Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(4):975-983. doi:10.7150/ijms.50197 This issue Cite
Research Paper
Angiotensin-Converting Enzyme Inhibitor, Captopril, Improves Scar Healing in Hypertensive Rats
Eun Young Rha, Jae Won Kim, Jun Hyeok Kim, Gyeol Yoo ![]()
Department of Plastic and Reconstructive Surgery, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
Received 2020-7-1; Accepted 2020-12-22; Published 2021-1-1
Abstract
Pathological cutaneous scars, with aberrant extracellular matrix accumulation, have multiple origins. Antihypertensive medications, such as calcium channel blockers, have been used to treat pathological scars. However, a relationship between angiotensin-converting enzyme (ACE) inhibitors, pathological scars, and blood pressure (BP) has never been reported. Here, we aimed to compare the differences in scar development and the effects of the administration of systemic ACE inhibitor on scar tissue in a normotensive rat, the Wistar Kyoto rat (WKY), a hypertensive rat, and the spontaneously hypertensive rat (SHR). Using an 8-mm punch, we created two full-thickness skin defects in a total of 32 rats (16 WKY and 16 SHR) to obtain a total of 64 wounds. We established control WKY (n = 16), captopril-treated WKY (n = 16), control SHR (n = 16), and captopril-treated SHR (n = 16) groups and started captopril (100 mg/g per day) treatment on day 21 in the appropriate groups. The BP of all groups was measured at 0, 3, and 5 weeks. The scar area was measured by histopathological examination, and scarring was expressed in terms of scar area and fibroblast and capillary counts. The expression of heat shock protein (HSP) 47, type I and III collagens, alpha-smooth muscle actin (α-SMA), Ki67, and vascular endothelial growth factor (VEGF) was investigated using immunohistochemistry. The scar area and fibroblast count were significantly higher in control SHR than in control WKY. The scar area, fibroblast count, and capillary count were significantly smaller in captopril-treated SHR than in control SHR. Immunostaining for α-SMA, Ki67, and VEGF also showed a noticeable decrease in scarring in the treated SHR compared with that in control SHR. Thus, BP affects scar development in a rat model, and an ACE inhibitor is more effective at reducing scars in hypertensive rats than in normotensive rats.
Keywords: angiotensin-converting enzyme inhibitor, Wistar Kyoto rat, spontaneously hypertensive rat, scar area, fibrosis, vascular endothelial growth factor
Introduction
Keloid and hypertrophic scars, which are pathological cutaneous scars, are characterized by prolonged, aberrant extracellular matrix (ECM) accumulation [1]. They are considered dermal fibro-proliferative lesions rather than an inevitable postoperative outcome in patients with high aesthetic expectations [2].
The development of pathological scars has multiple etiologies, including single nucleotide polymorphisms, local mechanical tension, systemic metabolic adenosine triphosphate levels, antinuclear antibodies, endocrinological sebum secretion, and nutritional fatty acids [3]. Recently, it was hypothesized that hypertension is a factor in pathological scar development [1]. Antihypertensive medications such as angiotensin-converting enzyme (ACE) inhibitors and calcium channel blockers have been used to treat pathological scars [4, 5]. Clinical and animal studies have described the effect of the systemic administration of ACE inhibitors on pathological scars [4, 6, 7]. Uzun et al. reported that early application of enalapril after dermal injury reduces scar formation using rabbit ear hypertrophic scar model [7]. Fang et al. [8] demonstrated that ACE inhibitors suppress the TGF-β1/SMAD2/3 and TGF-β1/TAK1 pathways in vivo and in vitro using mouse NIH 3T3 fibroblasts cultured with different concentrations of ACE inhibitors and a rat model of full-thickness skin wound. However, no studies have directly demonstrated a relationship between ACE inhibitors, pathological scars, and blood pressure (BP).
Given this background, we compared the differences in scar development and the effects of the systemic administration of an ACE inhibitor, captopril, on cutaneous scar tissues in Wistar Kyoto rats (WKY), a genetically related normotensive control strain, and spontaneously hypertensive rats (SHR), a genetically related hypertensive strain.
Materials and Methods
Experimental Setting and Animals
We used a total of 32 male rats (16 male WKY and 16 male SHR (SLC, Nakaizu, Japan)) at the age of 12-13 weeks, weighing 250-350 g. The animals were kept in individual cages in a temperature-controlled room with a 12 h light/12 h dark cycle and were provided food and water ad libitum. All animal experiments were performed to minimize suffering.
Experimental Rat Model
The WKY and SHR were anesthetized by an intraperitoneal injection of sodium pentobarbital (40 mg/kg). After shaving the hair on the dorsal surface, two full-thickness excisional wounds were created through the panniculus carnosus on the back using an 8-mm biopsy punch. After hemostasis was achieved by manual compression, the wounds were covered with polyurethane dressing (Tegaderm, 3M Health Care, St. Paul, MN). A total of 64 wounds were examined grossly for signs of infection, desiccation, and epithelialization at 1-day intervals. All wounds were epithelialized on day 14.
Treatment
The 16 WKY and 16 SHR were randomly assigned into four groups: Control WKY (WKY-C, n = 16), captopril-treated WKY (WKY-T, n = 16), control SHR (SHR-C, n = 16), and captopril-treated SHR (SHR-T, n = 16). Between days 21 and 35 after wounding, the WKY-T and SHR-T groups were treated with captopril (100 mg/kg per day added to drinking water) using a dosage regimen adapted from previous studies [9]. The WKY-C and SHR-C groups served as the normal control, receiving water only. The body weight and systolic BP were measured at weeks 0, 3, and 5 using a non-invasive tail-cuff system (IITC Life Science, Woodland Hills, CA).
Tissue Preparation
A total of 32 rats (8 rats in each group) were euthanized on day 36 after wounding, and a total of 64 tissue biopsies were performed to evaluate inflammatory cell counts and scar areas. Tissue samples (10 mm thick) were collected from the middle region of the wounds. All specimens were fixed in 10% neutral-buffered formalin, dehydrated, embedded in paraffin blocks, cut into 3-µm sections, and stained with hematoxylin and eosin (H&E) and Masson's trichrome stain for examination under a light microscope to measure the scar area, collagen deposition and neovasculature [7, 8, 10].
Histopathology
The severity of the scarring in the tissue specimens was determined using the scar area, fibroblast count, and capillary count. For histopathological examinations, digital images were captured using Leica Application Suite Software LAS V 4.1 (Leica Microsystems, Wetzlar, Germany). The scar area was defined as the area showing the destruction of the standard architecture compared with the surrounding area, as measured by tracing the scar margin using ImageJ software (ImageJ; National Institute of Health, Bethesda, MD, USA) [11]. The mean scar area for each wound was then converted from pixel number to square micrometers, calculated using the ratio of the number of pixels to the scale bar [12]. Blind measurements were performed on the scars, and two values were averaged. The number of fibroblasts in three randomly chosen fields of 1 mm2 was counted under 400x magnification, and the number of capillary lumens per field was counted under 40x magnification by two histologists blinded to the groups.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded (FFPE) 4-μm-thick sections using a Benchmark Ultra autostainer (Roche Tissue Diagnostics, Tucson, AZ, USA) and the UltraViewTM universal DAB detection kit (Ventana), according to the manufacturer's instructions. Paraffin sections were immunostained with primary antibodies against HSP47 (diluted 1:300, monoclonal EPR4217; GeneTex, CA, USA), collagen I (diluted 1:100, monoclonal 3G3; Abcam, Cambridge, MA, USA), collagen III (diluted 1:100, polyclonal ab7778; Abcam), Ki67 (clone 30-9, Ventana), actin (clone 1A4, Ventana), and vascular endothelial growth factor (VEGF) (ab1316) (Abcam). The scar tissue was scored using a semi-quantitative approach based on the proportion of positively stained cells to assess HSP47, collagen I, collagen III, and actin immunostaining as follows: (-) absence of staining; (1+) weak staining; (2+) moderate staining; (3+) strong staining. Basal cells (100 cells) were counted in three random locations in a tissue section (100× magnification) to calculate the Ki67 proliferation index, after which the number of positive cells along the length of the epidermis was determined. The proliferation index was defined as the average percentage of Ki67-positive nuclei, as previously described [13]. VEGF expression was evaluated in vessels and inflammatory cells (mainly lymphocytes, plasma cells, and neutrophils) and assessed semi-quantitatively based on the sum of the staining intensity (0, negative; 1, weak; 2, moderate; and 3, strong) and the distribution of positive cells in five random scar fields in each sample (0, <10%; 1, 11-50%; 2, >50%). A score exceeding 4 indicated strong VEGF expression; otherwise, it indicated weak expression.
Ethical Considerations
This study was approved by the Animal Care and Use Committee of Yeouido St. Mary's Hospital, Catholic University of Korea (YEO20141003T).
Statistical Analysis
Statistical analysis was performed using SPSS ver. 18.0 for Windows (SPSS, Chicago, IL, USA). All the data are expressed as the mean ± standard deviation (SD). The scar area, fibroblast count, and capillary count in all of the groups were compared using one-way analysis of variance (ANOVA). HSP47, type I and III collagen, and VEGF expression scores, as well as the Ki67 proliferation index, were compared using the Kruskal-Wallis test, followed by posthoc Bonferroni correction for multiple comparisons. We also used repeated-measures ANOVA to compare the systolic BP between 3 and 5 weeks in all of the groups. P- values < 0.05 were considered statistically significant.
Results
Captopril Treatment Reduces Systolic Blood Pressure in Normotensive and Hypertensive Rats (Systolic BP Values Changed at Different Timepoints)
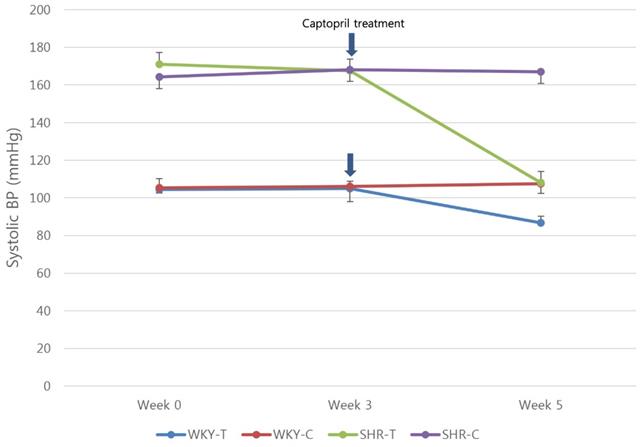
At 3 weeks (baseline), the mean systolic BPs of the WKY-C, WKY-T, SHR-C, and SHR-T groups were 106 ± 8, 104.9 ± 3.9, 168.1 ± 6.6, 167.6 ± 6.2 mmHg, respectively; at 5 weeks (2 weeks after treatment), they were 107.6 ± 5.1, 86.8 ± 3.4, 167.1 ± 6.9, 108 ± 6.2 mmHg, respectively. In WKY-T and SHR-T, the mean systolic BP at 5 weeks was significantly lower compared with that at 3 weeks (P < 0.0001) (Figure 1).
Captopril treatment reduces systolic blood pressure in normotensive and hypertensive rats. Serial systolic blood pressure in the Wistar Kyoto rat group (red and blue lines) and spontaneously hypertensive rat group (purple and green lines). Arrows show the starting point of captopril treatment. WKY-C, control Wistar Kyoto rats; WKY-T, captopril-treated Wistar Kyoto rats; SHR-C, control spontaneously hypertensive rats; SHR-T, captopril-treated spontaneously hypertensive rats. Statistical significance was considered at P < 0.05 based on repeated-measures ANOVA followed by a post hoc Bonferroni correction for multiple comparisons.
Captopril Treatment Inhibits Scar Area in Hypertensive Rats In Vivo
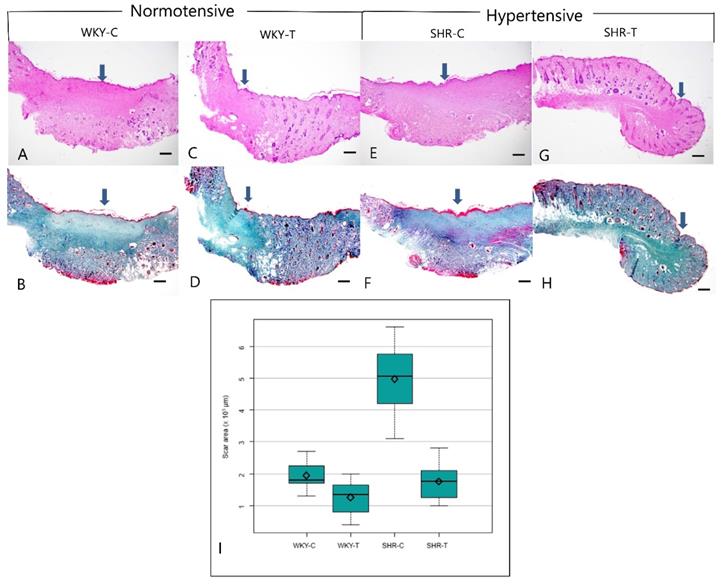
Histopathological analysis revealed larger scar areas, comprising a dense connective tissue, a less organized collagen structure, and many capillary lumens, in the WKY control group than in the captopril-treated WKY group. Compared with the SHR control group, the captopril-treated SHR group showed a focal scar area with less prevalent fibroblasts, loosely arranged collagen bundles, and fewer capillary lumens (Figure 2A-2H). The median scar area differed significantly among all four groups (P < 0.0001). In addition, the scar area was significantly smaller in the wounds of SHR-T and WKY-C than in those of SHR-C. However, the differences between the wounds of WKY-C and WKY-T and WKY-T and SHR-T rats were not significant (Figure 2I and Table 1).
Summary of the histopathological data.
| WKY-C | WKY-T | SHR-C | SHR-T | P-value | Post hoc | |
|---|---|---|---|---|---|---|
| Scar area (× 103 µm) | 1.9 ± 0.4 | 1.3 ± 0.5 | 5±1.2 | 1.8±0.6 | < 0.0001 | 1=2=4<3 |
| Fibroblast count | 91.5±19.1 | 67.1±20.6 | 212.1±51.1 | 64.8±20.1 | < 0.0001 | 1=2=4<3 |
| Capillary count | 27.9±8.7 | 11.5±9 | 25.6±10.3 | 12.1±5.4 | 0.0004 | 2=4<1=3 |
Abbreviations: WKY-C, control Wistar Kyoto rats; WKY-T, captopril-treated Wistar Kyoto rats; SHR-C, control spontaneously hypertensive rats; SHR-T, captopril-treated spontaneously hypertensive rats.
All data are expressed as the mean ± SD.
Statistical significance at P < 0.05 based on an ANOVA followed by a post hoc Bonferroni correction for multiple comparisons.
Histopathological change between normotensive and hypertensive rats after captopril treatment. Captopril treatment significantly inhibits scar area in hypertensive rats. (A, B) Control Wistar Kyoto rat. A prominent scar area (arrow) includes fibroblast proliferation and dense connective tissue (A, Hematoxylin & eosin, B, Masson trichrome, 20×). (C, D) Captopril-treated Wistar Kyoto rat. The scar area (arrow) is less marked, and collagen is coarsely distributed (C, Hematoxylin & eosin; D, Masson trichrome, 20×). (E, F) Control spontaneously hypertensive rat. A prominent scar (arrow), characterized by fibroblast proliferation, dense connective tissue, and many capillary lumens, occupies much of the total area (E, Hematoxylin & eosin; F, Masson trichrome, 20×). (G, H) Captopril-treated spontaneously hypertensive rat. The scar area (arrow) makes up a smaller part of the total area and contains sparsely distributed collagen (G, Hematoxylin & eosin; H, Masson trichrome stain, 20×). (I) The mean scar area differed significantly among the four groups. Captopril treatment significantly inhibits scar area in hypertensive rats. Scale bars in all images are 500 μm. WKY-C, control Wistar Kyoto rats; WKY-T, captopril-treated Wistar Kyoto rats; SHR-C, control spontaneously hypertensive rats; SHR-T, captopril-treated spontaneously hypertensive rats. P < 0.0001 with posthoc Bonferroni correction.
Captopril Treatment Inhibits Fibrosis in Hypertensive Rats In Vivo
The α-SMA expression, however, showed a significant difference among the four groups (P = 0.0000); it was significantly lower in the wounds of WKY-T and SHR-T than in those of WKY-C and SHR-C, respectively (Figure 3A, 3B and Table 2). The Ki67 proliferation index differed significantly among the four groups (P < 0.0000) and was significantly lower in the wounds of SHR-T and WKY-C than in those of SHR-C. There were no significant differences in KI67 proliferation index between the wounds of WKY-C and WKY-T or between those of WKY-T and SHR-T (Figure 3C, 3D, and Table 2).
Summary of the immunohistochemical data.
| WKY-C | WKY-T | SHR-C | SHR-T | P-value | Post hoc | |
|---|---|---|---|---|---|---|
| HSP47 expression score | 2.6±0.5 | 2.5±0.5 | 2.7±0.5 | 2.6±0.5 | 0.746 | |
| Collagen type I expression score | 1.1±0.5 | 0.9±0.7 | 1.1±0.7 | 0.7±0.7 | 0.227 | |
| Collagen type III expression score | 2.6±0.5 | 2.4±0.5 | 2.8±0.4 | 2.4±0.5 | 0.159 | |
| α-SMA expression score | 2.3±0.5 | 1.5±0.5 | 2.7±0.5 | 1.3±0.5 | 0.0000 | 2=4<1=3 |
| Ki67 proliferation index | 2.4±1.7 | 1.1±0.5 | 5.6±2.4 | 1.2±0.5 | 0.0000 | 1=2=4<3 |
| VEGF expression score | 3.1±1.1 | 2.6±1.0 | 3.7±1.3 | 1.6±1.3 | 0.0000 | 2=4<1=3 |
Abbreviations: WKY-C, control Wistar Kyoto rats; WKY-T, captopril-treated Wistar Kyoto rats; SHR-C, control spontaneously hypertensive rats; SHR-T, captopril-treated spontaneously hypertensive rats.
All data are expressed as the mean ± SD.
Statistical significance at P < 0.05 based on Kruskal-Wallis test followed by post hoc Bonferroni correction for multiple comparisons.
Immunostaining for α-SMA and Ki67 proliferation index. Captopril treatment inhibits fibrosis in hypertensive rats. (A) Intense α-SMA staining is seen in the control spontaneously hypertensive rat tissues, and (B) weak staining in the captopril-treated spontaneously hypertensive rat tissues (100×). (C) Strong Ki67 immunoreactivity is seen in the control spontaneously hypertensive rat tissues and (D) weak immunoreactivity in the captopril-treated spontaneously hypertensive rat tissues (100×). (E) The mean fibroblast count differed significantly among the four groups. Scale bars in all images are 50 μm. WKY-C, control Wistar Kyoto rats; WKY-T, captopril-treated Wistar Kyoto rats; SHR-C, control spontaneously hypertensive rats; SHR-T, captopril-treated spontaneously hypertensive rats. P < 0.0001 with posthoc Bonferroni correction.
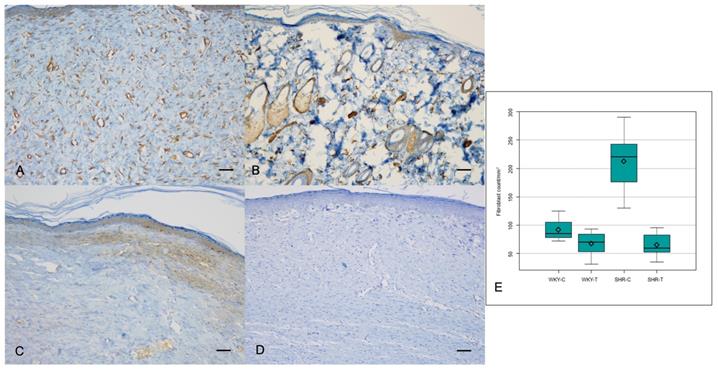
The median fibroblast count differed significantly among all four groups (P < 0.0001). The fibroblast counts were significantly lower in the wounds of SHR-T and WKY-C than in those of SHR-C. Furthermore, the difference in fibroblast counts between the wounds of WKY-C and WKY-T and between those of WKY-T and SHR-T was not significant (Figure 3E and Table 1).
Captopril Treatment Inhibits Scar Deposition by Inhibiting Capillary Count and VEGF Expression in Normotensive and Hypertensive Rats
The VEGF expression score differed significantly among the four groups (P = 0.000) and was significantly lower in the wounds of WKY-T than those of WKY-C and in the wounds of SHR-T than those of SHR-C. There was no significant difference between the wounds of WKY-C and SHR-C (Figure 4A, 4B and Table 2).
Immunostaining for VEGF and mean capillary count in each group. Captopril treatment inhibits scar deposition by inhibiting capillary count and VEGF in normotensive and hypertensive rats. (A) Strong VEGF immunoreactivity is seen in the control spontaneously hypertensive rat tissues and (B) weak VEGF immunoreactivity in the captopril-treated spontaneously hypertensive rat tissues (200×). (C) The mean capillary count differed significantly among the four groups. Scale bars in all images are 50 μm. WKY-C, control Wistar Kyoto rats; WKY-T, captopril-treated Wistar Kyoto rats; SHR-C, control spontaneously hypertensive rats; SHR-T, captopril-treated spontaneously hypertensive rats. P = 0.0004 with post hoc Bonferroni correction.
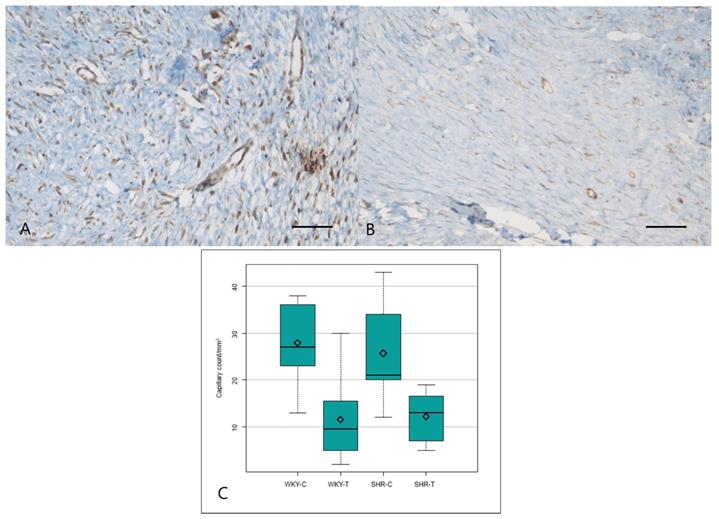
The median capillary count also differed significantly among all four groups (P = 0.0004). Capillary counts were significantly lower in the wounds of WKY-T and SHR-T than in those of WKY-C and SHR-C, respectively. The differences between the wounds of WKY-C and SHR-C were not significant (Figure 4C and Table 1).
Captopril Treatment Has No Effect on Collagen I, Collagen III, and HSP47 Expression in Hypertensive Rats In Vivo
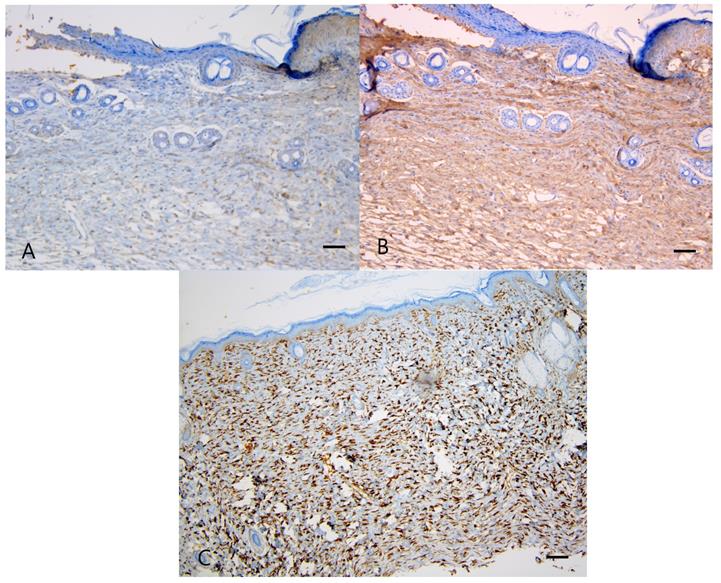
The expression of collagen types I and III also did not significantly differ among the groups (P = 0.227, 0.159, respectively), as shown in Figure 5A, 5B and Table 2. Similarly, the HSP47 expression did not significantly differ among the groups (P = 0.746), as shown in Figure 5C and Table 2.
Immunostaining for collagen type I, III, and HSP47. Captopril treatment had no significant effect on collagen type I, III, and HSP47 in normotensive and hypertensive rats. (A) Weak collagen type I staining is seen in the control spontaneously hypertensive rat tissues, and (B) strong collagen type III staining in the control spontaneously hypertensive rat tissues (100×). (C) Intense staining of HSP47 is seen in the control spontaneously hypertensive rat tissues (100×). Scale bars in all images are 50 μm.
Discussion
This study was conducted to assess scar development and the effect of the ACE inhibitor, captopril, on pathological scars in genetically normotensive and hypertensive rat models. Hypertension affects the profibrotic functional changes in endothelial cells, myofibroblasts, dermal fibroblasts, and mast cells, which form keloid and hypertrophic scars. These functional changes are mediated by inflammation-induced tissue hypoxia and the renin-angiotensin system (RAS) [1]. Recent research has demonstrated that a local RAS is present within healthy skin and plays a vital role in wound repair and tissue reconstruction through Ang II [14, 15], which stimulates proliferation, migration, and matrix production of human dermal fibroblasts [16, 17], as well as in keloid pathogenesis via the activation of angiotensin type 1 receptor (AT1)-dependent signaling pathway [18].
Hypertension-induced endothelial dysfunction promotes cutaneous fibrosis [19]. For example, in hypertensive heart disease, elevated blood pressure causes activation of the RAS, which is followed by transforming growth factor-β (TGF-β)/Smad3 signaling activation and increased local inflammation, ECM production, and fibrosis [1, 20]. Keloid induces interstitial hypertension, thereby increasing local cellular metabolic demands, decreasing metabolic substrate levels, and ultimately leading to tissue hypoxia [21, 22]. This inflammation-induced hypoxic state in keloid may be caused by endothelial hypertrophy, which induces partial occlusion of microvessels. Therefore, endothelial dysfunction caused by vascular hypertension may promote hypoxia-induced fibrosis in keloids [23-25].
Several recent reports described the effect of the pharmacological antagonism of Ang II by an ACE inhibitor or AT1 antagonist, which are widely used as antihypertensive drugs, in suppressing skin wound repair, hypertrophic scars, or keloid [15, 26]. Nevertheless, few studies have demonstrated the relationship between scar development and hypertension in an animal model. In the present study, we used captopril among the various ACE inhibitors because its scar reducing effect has been determined in previous in vivo studies, and the effect of lowering blood pressure in SHR has also been investigated previously [4, 9, 27]. We determined the relationship between the systemic effect (lowering blood pressure) and topical effect (cutaneous scar reduction). Captopril treatment was initiated after the termination of wound healing to exclude its effect on the inflammatory phase during wound healing.
In this study, the scar area, fibroblast count, and capillary count of SHR-T were lower than that of SHR-C in hypertensive rats, while in normotensive rats only the capillary count was significantly lower in WKY-T than in WKY-C. These results suggest that the effect of the ACE inhibitor on scar development was more prominent in the hypertensive rat group than in the normotensive rat group. ACE inhibitor treatment significantly lowered the systolic BP in both SHR and WKY. In hypertensive conditions, however, elevated circulatory Ang II levels might be related to the elevation of local Ang II in SHR.
Next, examining the difference between the control groups of normotensive and hypertensive rats, the scar area and fibroblast count were found to be significantly lower in WKY-C than in SHR-C, indicating that hypertension might be related to the cutaneous scar development in this rat model, which is consistent with other reports [1]. Dermal fibroblasts are mechanosensitive cells that actively interact with their microenvironment, expressed, for example, under increased fluid pressure and shear stress on the cells in the vascular lumen or walls due to hypertension [1]. In this study, the fibroblast count of hypertensive rats was higher than that of normotensive rats. Further studies should examine whether there is a correlation between circulating Ang II levels and the local Ang II type 1 receptor in the scar tissue in SHR.
The immunohistochemical analysis in this study showed that VEGF expression was more prominent in SHR-C than in SHR-T. VEGF is a vascular permeability factor that promotes neovascularization and cell growth [28, 29]. In addition to being a potent endothelial cell-specific cytokine, it is also a mitogenic, chemotactic factor in vitro and an inducer of angiogenesis in vivo [30-32]. Ang II accelerates neovascularization and promotes the release of VEGF and platelet-derived growth factor [15, 33]. The decrease in VEGF expression in SHR-T observed in this study may thus be related to the downregulation of Ang II in SHR-T. HSP47, a 47-kDa heat shock protein, is a collagen-specific molecular chaperone localized in the endoplasmic reticulum that plays an essential role in collagen biosynthesis in skin fibrosis [34]. In this study, the expression of HSP47 and collagen type I and III did not differ significantly among the groups, in contrast to the histopathology results. Protein non-specificity for rat wounds and our semi-quantitative measurement method are among the limitations of this study.
α-SMA is a marker for myofibroblasts and is detected in fibroblasts and vascular endothelial cells [35]. Myofibroblasts play a crucial role in wound contraction and are involved in the remodeling of granulation tissue. However, in cases of excessive scarring and fibrosis, myofibroblasts persist and contribute to excessive collagen production. In a mouse burn model, the level of α-SMA expression was shown to correlate with the severity of scarring [36]. In this study, α-SMA expression was more prominent in SHR-C and WKY-C than in SHR-T and WKY-T, respectively. The Ki67 proliferation index was higher in SHR-C than in SHR-T. Ki67 is a marker for epidermal proliferation [13], and the number of Ki67-positive proliferating keratinocytes was significantly decreased in SHR-T compared to SHR-C. This phenomenon could be related to the inhibition of keratinocyte migration inhibited by angiotensin II receptor blockade [17].
This study had several limitations. First, anesthesia might have affected the plasma renin activity and circulatory Ang II levels in the rats. However, any effect of anesthesia should have disappeared, because we started the captopril treatment 3 weeks after the creation of the wound under anesthesia. Second, the wound healing process in rats is different from that in humans; rats have loose skin and subcutaneous panniculus carnosus; therefore, wound healing occurs via contraction and collagen formation [37]. Consequently, the scars that developed in the rats might not have been pathological. However, the differences in the scar development between normotensive and hypertensive rats and between captopril-treated and control hypertensive rats were significant. We believe that this finding is relevant, although the issue of a "pathological scar" is unresolved. Third, we measured the blood pressure of spontaneously hypertensive and normotensive rats using a tail-cuff system because of its non-invasiveness, according to the previous studies on rat experiments [38-39]. However, the blood pressure measurement of rats using the telemetry system is a more precise and better method than the tail-cuff system.
In this study, large scars tended to develop more often in hypertensive rats than in normotensive rats. The ACE inhibitor significantly reduced the scars in the hypertensive rats, while it did not reduce the scars effectively in the normotensive rats. Based on these results, blood pressure might have affected scar development in the hypertensive rat wound model used in this study. Moreover, the ACE inhibitor would be effective at reducing scars that develop only in hypertensive conditions. Further studies are necessary to demonstrate the mechanisms of action of ACE inhibitors on pathological scars in an established animal scar model using multiple parameters, such as plasma Ang II levels and tissue AT1 receptor levels.
Abbreviations
ECM: extracellular matrix; ACE: angiotensin-converting enzyme; WKY: Wistar Kyoto rats; SHR: spontaneously hypertensive rats; H&E: hematoxylin and eosin; FFPE: formalin-fixed paraffin-embedded; ANOVA: one-way analysis of variance; RAS: renin-angiotensin system; Ang II: angiotensin II; AT1: angiotensin type 1 receptor; VEGF: vascular endothelial growth factor; BP: blood pressure.
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2018R1C1B6005381) and by the financial support of the Catholic Medical Center Research Foundation provided in the program year of 2018.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huang C, Ogawa R. The link between hypertension and pathological scarring: does hypertension cause or promote keloid and hypertrophic scar pathogenesis? Wound Repair Regen. 2014;22(4):462-6
2. Ahuja RB, Chatterjee P. Comparative efficacy of intralesional verapamil hydrochloride and triamcinolone acetonide in hypertrophic scars and keloids. Burns. 2014;40(4):583-8
3. Huang C, Murphy GF, Akaishi S. et al. Keloids and hypertrophic scars: update and future directions. Plast Reconstr Surg Glob Open. 2013;1(4):e25
4. Ardekani G.S, Aghaei S, Nemati MH. et al. Treatment of a postburn keloid scar with topical captopril: report of the first case. Plast Reconstr Surg. 2009;123(3):112e-3e
5. D' Andrea F, Brongo S, Ferraro G. et al. Prevention and treatment of keloids with intralesional verapamil. Dermatology. 2002;204(1):60-2
6. Iannello S, Milazzo P, Bordonaro F. et al. Low-dose enalapril in the treatment of surgical cutaneous hypertrophic scar and keloid-two case reports and literature review. MedGenMed. 2006;8(4):60
7. Uzun H, Bitik O, Hekimoglu R. et al. Angiotensin-converting enzyme inhibitor enalapril reduces formation of hypertrophic scars in a rabbit ear wounding model. Plast Reconstr Surg. 2013;132(3):361e-71e
8. Fang QQ, Wang XF, Zhao WY. et al. Angiotensin-converting enzyme inhibitor reduces scar formation by inhibiting both canonical and noncanonical TGF-β1 pathways. Sci Rep. 2018;8(1):3332
9. Bolterman RJ, Manriquez MC, Ortiz Ruiz MC. et al. Effects of captopril on the renin angiotensin system, oxidative stress, and endothelin in normal and hypertensive rats. Hypertension. 2005;46(4):943-7
10. Tan WQ, Fang QQ, Shen XZ. et al. Angiotensin-converting enzyme inhibitor works as a scar formation inhibitor by down-regulating Smad and TGF-β-activated kinase 1 (TAK1) pathways in mice. Br J Pharmacol. 2018;175(22):4239-52
11. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671-5
12. Jeong W, Yang CE, Roh TS. et al. Scar prevention and enhanced wound healing induced by polydeoxyribonucleotide in a rat incisional wound-healing model. Int J Mol Sci. 2017;18(8):1698
13. Limandjaja GC, van den Broek LJ, Waaijman T. et al. Increased epidermal thickness and abnormal epidermal differentiation in keloid scars. Br J Dermatol. 2017;176(1):116-26
14. Steckelings UM, Wollschlager T, Peters J. et al. Human skin: source of and target organ for angiotensin II. Exp Dermatol. 2004;13(3):148-54
15. Takeda H, Katagata Y, Hozumi Y. et al. Effects of angiotensin II receptor signaling during skin wound healing. Am J Pathol. 2004;165(5):1653-62
16. Kawaguchi Y, Takagi K, Hara M. et al. Angiotensin II in the lesional skin of systemic sclerosis patients contributes to tissue fibrosis via angiotensin II type 1 receptors. Arthritis Rheum. 2004;50(1):216-26
17. Yahata Y, Shirakata Y, Tokumaru S. et al. A novel function of angiotensin II in skin wound healing. Induction of fibroblast and keratinocyte migration by angiotensin II via heparin-binding epidermal growth factor (EGF)-like growth factor-mediated EGF receptor transactivation. J Biol Chem. 2006;281(19):13209-16
18. Bond JE, Bergeron A, Thurlow P. et al. Angiotensin-II mediates nonmuscle myosin II activation and expression and contributes to human keloid disease progression. Mol Med. 2011;17(11-12):1196-203
19. Virdis A, Taddei S. How to evaluate microvascular organ damage in hypertension: assessment of endothelial function. High Blood Press Cardiovasc Prev. 2011;18(4):163-7
20. Graf K, Schaefer-Graf UM. Is Smad3 the key to inflammation and fibrosis in hypertensive heart disease? Hypertension. 2010;55(5):1088-9
21. Kelly AP. Keloids. Dermatol Clin. 1988;6(3):413-24
22. Ogihara T, Kikuchi K, Matsuoka H. et al. The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH 2009). Hypertens Res. 2009;32(1):3-107
23. Arima J, Huang C, Rosner B. et al. Hypertension: a systemic key to understanding local keloid severity. Wound Repair Regen. 2015;23(2):213-21
24. Karna E, Szoka L, Palka JA. Captopril-dependent inhibition of collagen biosynthesis in cultured fibroblasts. Pharmazie. 2010;65(8):614-7
25. Stawski L, Han R, Bujor AM. et al. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res Ther. 2012;14(4):R194
26. Qiu JG, Factor S, Chang TH. et al. Wound healing: captopril, an angiogenesis inhibitor, and Staphylococcus aureus peptidoglycan. J Surg Res. 2000;92(2):177-85
27. Akershoek JJJ, Brouwer KM, Vlig M. et al. Early intervention by Captopril does not improve wound healing of partial thickness burn wounds in a rat model. Burns. 2018;44(2):429-435
28. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249-57
29. Sayah DN, Soo C, Shaw WW. et al. Downregulation of apoptosis-related genes in keloid tissues. J Surg Res. 1999;87(2):209-16
30. Nissen NN, Polverini PJ, Koch AE. et al. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am J Pathol. 1988;152(6):1445-52
31. Taub PJ, Silver L, Weinberg H. Plastic surgical perspectives on vascular endothelial growth factor as gene therapy for angiogenesis. Plast Reconstr Surg. 2000;105(3):1034-42
32. Le AD, Zhang Q, Wu Y. et al. Elevated vascular endothelial growth factor in keloids: relevance to tissue fibrosis. Cells Tissues Organs. 2004;176(1-3):87-94
33. Rodgers K, Abiko M, Girgis W. et al. Acceleration of dermal tissue repair by angiotensin II. Wound Repair Regen. 1997;5(2):175-83
34. Kuroda K, Tajima S. HSP47 is a useful marker for skin fibroblasts in formalin-fixed, paraffin-embedded tissue specimens. J Cutan Pathol. 2004;31(3):241-6
35. Carthy JM, Sundqvist A, Heldin A. et al. Tamoxifen Inhibits TGF-beta-Mediated Activation of Myofibroblasts by Blocking Non-Smad Signaling Through ERK1/2. J Cell Physiol. 2015;230(12):3084-92
36. Wang XQ, Kravchuk O, Winterford C. et al. The correlation of in vivo burn scar contraction with the level of alpha-smooth muscle actin expression. Burns. 2011;37(8):1367-77
37. Grada A, Mervis J, Falanga F. Research Techniques Made Simple: Animal Models of Wound Healing. J Invest Dermatol. 2018;138(10):2095-2105.e1
38. Lipták B, Kaprinay B, Gáspárová Z. A rat-friendly modification of the non-invasive tail-cuff to record blood pressure. Lab Anim (NY). 2017;46(6):251-3
39. Li H, Zhang X, Wang F. et al. MicroRNA-21 Lowers Blood Pressure in Spontaneous Hypertensive Rats by Upregulating Mitochondrial Translation. Circulation. 2016;134(10):734-51
Author contact
![]() Corresponding author: Gyeol Yoo, MD, PhD. Department of Plastic and Reconstructive Surgery, Incheon St. Mary's Hospital, College of Medicine, The Catholic University of Korea, 56, Dongsu-ro, Bupyeong-gu, Incheon, 21431, Republic of Korea. Tel: +82-32-280-5184, Fax: +82-32-280-6290, E-mail: psygac.kr
Corresponding author: Gyeol Yoo, MD, PhD. Department of Plastic and Reconstructive Surgery, Incheon St. Mary's Hospital, College of Medicine, The Catholic University of Korea, 56, Dongsu-ro, Bupyeong-gu, Incheon, 21431, Republic of Korea. Tel: +82-32-280-5184, Fax: +82-32-280-6290, E-mail: psygac.kr