrichment results are presented in Figure 5C. In addition, a topGO-directed acyclic graph was constructed with the enriched terms. Ultimately, 887 miRNA target genes were annotated with the GO terms “positive regulation of transcription” (GO: 0045944), “G-protein coupled receptor signaling pathway” (GO: 0007186), “G-protein coupled receptor activity” (GO: 0004930) and “nucleolus” (GO: 0005730). The results of the KEGG pathway enrichment analysis of the differentially expressed genes are shown in Figure 5D. Five hundred forty target genes were enriched in the KEGG pathways Jak-STAT signaling pathway, mRNA surveillance pathway, spliceosome and protein processing in the endoplasmic reticulum. All data for differentially expressed miRNAs and the analysis of the miRNAs are provided in Support Tables S10-12.
The top 10 miRNAs with the most significant differentially expressed
| #ID | Pvalue | FDR | log2FC | regulated |
|---|---|---|---|---|
| novel_miR_581 | 0.000 | 0.000 | 2.270 | night-up |
| rno-miR-341 | 0.000 | 0.000 | 0.926 | night-up |
| rno-miR-1298 | 0.000 | 0.000 | 1.266 | night-up |
| rno-miR-154-3p | 0.000 | 0.000 | 0.996 | night-up |
| rno-miR-122-5p | 0.000 | 0.000 | 1.485 | night-up |
| rno-miR-124-3p | 0.000 | 0.000 | 0.870 | night-up |
| rno-miR-431 | 0.000 | 0.000 | 0.706 | night-up |
| rno-miR-101a-3p | 0.000 | 0.000 | 0.535 | night-up |
| novel_miR_644 | 0.000 | 0.000 | 1.428 | night-up |
| rno-miR-666-5p | 0.000 | 0.000 | 0.621 | night-up |
| rno-miR-376c-3p | 0.000 | 0.000 | -0.578 | day-up |
| rno-miR-342-3p | 0.000 | 0.000 | -0.621 | day-up |
| rno-miR-191a-5p | 0.000 | 0.000 | -0.486 | day-up |
| rno-miR-129-1-3p | 0.000 | 0.000 | -0.494 | day-up |
| rno-miR-129-2-3p | 0.000 | 0.000 | -0.494 | day-up |
| rno-miR-125b-1-3p | 0.000 | 0.000 | -0.476 | day-up |
| rno-miR-365-3p | 0.000 | 0.000 | -0.755 | day-up |
| rno-miR-494-3p | 0.000 | 0.000 | -0.543 | day-up |
| rno-miR-451-5p | 0.000 | 0.000 | -0.491 | day-up |
| rno-miR-10b-5p | 0.000 | 0.000 | -0.577 | day-up |
Differential expression of miRNAs between day and night. (A) Cluster analysis of the differentially expressed miRNAs. The different columns represent different samples, and the different rows represent different miRNAs. The color represents the level of miRNA expression in the sample (log2TPM+1). (B) Volcano plot of miRNA expression between the two groups. The abscissa represents the logarithm of the difference in miRNA expression between two groups. The ordinate represents the negative logarithm of the FDR. The blue dots represent miRNAs with day-up expression, the red dots represent miRNAs with night-up expression, and the black dots represent miRNAs with no significant differences in expression. (C) GO map of all the miRNA target genes and the differentially expressed genes. The abscissa indicates the GO classification, the left ordinate indicates the percentage of genes, and the right ordinate indicates the number of genes. DE miRNA Target: the miRNA targeting genes with differential expression. All miRNA Target: all the miRNA targeting genes. (D) KEGG classification map of the miRNA target genes. The ordinate lists the name of the KEGG metabolic pathway, and the abscissa shows the number of miRNA target genes annotated to the pathway and the proportion among the total number of genes annotated.
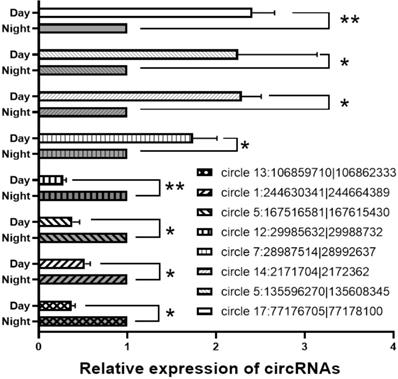
Relative expression of circRNAs
For verification of the circRNA data generated using RNA-seq analysis, we selected 8 circRNAs and then detected their relative expression levels in the pineal gland during the day and night using qPCR. Since the two side sequences of the circRNA splicing site cannot match on the genome, the primers for circRNAs were designed to amplify across circRNA splicing sites with specificity (Support Table S13). As shown in Figure 6, these circRNAs were significantly differentially expressed in the rat pineal gland between day and night.
qPCR results for differentially expressed circRNA. *, P<0.05. **, P<0.01.
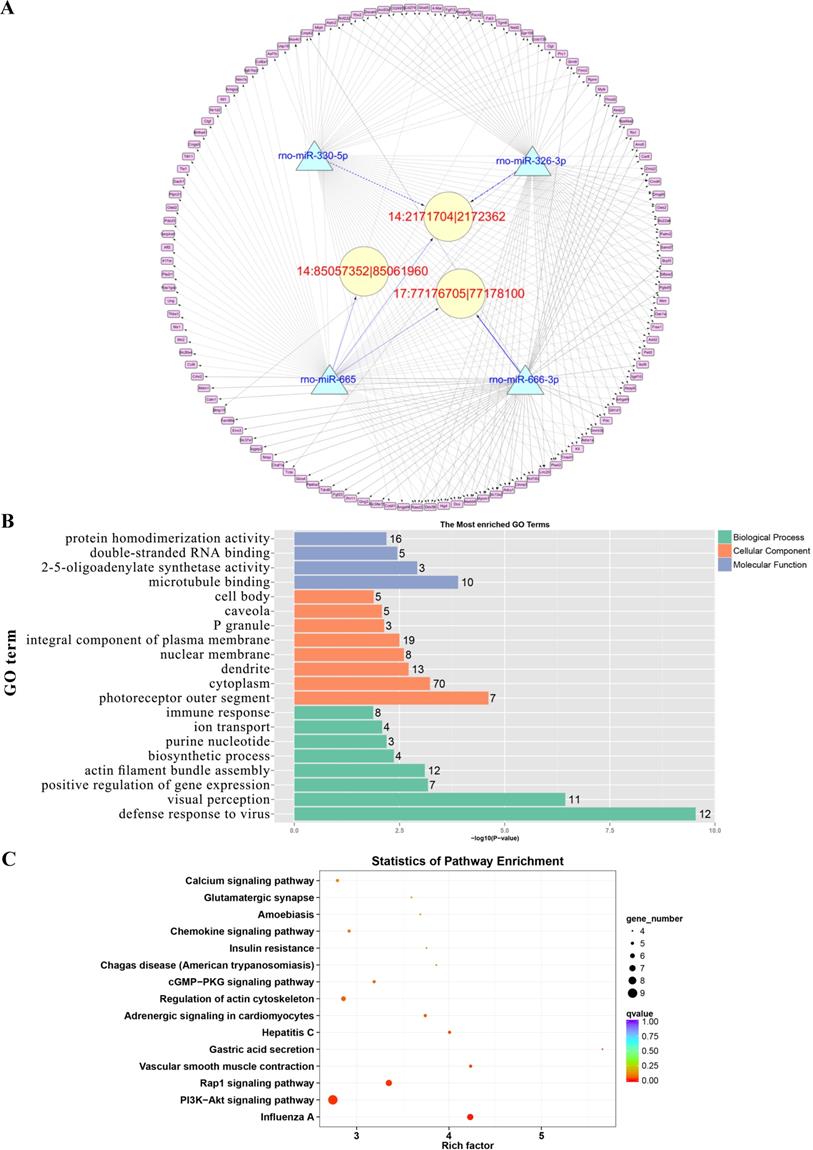
Interactions between circRNAs, mRNAs and miRNAs
Since circRNAs and mRNAs contain multiple miRNA binding sites, miRNAs that may target circRNAs and mRNAs were predicted by the software. The circRNAs, miRNAs and mRNAs we used were identified as differentially expressed. Notably, 6222 interactions among 20 night upregulated circRNAs, 37 night downregulated miRNAs and 205 night upregulated mRNAs were predicted as the ceRNA network in the rat pineal gland at night, and 9718 interactions among 20 day upregulated circRNAs, 53 day downregulated miRNAs and 195 day upregulated mRNAs were predicted as the ceRNA network during the day time. The day and night circRNA-miRNA-mRNA regulatory networks were generated using Cytoscape software to further elucidate the underlying mechanism of the differentially expressed circRNAs we identified (Figure 7A and D).
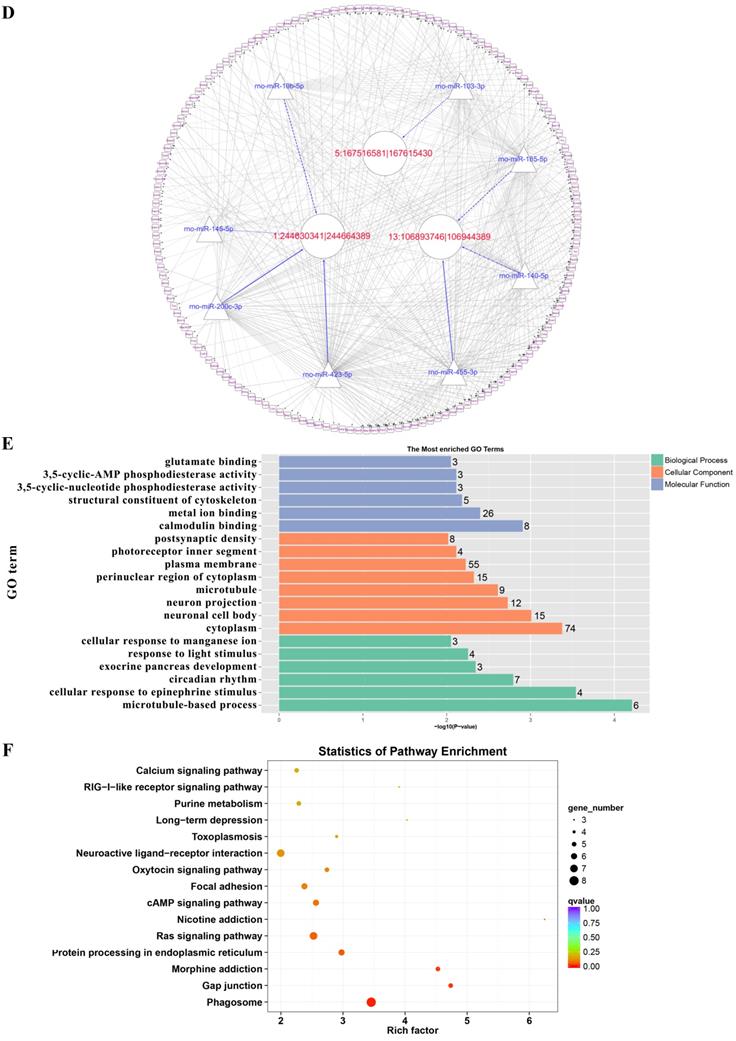
Prediction of the targeting relationships among miRNAs, mRNAs and circRNAs with differential expression. (A) The daytime ceRNA network. The triangles represent miRNAs, the circles represent circRNAs, the rectangles represent mRNAs, the blue lines indicate targeting relationships between miRNAs and circRNAs, and the black lines indicate targeting relationships between miRNAs and mRNAs. (B) GO map of all the miRNA target genes in the daytime ceRNA network. (C) KEGG classification map of the miRNA target genes in the daytime ceRNA network. (D) The night-time ceRNA network. (E) GO map of all the miRNA target genes in the night-time ceRNA network. The left ordinate lists the GO classification, the abscissa shows the percentage of genes, and the right ordinate indicates the number of genes. (F) KEGG classification map of the miRNA target genes in the night-time ceRNA network. The ordinate lists the names of the KEGG metabolic pathways, and the abscissa shows the number of miRNA target genes annotated to the pathway and the proportion among the total number of genes annotated.
GO analysis and KEGG pathway analysis were conducted to explore the biological functions of the ceRNA target genes
For the daytime ceRNA network, the GO analysis revealed that the terms with the most genes and the terms with the highest enrichment scores were both “response to virus” (GO: 0009615) in the biological process category. For the cellular component category, the term “cytoplasm” (GO: 0005737) included the most genes, and “photoreceptor outer segment” (GO: 0001750) was the most significantly enriched term. Furthermore, “protein homodimerization activity” (GO: 0042803) was related to the most genes, and the term with the most significant enrichment was “microtubule binding” (GO: 0008017) in the molecular function category (Figure 7B). Moreover, the enriched KEGG pathways were the PI3K-Akt signaling pathway, Rap1 signaling pathway, adrenergic signaling in cardiomyocytes, and cGMP-PKG signaling pathway, among others. These pathways were related to the transmission of light signals and regulation of neuroendocrine signals (Figure 7C).
For the night ceRNA network, the GO analysis revealed that the term with most genes and the term with the highest enrichment score were both “cytoplasm” (GO: 0005737). For the biological process and molecular function categories, the terms “cellular response to epinephrine stimulus” (GO: 0071872), “circadian rhythm” (GO: 0007623), “response to light stimulus” (GO: 0009416), and “3',5'-cyclic-AMP phosphodiesterase activity” (GO: 0004115) were related to melatonin secretion and circadian rhythm in the pineal gland (Figure 7E). Moreover, the enriched KEGG pathways were phagosome, gap junction, morphine addiction, protein processing in endoplasmic reticulum, Ras signaling pathway, and cAMP signaling pathway, among others (Figure 7F).
Discussion
The pineal gland is the main endocrine organ that secretes melatonin, a biological circadian signal that plays an important regulatory role in animal fitness [39]. Changes in noncoding RNAs in the pineal gland are considered an important component of the circadian rhythm mechanism [40]. According to previous studies, lncRNAs exhibit differential expression in the pineal gland between day and night and are believed to be involved in circadian biology [41]. miR-483 and miR-325-3p were also found to be involved in circadian rhythm regulation by inhibiting AANAT expression [13, 14]. At the same time, as a member of the ncRNA family, circRNAs have been increasingly shown to play important roles in various cellular functions [42]. However, studies of circRNAs in the rat pineal gland have rarely been reported. Only one study reported significant differences in circRNA expression in the pineal gland of AD mice [43]. Therefore, in our study, RNA-seq was used to identify and detect the expression of mRNAs, circRNAs and miRNAs in the rat pineal gland between day and night. Finally, we identified 40 circRNAs and 97 miRNAs that were differentially expressed. And at the same time, the lengths and distributions of all circRNAs on chromosomes were sort out, which may provide the data basis for further targeted studies of circRNAs.
For the miRNAs in the pineal gland, previous study showed that miR-182, miR-183, miR-127 and miR-127 was the 4 most abundant pineal miRNAs. And pinealocytes transfected with these miRNAs showed no effect on melatonin synthesis. In our research, miR-182 and miR-183-5p are also the most abundant miRNAs. For more and more miRNAs are annotated in the miRBase, other miRNAs are found with a high level of expression in our research. Such as the let-7 fimally, which is mainly expressed in mammalian brains, more members of it are found in the pineal gland than before. And some of them are significantly changed between day and night. As miRNAs are involved in transcriptional regulation of mRNA, the potential functions of miRNAs can be interpreted by predicting the target genes. According the predicted results, we can purposefully transfect miRNAs and verify their physiological functions.
Typically, circRNAs function as miRNA sponges to competitively adsorb microRNAs and regulate intracellular gene expression, which is known as the ceRNA mechanism [44]. We examined circRNA-miRNA and miRNA-mRNA interactions and discovered that each selected circRNA that contained at least one miRNA binding site was able to interact with several miRNAs. Then, the differentially expressed circRNAs, miRNAs and mRNAs were selected to draw a complete picture of ceRNA regulatory networks. This circRNA-miRNA-mRNA network may provide insights into the regulatory pathways in the pineal gland or the cascade-amplifying synergistic effects of circRNA-miRNA and miRNA-mRNA networks. This network provided theoretical support that circRNAs probably played a role in circadian rhythms by indirectly targeting certain mRNAs.
Furthermore, a GO enrichment analysis and KEGG pathway analysis were also performed to functionally annotate the predicted target genes. For the daytime ceRNA network, GO analysis revealed the enrichment of the terms “photoreceptor outer segment” and “protein homodimerization activity”, which are related to adrenergic receptors [45] and rhythmic pineal protein/DNA binding transcription factors [46]. The KEGG pathway analysis revealed several important pathways related to the circadian clock, including the PI3K-Akt signaling pathway, adrenergic signaling in cardiomyocytes and cGMP-PKG signaling pathway. The PI3K-Akt signaling pathway has been shown to be regulated by the clock proteins Cry1, Cry2 and Bmal1, which are part of the oscillations of the transcriptional-translational negative feedback system and regulate the circadian rhythm [43, 47, 48]. Mustafa C Beker reported an interaction of melatonin and Bmal1 in the regulation of PI3K/AKT pathway components and cellular survival [49]. Moreover, the inhibition of cGMP phosphodiesterase enhances photic responses and the synchronization of the biological circadian clock in rodents [50]. For the night-time ceRNA network, the GO analysis revealed enrichment of the terms “cellular response to epinephrine stimulus”, “circadian rhythm” and “3',5'-cyclic-AMP phosphodiesterase activity”, which are related to melatonin secretion. KEGG pathway analysis revealed several important pathways related to melatonin secretion, including the cAMP signaling pathway [51, 52]. Altogether, GO and KEGG analyses suggested that circRNAs with different expression may participate in the process of pineal biological function via different pathways.
In summary, this is the first study to profile the transcriptome of the rat pineal gland from day and night. We constructed a ceRNA network of circRNAs, miRNAs, and mRNAs, which may play a key regulatory role in the circadian rhythm and melatonin secretion of the pineal gland. We expect that our study will be helpful for researchers interested in the gene expression change of the pineal gland with circadian rhythm. Although we identified differentially expressed RNAs between day and night, more experiments are needed at different time points to prove circadian changes. In the future, additional deep-seated regulatory mechanisms await discovery, and further research on them may expose potential therapeutic targets for circadian rhythm diseases. Ideally, our future studies will construct circRNA regulation pathways.
Abbreviations
MT: melatonin; cAMP: cyclic adenosine monophosphate; NE: norepinephrine; AANAT: arylalkylamine N-acetyltransferase; ASMT: acetylserotonin O-methyltransferase; ceRNA: competitive endogenous RNA; CIRI: CircRNA identifier; TPM: transcripts per kilobase million; FDR: false discovery rate; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Supplementary Material
Supplementary figures and table S1.
Supplementary tables.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31872349).
Author contributions
B.Y. and J.B.Z. were responsible for the main experimental concept and design; the experiments were performed by Y.Z., H.Q.W., H.X.G., Y.J.H. and D.X.H. Y.Z., Y.G. and H.J. performed the data analyses and contributed reagents, and the manuscript was written by Y.Z. All of the authors approved the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Simonneaux V, Ribelayga C. Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol Rev. 2003;55:325-95
2. Roseboom PH, Klein DC. Norepinephrine stimulation of pineal cyclic AMP response element-binding protein phosphorylation: primary role of a beta-adrenergic receptor/cyclic AMP mechanism. Mol Pharmacol. 1995;47:439-49
3. Maronde E, Pfeffer M, Olcese J, Molina CA, Schlotter F, Dehghani F. et al. Transcription factors in neuroendocrine regulation: rhythmic changes in pCREB and ICER levels frame melatonin synthesis. J Neurosci. 1999;19:3326-36
4. Bailey MJ, Coon SL, Carter DA, Humphries A, Kim JS, Shi Q. et al. Night/day changes in pineal expression of >600 genes: central role of adrenergic/cAMP signaling. The Journal of biological chemistry. 2009;284:7606-22
5. Chik CL, Mackova M, Price D, Ho AK. Adrenergic regulation and diurnal rhythm of p38 mitogen-activated protein kinase phosphorylation in the rat pineal gland. Endocrinology. 2004;145:5194-201
6. Baler R, Covington S, Klein DC. The rat arylalkylamine N-acetyltransferase gene promoter. cAMP activation via a cAMP-responsive element-CCAAT complex. The Journal of biological chemistry. 1997;272:6979-85
7. Garcia RA, Afeche SC, Scialfa JH, do Amaral FG, dos Santos SH, Lima FB. et al. Insulin modulates norepinephrine-mediated melatonin synthesis in cultured rat pineal gland. Life Sci. 2008;82:108-14
8. Ribelayga C, Gauer F, Calgari C, Pevet P, Simonneaux V. Photoneural regulation of rat pineal hydroxyindole-O-methyltransferase (HIOMT) messenger ribonucleic acid expression: an analysis of its complex relationship with HIOMT activity. Endocrinology. 1999;140:1375-84
9. Korf HW, Schomerus C, Stehle JH. The pineal organ, its hormone melatonin, and the photoneuroendocrine system. Adv Anat Embryol Cell Biol. 1998;146:1-100
10. Man GC, Wang WW, Yim AP, Wong JH, Ng TB, Lam TP. et al. A review of pinealectomy-induced melatonin-deficient animal models for the study of etiopathogenesis of adolescent idiopathic scoliosis. Int J Mol Sci. 2014;15:16484-99
11. Cos S, Sanchez-Barcelo EJ. Melatonin and mammary pathological growth. Front Neuroendocrinol. 2000;21:133-70
12. Rudnitskaya EA, Maksimova KY, Muraleva NA, Logvinov SV, Yanshole LV, Kolosova NG. et al. Beneficial effects of melatonin in a rat model of sporadic Alzheimer's disease. Biogerontology. 2015;16:303-16
13. Yang Y, Sun B, Huang J, Xu L, Pan J, Fang C. et al. Up-regulation of miR-325-3p suppresses pineal aralkylamine N-acetyltransferase (Aanat) after neonatal hypoxia-ischemia brain injury in rats. Brain Res. 2017;1668:28-35
14. Clokie SJ, Lau P, Kim HH, Coon SL, Klein DC. MicroRNAs in the pineal gland: miR-483 regulates melatonin synthesis by targeting arylalkylamine N-acetyltransferase. The Journal of biological chemistry. 2012;287:25312-24
15. Ding X, Sun B, Huang J, Xu L, Pan J, Fang C. et al. The role of miR-182 in regulating pineal CLOCK expression after hypoxia-ischemia brain injury in neonatal rats. Neurosci Lett. 2015;591:75-80
16. Li H, Xu LX, Yu J, Tan L, Miao P, Yang X. et al. The role of a lncRNA (TCONS_00044595) in regulating pineal CLOCK expression after neonatal hypoxia-ischemia brain injury. Biochemical and biophysical research communications. 2020;528:1-6
17. Kleaveland B, Shi CY, Stefano J, Bartel DP. A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell. 2018;174:350-62 e17
18. Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: Identification, biogenesis and function. Biochim Biophys Acta. 2016;1859:163-8
19. Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J. et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141-57
20. Liu X, Wei S, Deng S, Li D, Liu K, Shan B. et al. Genome-wide identification and comparison of mRNAs, lncRNAs and circRNAs in porcine intramuscular, subcutaneous, retroperitoneal and mesenteric adipose tissues. Anim Genet. 2019;50(3):228-241
21. Huang J, Zhou Q, Li Y. Circular RNAs in Gynecological Disease: promising biomarkers and diagnostic targets. Biosci Rep. 2019;17:39 (5)
22. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK. et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384-8
23. Zhang H, Wang X, Hu B, Zhang F, Wei H, Li L. Circular RNA ZFR accelerates non-small cell lung cancer progression by acting as a miR-101-3p sponge to enhance CUL4B expression. Artif Cells Nanomed Biotechnol. 2019;47:3410-6
24. Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L. et al. Translation of CircRNAs. Mol Cell. 2017;66:9-21 e7
25. Ragan C, Goodall GJ, Shirokikh NE, Preiss T. Insights into the biogenesis and potential functions of exonic circular RNA. Scientific reports. 2019;9:2048
26. Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186-94
27. Ly A, Marsman M, Wagenmakers EJ. Analytic posteriors for Pearson's correlation coefficient. Stat Neerl. 2018;72:4-13
28. Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:4
29. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333-8
30. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic acids research. 2019;47:D155-D62
31. Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic acids research. 2012;40:37-52
32. Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS. et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2:e219
33. Deng Y, Li J, Wu S, Zhu Y, Chen Y, He FJCE. Integrated nr database in protein annotation system and its localization. 2006; 32: 71-4.
34. UniProt Consortium T. UniProt: the universal protein knowledgebase. Nucleic acids research. 2018;46:2699
35. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25-9
36. Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic acids research. 2000;28:33-6
37. Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7
38. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic acids research. 2004;32:D277-80
39. Amaral FGD, Cipolla-Neto J. A brief review about melatonin, a pineal hormone. Archives of endocrinology and metabolism. 2018;62:472-9
40. Su SC, Reiter RJ, Hsiao HY, Chung WH, Yang SF. Functional Interaction between Melatonin Signaling and Noncoding RNAs. Trends Endocrinol Metab. 2018;29:435-45
41. Coon SL, Munson PJ, Cherukuri PF, Sugden D, Rath MF, Moller M. et al. Circadian changes in long noncoding RNAs in the pineal gland. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13319-24
42. Chen LL. The biogenesis and emerging roles of circular RNAs. Nature reviews Molecular cell biology. 2016;17:205-11
43. Nam KI, Yoon G, Kim YK, Song J. Transcriptome Analysis of Pineal Glands in the Mouse Model of Alzheimer's Disease. Front Mol Neurosci. 2019;12:318
44. Wang R, Zhang S, Chen X, Li N, Li J, Jia R. et al. CircNT5E Acts as a Sponge of miR-422a to Promote Glioblastoma Tumorigenesis. Cancer Res. 2018;78:4812-25
45. Pfeffer M, Kuhn R, Krug L, Korf HW, Stehle JH. Rhythmic variation in beta1-adrenergic receptor mRNA levels in the rat pineal gland: circadian and developmental regulation. Eur J Neurosci. 1998;10:2896-904
46. Davies JS, Carter DA. Protein/DNA interaction profiling reveals novel regulators of the pineal transcriptome. Mol Cell Endocrinol. 2006;252:19-26
47. Huang WY, Zou X, Lu FE, Su H, Zhang C, Ren YL. et al. Jiao-tai-wan Up-regulates Hypothalamic and Peripheral Circadian Clock Gene Cryptochrome and Activates PI3K/AKT Signaling in Partially Sleep-deprived Rats. Curr Med Sci. 2018;38:704-13
48. Zhao X, Cho H, Yu RT, Atkins AR, Downes M, Evans RM. Nuclear receptors rock around the clock. EMBO Rep. 2014;15:518-28
49. Beker MC, Caglayan B, Caglayan AB, Kelestemur T, Yalcin E, Caglayan A. et al. Interaction of melatonin and Bmal1 in the regulation of PI3K/AKT pathway components and cellular survival. Scientific reports. 2019;9:19082
50. Plano SA, Agostino PV, de la Iglesia HO, Golombek DA. cGMP-phosphodiesterase inhibition enhances photic responses and synchronization of the biological circadian clock in rodents. PLoS One. 2012;7:e37121
51. Gupta BB, Spessert R, Vollrath L. Molecular components and mechanism of adrenergic signal transduction in mammalian pineal gland: regulation of melatonin synthesis. Indian journal of experimental biology. 2005;43:115-49
52. Karolczak M, Korf HW, Stehle JH. The rhythm and blues of gene expression in the rodent pineal gland. Endocrine. 2005;27:89-100
Author contact
![]() Corresponding authors: Jia-Bao Zhang, E-mail: zjbedu.cn; Tel.: +86-431-8783-6551; Bao Yuan, E-mail: yuan_baoedu.cn; Tel.: +86-431-8783-6536.
Corresponding authors: Jia-Bao Zhang, E-mail: zjbedu.cn; Tel.: +86-431-8783-6551; Bao Yuan, E-mail: yuan_baoedu.cn; Tel.: +86-431-8783-6536.