Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(16):2454-2467. doi:10.7150/ijms.47598 This issue Cite
Research Paper
An α2-adrenoceptor agonist: Dexmedetomidine induces protective cardiomyocyte hypertrophy through mitochondrial-AMPK pathway
Xiaojian Weng1*, Hua Liu2*, Xiaodan Zhang3, Qianqian Sun1, Cheng Li1, Minglu Gu1, Yanyifang Xu1, Shitong Li4, Weiwei Li1 ![]() , Jianer Du1
, Jianer Du1 ![]()
1. Department of Anesthesiology and SICU, Xinhua Hospital, Shanghai, China.
2. Department of Anesthesiology, The Ninth People's Hospital, Shanghai, China.
3. Department of ICU, Shanghai General Hospital, Shanghai, China.
4. Department of Anesthesiology, Shanghai General Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China.
*These authors contributed equally to this work.
Received 2020-4-29; Accepted 2020-8-27; Published 2020-9-9
Abstract
Aims: Dexmedetomidine (Dex) as a highly selective α2-adrenoceptor agonist, was widely used anesthetic in perioperative settings, whether Dex induces cardiac hypertrophy during perioperative administration is unknown.
Methods: The effects of Dex on cardiac hypertrophy were explored using the transverse aortic constriction model and neonatal rat cardiomyocytes.
Results: We reported that Dex induces cardiomyocyte hypertrophy with activated ERK, AKT, PKC and inactivated AMPK in both wild-type mice and primary cultured rat cardiomyocytes. Additionally, pre-administration of Dex protects against transverse aortic constriction induced-heart failure in mice. We found that Dex up-regulates the activation of ERK, AKT, and PKC via suppression of AMPK activation in rat cardiomyocytes. However, suppression of mitochondrial coupling efficiency and membrane potential by FCCP blocks Dex induced AMPK inactivation as well as ERK, AKT, and PKC activation. All of these effects are blocked by the α2-adrenoceptor antagonist atipamezole.
Conclusion: The present study demonstrates Dex preconditioning induces cardiac hypertrophy that protects against heart failure through mitochondria-AMPK pathway in perioperative settings.
Keywords: anesthesia, cardiac preconditioning, dexmedetomidine, cardiac hypertrophy, heart failure, mitochondria
Introduction
Considerable compelling clinical evidence and experiment results have suggested an important association between anesthetics and cardiac preconditioning in the perioperative setting [1, 2]. Cardiac hypertrophy is a common preconditioning morphologic adaptation to various pharmacologic stimuli [3]. However, whether an association exists between dexmedetomidine (Dex) preconditioning and cardiac hypertrophy is unknown.
Dex, which has been approved for anesthetic use in Europe since 2011, is a highly selective a2-adrenoceptor agonist with systemic sedative, analgesic, and anxiolytic effects that is suitable for short- and long-term sedation in intensive care settings [4, 5]. In a recent study modeling sepsis, Dex was shown to attenuate systemic inflammation [6], whereas other studies demonstrated that Dex protects tissues against hypoxia-ischemia injury by suppressing apoptosis and improving adaptation to hypoxia [7-9]. Other studies have shown that Dex enhances cell viability by decreasing production of harmful metabolites, such as reactive oxygen species (ROS) and lactate [10, 11]. Collectively, these data suggest that Dex plays a protective role in various organs in perioperative settings via different molecular mechanisms. However, few reports have described the effect of Dex on various morphologic parameters.
Three highly homologous subtypes of the a2-adrenoceptor, a G protein-coupled receptor, have been described to date (a2A-, a2B-, and a2C-adrenergic receptor), and a fourth subtype, the a2D-adrenergic receptor, has been identified in several non-human species [12]. Although a2-adrenoceptors are widely distributed in the central nervous system and play a role in presynaptic inhibition of neurotransmitter release, some animal experimental studies indicated that it is also expressed in the kidney, vasculature, and heart [11, 13, 14]. Some G protein-coupled receptor agonists, such as endothelin-1 and phenylephrine, reportedly induce cardiomyocyte hypertrophy [15]. However, whether Dex induces similar cardiac hypertrophy is unknown.
Cardiac hypertrophy is a regulated, adaptive morphologic response to excessive pressure or high output demand. Excessive pressure on the heart, as often seen in hypertension, causes pathologic cardiac hypertrophy; whereas long-term exercise causes physiologic cardiac hypertrophy, as seen in cases of athletic heart [3]. Although both types of cardiac hypertrophy exhibit a similar phenotype of increased cardiac myocyte mass, they differ considerably in terms of molecular phenotype, cellular conductive pathway, and clinical consequences [16]. The present study investigated the protective effect of Dex on cardiac hypertrophy, and involved in vivo experiments in mice and in vitro experiments using neonatal rat cardiomyocytes (NRCMs). Our results suggest that Dex-induced cardiac hypertrophy potentially protects the heart against transverse aortic constriction (TAC)-induced heart failure. Dex-induced hypertrophy of cardiac myocytes (cardiomyocytes) in both the in vivo and in vitro experiments via inactivation of AMPK-dependent activation of the ERK, AKT, and PKC signaling pathways. Furthermore, Dex-induced inactivation of AMPK is dependent on high mitochondrial coupling efficiency and membrane potential. Our present study for the first time investigated a novel effect of Dex on cardiomyocyte and provided a novel mechanism of Dex-induced the protective cardiac myocyte hypertrophy.
Material and Methods
Ethics approval
All experiments were approved by the Ethics Committee (Approval No.: XHEC-F-2017-076) and performed in accordance with relevant guidelines and regulations.
Experimental animals
Male C57BL/6 mice were purchased from Shanghai Laboratory Animals Center (SLAC, Shanghai, China). Mice were provided free access to a normal chow diet and water under the conditions of the 23°C temperature and 50‑55% humidity with a 12-h light (08:00-20:00)/12-h dark (20:00-08:00) cycle.
TAC model
Male mice (10~12 weeks old) weighing an average of 22-25 g were used to establish a TAC model, as reported previously [17]. In brief, mice were anaesthetized by intraperitoneal injection of pentobarbital salt (50 mg/kg). The aortic arch was accessed via a left parasternal thoracotomy, and the thoracic aorta at the arch between the brachiocephalic trunk and left common carotid artery was surgically constricted using a 27-gauge needle to generate excessive pressure and progression to heart failure. Post-modeling, the Dex was administered for 2 consecutive weeks and observed mice survival for further 6-8 weeks.
Cardiac echography
Mice were kept anaesthetized via inhalation of 2~3% isoflurane during the entire cardiac echography procedure. Transthoracic cardiac echography was performed using a Vevo 2100 Ultra-High-Frequency ultrasound device for small animal imaging (FujiFilm VisualSonics, Toronto, Canada) equipped with an MS 400 linear array transducer (18-38 MHz). M-mode recordings at the midventricular level were taken, and images were analyzed using dedicated software (Vevo 2100, version 1.4.0). Internal ventricular septum thickness, left ventricular wall thickness, and internal diameters were measured. Left ventricular volume, ejection fraction (EF), and percent left ventricular fractional shortening (%FS) were calculated from the M-mode measurements. All measurements and calculations were performed under double-blind conditions with regarding to either vehicle or Dex administration.
NRCM culture
NRCMs were cultured as described in our previous report [11]. In brief, 1~3-day-old neonatal Wistar rats were purchased from SLAC (Shanghai, China). The entire heart was removed from each rat and cut into small pieces, which were incubated in digestion solution (0.025% collagen type II, 0.06% trypsin, and 20 μg/ml DNase) at 37°C three times (15 min per time). Cardiomyocytes were separated using a Percoll gradient (45.5% + 58.5%) system. The collected cardiomyocytes were seeded for culture at 1×105 cells per cm2 in Dulbecco's modified Eagle medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (Thermo Fisher Scientific) and incubated at 37°C in a 5% CO2/95% O2 atmosphere.
Reagents use
Dex (Sigma; Merck Millipore, Darmstadt, Germany) was solubilized in sterile water. Based on the maximum Dex clinical plasma concentration (1.25 ng/ml), in in vivo experiments, Dex (0.7 μg/kg/h) was administered subcutaneously for 2 weeks via an osmotic pump (Alzet, Model 2002, Cupertino, CA, USA). For all in vitro experiments, cardiomyocytes were treated with Dex at 37°C for 24 h. For AMPK activation, cardiomyocytes were incubated with AICAR (1 mM) (Sigma, Shanghai, China) for 24 h simultaneously with Dex treatment. For suppression of mitochondrial coupling efficiency and membrane potential, cardiomycytes were incubated with FCCP (50 mM) (Sigma, Shanghai, China) for 24 h simultaneously with Dex treatment. For α2-adrenoceptor blocking, cardiomyocytes were pre-incubated with atipamezole (10 nM) (Sigma, Shanghai, China) for 30 min before Dex treatment.
Mitochondrial TMRE assay and cellular flux analyzer
Cardiomyocytes mitochondrial coupling efficiency were calculated after measurement of cardiomyocyte OCR by Seahorse XF 24 extracellular analyzer (Agilent Technologies, Inc.) after Dex incubation for 24 h. Cardiomyocytes were cultured in collagen-coated 96-well dishes for TMRE assay (cat. no. ab113852; Abcam; Cambridge, UK) to detect mitochondrial membrane potential after Dex incubation for 24 h. The details have been reported in our previous study [11].
Observation of live NRCMs and cellular area calculation
Cardiomyocytes were cultured in 12-well, collagen-coated multi-well dishes. Cardiomyocytes were observed and images were captured using a fluorescence microscope (Olympus FluoView™ FV1000; Olympus Corp., Tokyo, Japan). The area of cardiomyocytes was measured using ImageJ software (version 1.47; National Institutes of Health) from five randomly selected visual fields in each well. Three wells (15 visual fields) of cardiomyocytes were measured and calculated.
[3H]-Leucine incorporation
NRCMs were pretreated with Dex for 24 h, washed with ice-cold sterile PBS three times after incubation with [3H]-leucine (1 μCi/ml) for 12 h, then incubated with ice-cold trichloroacetic acid (5% [v/v]) at 4°C for 1 h. After another wash with ice-cold, sterile PBS, NRCMs were suspended in 0.1 mol/l NaOH. Radioactivity was measured using a Beckman LS6500 liquid scintillation counter (Beckman Instrument, Inc. Fullerton, CA, USA).
Cellular immunofluorescence staining
Cardiomyocytes were cultured in 4-well, multi-chamber glass slides (MatTek Corp., Tokyo, Japan). Following Dex treatment for 24 h, cells were washed with sterile PBS three times (5 min each) and fixed in cold acetone for 20 min, then washed with TBS three times. Following blocking with BSA (Sigma; Merck Millipore) for 1 h at room temperature (24-26°C), immunofluorescence staining was performed using an anti-Myh6 antibody (goat pAb; 1:600; sc-168676; Santa Cruz Biotechnology, Inc.) and donkey anti-goat IgG (red) (AlexaFluor® 488, Abcam, Shanghai, China). Counterstaining with fluorescence mounting medium containing DAPI (blue; Thermo Fisher Scientific, Inc.) was performed to visualize normal nuclei. Sections were observed using a fluorescence microscope (Olympus FluoView™ FV1000; Olympus Corp.).
Histologic analysis
Mouse heart tissue was fixed in 4% formaldehyde (Sinopharm Chemical Reagent Co., Ltd, Shanghai, China) at room temperature for more than 12 h, then embedded in paraffin after complete dehydration using ethyl alcohol. Blocks were cut into 4-μm-thick slices, air-dried, dewaxed in xylene, and stained with hematoxylin and eosin (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China). Heart tissue images were captured using a fluorescence microscope (Olympus FluoView™ FV1000; Olympus Corp.) and light-field observation model.
RT-qPCR
Total RNA extraction from cultured cardiomyocytes or heart tissue, cDNA synthesis, and real-time PCR amplification were performed according to routine methods described in our previous report [11]. Results were normalized to expression of the 18s rRNA gene. The primers used in the present study were reported previously [17] and are shown in Supplementary Table 1.
Western blot analysis
Whole protein from NRCMs or mouse heart tissue was extracted as previously reported [11]. Intercellular signaling was analyzed using a Pathscan Intercellular Signaling Array kit (#7323 and #12856; Cell Signaling Technology, Inc.) following the protocol provided, and routine western blotting procedures were performed according to our previous report [11]. The primary antibodies used were as follows: anti-ANP (mouse mAb; 1:300; sc-515701; Santa Cruz Biotechnology, Inc.), anti-BNP (rabbit pAb; 1:500; ab19645; Abcam), anti-MYH7 (mouse mAb; 1:300; sc-53089; Santa Cruz Biotechnology, Inc.), anti-Erk1/2 (rabbit mAb; 1:1000; #9102), anti-phospho-Erk1/2 (Thr202/Thr204) (rabbit mAb; 1:1000; #9102), anti-Akt (rabbit mAb; 1:1000; #9272), anti-phospho-Akt (Ser473) (rabbit mAb; 1:1000; #4060), anti-PKC (rabbit mAb; 1:1000; #2056), anti-phospho-PKC pan (Thr514) (rabbit mAb; 1:1000; #9379), anti-AMPK (rabbit mAb; 1:1000; #5831), anti-phospho-AMPK (Thr172) (rabbit mAb; 1:1000; #2535), and anti-GAPDH (rabbit mAb; 1:2000; #2118;Cell Signaling Technology, Inc.). The secondary antibodies used were horseradish peroxidase (HRP) conjugated (GE Healthcare Life Sciences, Beijing, China): anti-mouse IgG, HRP-conjugated whole Ab sheep (NA931) or anti-rabbit IgG, HRP-linked whole Ab donkey (NA934).
Statistical analysis
All experiments were repeated two or three times. All results are reported as the mean ± standard error of the mean. The normality of distribution was analyzed using the D'Agostino-Pearson omnibus normality test (version 6.0; GraphPad Software, Inc., La Jolla, CA, USA). Comparisons between two groups were analyzed using the Student's t‑test. Multiple comparisons between groups were performed using one‑way analysis of variance with Tukey's multiple comparisons test. Mouse survival was analyzed using the Kaplan-Meier log-rank test. Both one-way analysis of variance and Kaplan-Meier tests were performed using GraphPad Prism software (version 6.0). A P value of <0.05 was considered indicative of a statistically significant difference.
Results
Dex induces cardiac hypertrophy and protects against heart failure in mice
Firstly, to know whether Dex induces cardiac hypertrophy in vivo, mice were administered Dex subcutaneously for 2 weeks, after which cardiac echography was performed. We found that Dex significantly increased cardiac left ventricular posterior wall thickness (LVPW) but had no effect on left ventricular internal dimension (LVID) or %FS (Fig. 1A, B and Table 1). Consistently, we also observed enlarged cardiomyocytes in mice administered Dex compared to mice that received vehicle (Fig. 1C and D), accompanied with a significant increase in Anp mRNA levels (Fig. 1E). Dex did not increase the expression of several cardiac fibrosis markers (Ctgf, Col1, and Col3a) (Fig. 1E), indicating that Dex induces cardiac myocyte hypertrophy without cardiac fibrosis.
Echography parameters of wild-type mice administered either Dex or vehicle
| Measurement | Units | Administration group | P value | |
|---|---|---|---|---|
| Vehicle | Dexmedetomidine | |||
| IVST | ||||
| diastole | mm | 0.87±0.04 | 0.83±0.05 | 0.59 |
| systole | mm | 1.31±0.10 | 1.26±0.06 | 0.69 |
| LVID | ||||
| diastole | mm | 3.89±0.07 | 3.62±0.14 | 0.12 |
| systole | mm | 2.67±0.16 | 2.50±0.14 | 0.44 |
| LVPW | ||||
| diastole | mm | 0.73±0.02 | 0.92±0.04 | 0.0032 |
| systole | mm | 1.15±0.06 | 1.20±0.05 | 0.50 |
| LV volume | ||||
| diastole | μL | 65.6±2.9 | 55.9±5.2 | 0.14 |
| systole | μL | 27.1±4.0 | 22.9±3.1 | 0.43 |
| EF | % | 59.4±4.6 | 59.3±3.4 | 0.98 |
| FS | % | 31.5±3.1 | 31.0±2.3 | 0.91 |
IVST: interventricular septum thickness; LVID: left ventricular internal dimension; LVPW: left ventricular posterior wall; LV volume: left ventricular volume; EF: ejection fraction; FS: fractional shortening.
Dex induces cardiomyocyte hypertrophy and protects against heart failure in mice. (A) Representative cardiac echographs of vehicle- and Dex-treated mice. (B) LVPW, LVID, and FS% values determined from cardiac echographs shown in panel (A). (C) Representative images of hematoxylin and eosin-stained heart tissues from mice administered vehicle or Dex (scale bar: 50 μm). (D) Distribution of cardiomyocyte area in heart tissues from mice shown in panel (C). (E) Relative expression of mRNAs for cardiomyocyte hypertrophy markers (Anp, Bnp, Myh7) and cardiac fibrosis markers (Ctgf, Col1, Col3a) in vehicle and Dex-treated mice. Values for vehicle were set to 1. (F) Schedule of Dex administration in mice with TAC. (G) Kaplan-Meier survival curves for vehicle- and Dex-treated mice with TAC. (H) Representative cardiac echographs in vehicle- and Dex-treated mice with TAC. (I) LVPW, LVID, and FS% values were determined from cardiac echographs of H. Data are mean ± SEM (n=5 per group in each group. * P<0.05; ** P<0.01).
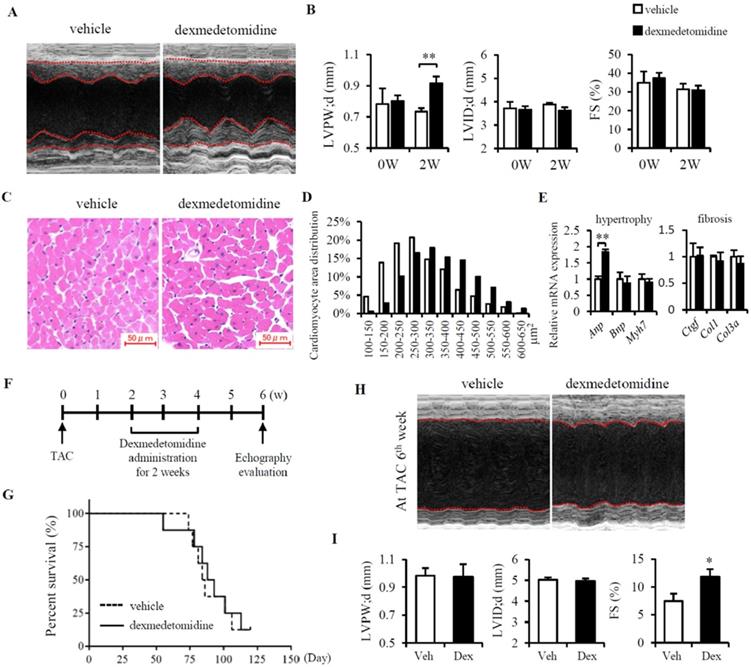
After this, we evaluated whether Dex-induced cardiac myocyte hypertrophy is beneficial or detrimental by using TAC model in mice. At 2 weeks after the TAC operation, the mice were administered Dex subcutaneously for 2 weeks. After another 2 weeks, cardiac function was evaluated using cardiac echography (Fig. 1F). Although we didn't find any difference in Mice survival between vehicle and Dex administered mice (Fig. 1G), mice administered Dex exhibited an increase in %FS but no changes in LVPW and LVID compared with mice administered with vehicle (Fig. 1H, I and Table 2). To further determine whether this attenuation in the severity of heart failure induced by Dex was associated with attenuated cardiac fibrosis, we evaluated the degree of cardiac fibrosis in the two groups of mice by examining the levels of several fibrosis markers (Ctgf, Col1, and Col3a) using RT-PCR and heart Masson trichrome staining. However, no significant differences were observed between mice administered Dex and vehicle (data not shown).
Echography parameters of wild-type mice with TAC administered either Dex or vehicle
| Measurement | Units | Administration groups | P value | |
|---|---|---|---|---|
| Vehicle | Dexmedetomidine | |||
| IVST | ||||
| diastole | mm | 1.02±0.05 | 1.04±0.06 | 0.83 |
| systole | mm | 1.26±0.09 | 1.38±0.11 | 0.43 |
| LVID | ||||
| diastole | mm | 5.03±0.12 | 4.96±0.13 | 0.71 |
| systole | mm | 4.77±0.15 | 4.27±0.07 | 0.034 |
| LVPW | ||||
| diastole | mm | 0.98±0.05 | 0.98±0.09 | 0.94 |
| systole | mm | 1.03±0.05 | 1.07±0.07 | 0.68 |
| LV volume | ||||
| diastole | μL | 120.1±6.9 | 116.4±6.8 | 0.71 |
| systole | μL | 106.3±7.8 | 81.9±3.0 | 0.041 |
| EF | % | 16.4±2.9 | 25.5±2.5 | 0.046 |
| FS | % | 7.44±1.4 | 11.9±1.3 | 0.048 |
IVST: interventricular septum thickness; LVID: left ventricular internal dimension; LVPW: left ventricular posterior wall; LV volume: left ventricular volume; EF: ejection fraction; FS: fractional shortening.
In total, TAC induced LVPW increased from 0.73 mm to 0.98mm (increased about 34%). In Dex group, after TAC, LVPW increased from 0.92 mm to 0.98 mm (increased only 6.5%) (Fig. 1B, I). This might indicates that Dex preconditioning helps the heart resist hypertrophy induced by TAC surgery. Additionally, Dex preconditioning did not affect heart FS% compared to the vehicle group (31.0% vs. 31.5%), but Dex preconditioning did prevent a decrease in FS% compared to the vehicle group (11.85% vs. 7.44%) (Fig. 1B and I). Dex preconditioning induces protective hypertrophy, providing protection against TAC-induced heart failure that might be a beneficial morphologic change.
Dex induces NRCMs growth with high protein synthesis
To investigate the primary effect of Dex on cardiomyocyte hypertrophy, we prepared primary cultured NRCMs and exposed them to Dex at three different doses (10-7, 10-6, and 10-5 M). After 24 h of Dex exposure on NRCMs, the morphology of live and MYH6-stained NRCMs was observed under an optical or fluorescence microscope, and the area of the NRCMs was calculated. Dex-treated NRCMs were much larger than NRCMs in the vehicle treated group in a dose dependent manner (Fig. 2A and B). To further confirm that the observed increases in NRCMs size was induced by Dex, we evaluated cardiomyocyte hypertrophy markers using RT-PCR and western blotting. We found that levels of Anp and Bnp were significantly up-regulated in cells treated with both the 10-6 and 10-5 M doses of Dex, whereas levels of Myh7 were significantly up-regulated only in cells treated with 10-5 M Dex (Fig. 2C and D).
Dex induces NRCMs enlargement with up-regulated protein synthesis. (A) Representative live NRCMs (top) observed by light microscopy and MYH6-stained NRCMs (bottom) observed by fluorescence microscopy in control cells and cells treated with three concentrations (10-7, 10-6, and 10-5 M) of Dex (red: MYH6-stained NRCMs; blue: DAPI-stained NRCM nuclei. Scale bars: 100 μm). (B) Cardiomyocyte area of NRCMs shown in panel (A).(C and E) Relative expression of Anp, Bnp, Myh7 (C) and c-jun and c-fos (E) mRNAs in NRCMs treated with vehicle and three concentrations (10-7, 10-6, and 10-5 M) of Dex. Values for the vehicle group were set to 1. (D) Representative western blot results for expression of ANP, BNP, and MYH7 in vehicle and Dex-treated cardiomyocytes. GAPDH was used as a loading control. (F) [3H]-Leucine incorporation rate in vehicle and Dex-treated cardiomyocytes. Value for the vehicle group was set to 100%. Data are mean ± SEM (* P<0.05; ** P<0.01; † p<0.001).
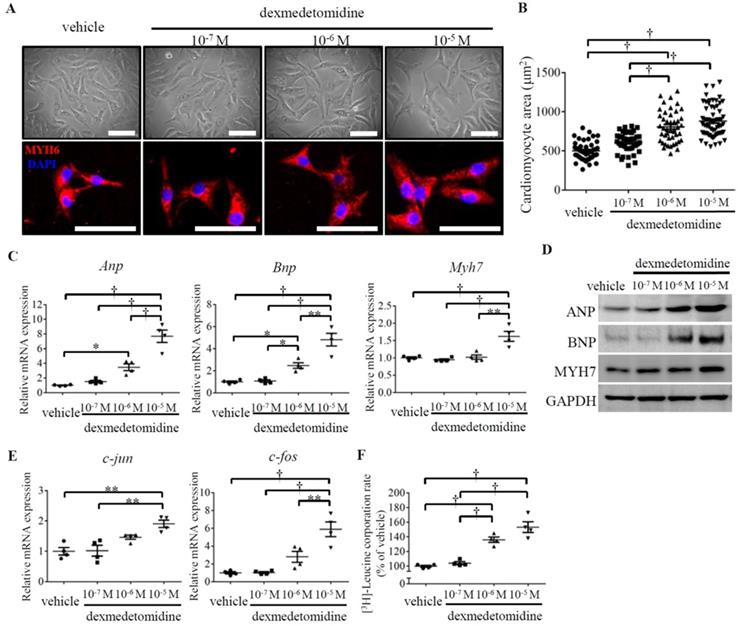
Proto-oncogenes are known to regulate cell proliferation and growth [18]. To evaluate whether Dex induces NRCMs growth, we investigated the expression of two proto-oncogenes: c-jun and c-fos. The expression of both proto-oncogenes was significantly up-regulated in NRCMs treated with 10-5 M Dex compared with vehicle treated NRCMs (Fig. 2E). As cardiomyocyte hypertrophy is typically associated with enhanced protein synthesis [19], we next investigated protein synthesis in Dex-treated NRCMs. The [3H]-leucine incorporation rate increased significantly, by 138% and 150%, in NRCMs treated with 10-6 M and 10-5 M Dex, respectively (Fig. 2F). Collectively, these results indicate that Dex promotes NRCM hypertrophy, as evidenced by increases in cell area, cell growth marker expression, and protein synthesis.
However, at 10-7 M, Dex did not affect the expression of cardiomyocyte hypertrophy markers (Anp, Bnp, and Myh7), cell growth markers (c-jun and c-fos), or protein synthesis (Fig. 2C-F), which could explain why Dex treatment at 10-7 M did not promote NRCMs hypertrophy (Fig. 2A and B).
ERK, AKT, PKC, and AMPK are involved in Dex-induced NRCMs hypertrophy
We then sought to determine what signaling pathway mediates cardiomyocyte hypertrophy induced by Dex. We examined lots of key protein by using western blotting and finally we found phosphorylation of ERK, AKT, and PKC were up-regulated whereas phosphorylation of AMPK was down-regulated in heart tissue of mice administered Dex compared to tissue from mice administered vehicle (Fig. 3A and B). These results indicate that the inactivation of AMPK and activation of ERK, AKT and PKC maybe involved in Dex promoted cardiac hypertrophy. We furtherly investigated these proteins in NRCMs, we found the same results showed inactivated AMPK and activated ERK, AKT and PKC in NRCMs after Dex treatment for 24 h (Fig. 3C and D).
Dex activates ERK, AKT, PKC and inactivates AMPK in mice heart tissue and NRCMs. (A) Representative western blots of expression of ERK, AKT, PKC, and AMPK and their phosphorylated forms in vehicle- and Dex-treated mice heart tissue. GAPDH was used as a loading control. (B) Relative quantification of the ratios of p-ERK/ERK, p-AKT/AKT, p-PKC/PKC, and p-AMPK/AMPK as determined from western blot bands shown in panel (A). Values of vehicle group were set to 1. (C) Representative western blots of expression of ERK, AKT, PKC, and AMPK and their phosphorylated forms. GAPDH was used as a loading control. (D) Relative quantification of the ratios of p-ERK/ERK, p-AKT/AKT, p-PKC/PKC, and p-AMPK/AMPK as determined from western blot bands in panel (C). Values for the vehicle group were set to 1. (E) Linear relationship between p-AMPK and p-ERK, p-AKT, or p-PKC in each NRCM group. Data are mean ± SEM (* P<0.05; ** P<0.01).
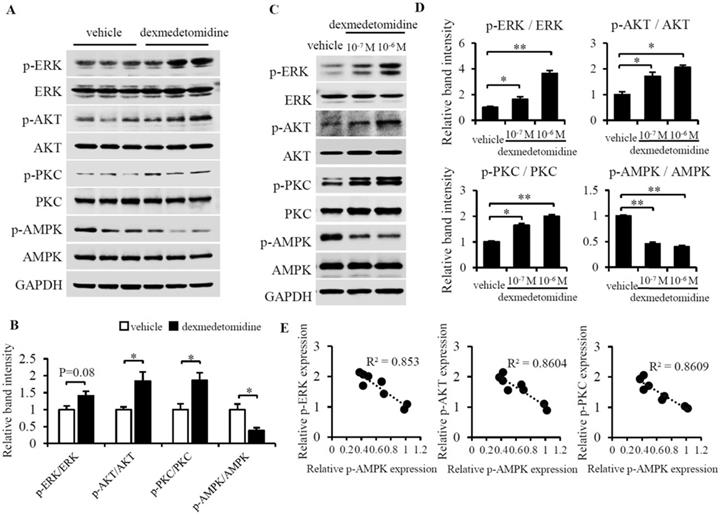
To further examine the potential protein involved in Dex-induced cardiomyocytes hypertrophy, we investigated cyclopedic conductive signaling proteins using intracellular signaling pathway array analysis (Supplementary Figure 1), which included 18 protein kinases. We found that the expression of 7 of the protein kinases changed in response to Dex. Of these kinases, except the most changed four proteins (p-ERK1/2, p-AKT, p-PKC and p-AMPK), we also found the expression of p-PTEN, p-mTOR and p-Stat3 [s727] were also increased in Dex-treated NRCMs compared to control NRCMs (Supplementary Figure 1). This analysis indicated p-PTEN, p-mTOR and p-Stat3 [s727] also play some potential role on Dex-induced cardiomyocyte hypertrophy.
As it was reported that overexpression of either ERK1/2, AKT, or PKC can cause compensated or concentric cardiac hypertrophy [20-22] and AMPK activation can prevent cardiac hypertrophy [23], we hypothesized that activation of ERK1/2, AKT, and PKC and inactivation of AMPK mainly mediates Dex-induced cardiomyocyte hypertrophy.
Dex-mediated up-regulation of p-ERK, p-AKT, and p-PKC is dependent on inactivation of p-AMPK
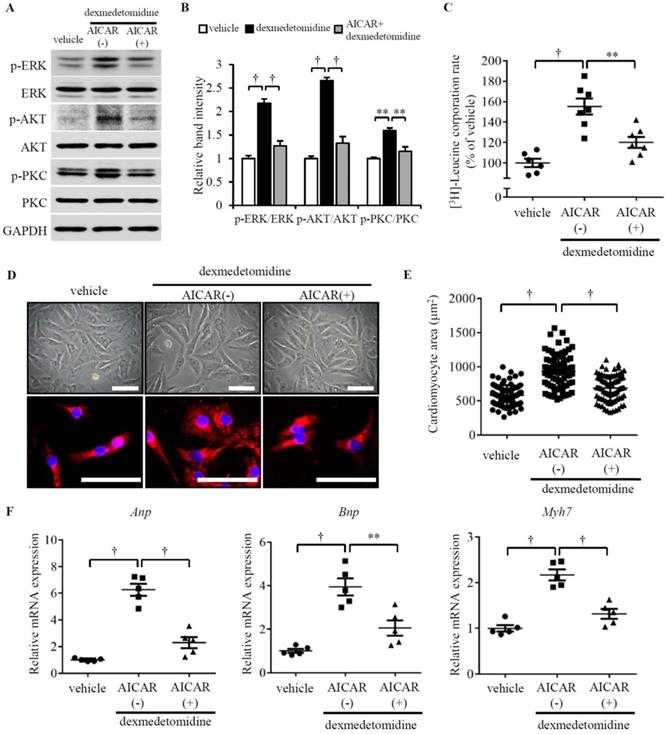
As we observed that ERK, AKT, and PKC were up-regulated, while AMPK was down-regulated, we sought to determine whether there is a relationship between the reversed changes in Dex-treated cardiomyocytes. Reports have indicated that AMPK activation suppresses cardiac protein synthesis and hypertrophy via inhibition of ERK, AKT, and PKC in cardiomyocytes [24-26] and the AMPK activator 5-aminoimidazole-4-carboxamide riboside (AICAR) inhibits activation of AKT in neurocytes [27]. We indeed found down-regulated p-AMPK is linear related with up-regulated p-ERK, p-AKT, and p-PKC (Fig. 3E). These reports and observations suggest a likelihood that Dex-induced up-regulation of p-ERK, p-AKT, and p-PKC may be associated with inactivation of AMPK. To test this hypothesis, we used AICAR to activate AMPK and investigated whether AMPK activation blocks Dex-induced NRCM hypertrophy. Interestingly, as we hypothesized, AICAR blocks Dex-induced activation of ERK, AKT, and PKC (Fig. 4A and B) as well as Dex-induced protein synthesis (Fig. 4C) in NRCMs. Additionally, AICAR-preconditioned NRCMs exhibited lower cardiomyocyte area compared to vehicle preconditioned NRCMs treated with Dex (Fig. 4D and E). To confirm this result, we also investigated the expression of cardiomyocyte hypertrophy markers. The expression of Anp, Bnp, and Myh7 mRNAs did not increase after Dex treatment in AICAR-preconditioned NRCMs, compared with vehicle-conditioned NRCMs (Fig. 4F). Collectively, these results suggest that AMPK activation blocks ERK, AKT, and PKC activation, protein synthesis, and cardiomyocyte hypertrophy, indicating that AMPK inactivation plays a crucial role.
Dex-induced NRCMs hypertrophy depends on the inactivation of p-AMPK. (A) Representative western blots of expression of ERK, AKT, and PKC and their phosphorylated forms in control and Dex-treated cells with and without AICAR pre-incubation. GAPDH was used as a loading control. (B) Relative quantification of the ratios of p-ERK/ERK, p-AKT/AKT, and p-PKC/PKC as determined from western blot bands shown in panel (A). Values of vehicle group were set to 1. (C) [3H]-Leucine incorporation rate in vehicle and Dex-treated NRCMs with and without AICAR pre-incubation. Value of vehicle group was set to 100%. (D) Representative live NRCMs (top) and MYH6-stained NRCMs (bottom) in vehicle and Dex-treated groups with and without AICAR (AMPK activator) pre-incubation (red: MYH6-stained NRCMs; blue: DAPI-stained NRCM nuclei. Scale bars: 100 μm). (E) Area of NRCMs shown in panel (D). (F) Relative expression of Anp, Bnp, and Myh7 mRNAs in vehicle and Dex-treated cardiomyocytes with and without AICAR pre-incubation. Values of vehicle group were set to 1. Data are mean ± SEM (n=5-7 per group in C and F, n=3-4 per group in B; ** P<0.01; † P<0.001).
Dex induces inactivation of AMPK is dependent on elevated mitochondrial coupling efficiency and membrane potential
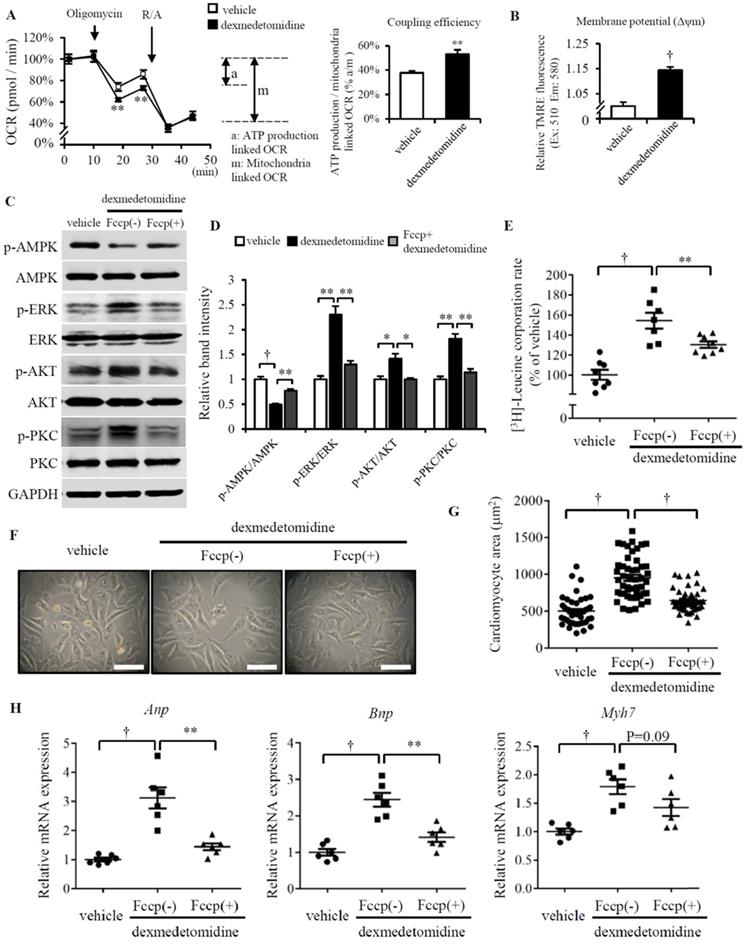
We next asked why AMPK is inactivated after Dex treatment. In our previous study, we reported Dex enhanced NRCMs mitochondrial coupling efficiency and membrane potential (Δψm) [11]. Other report also showed FCCP induced mitochondrial uncoupling increased AMPK activation [28]. So, these made us think about whether the inactivation of AMPK induced by Dex is resulted from the enhanced coupling efficiency and mitochondrial Δψm. Before examining this hypothesis, we firstly confirmed Dex elevates mitochondrial coupling efficiency and Δψm (Fig. 5A and B), the similar result with our previous report [11]. FCCP, a well-known mitochondrial potent uncoupler, suppresses mitochondrial coupling efficiency and Δψm [29, 30]. Next, we used FCCP to inhibit NRCMs mitochondrial coupling efficiency and Δψm, then we found FCCP increases AMPK activation and decreases ERK, AKT, PKC activation in Dex incubated NRCMs compared to that treated without FCCP (Fig. 5C and D). We also found the protein synthesis is blocked in Dex incubated NRCMs treated with FCCP compared with that treated without FCCP (Fig. 5E). Furtherly, paralleled with these results, FCCP suppresses the increased NRCMs area induced by Dex (Fig. 5F and G) and suppresses the increased mRNA expression (Anp, Bnp and Myh7) induced by Dex (Fig. 5H). Taken together, these results suggest the inhibition of mitochondrial coupling efficiency and Δψm facilitates the activation of AMPK, inactivation of ERK, AKT and PKC, as well as suppresses the following NRCMs protein synthesis and NRCMs hypertrophy.
Increased mitochondrial function facilities Dex-induced NRCMs hypertrophy. (A) Flux analysis indicates cellular OCR change in mitochondrial stress (left) and the calculated coupling efficiency determined by ATP production-/mitochondrial-linked OCR (right) in NRCMs incubated with or without Dex. (B) Mitochondrial membrane potential determined by mitochondrial TMRE assay in NRCMs incubated with or without Dex. (C) Representative western blots of expression of ERK, AKT, PKC, and AMPK and their phosphorylated forms in vehicle and Dex-treated groups with and without FCCP pre-incubation. GAPDH was used as a loading control. (D) Relative quantification of the ratios of p-ERK/ERK, p-AKT/AKT, p-PKC/PKC, and p-AMPK/AMPK as determined from western blot bands shown in panel (C). Values of vehicle were set to 1. (E) [3H]-Leucine incorporation rate in vehicle and Dex-treated NRCMs with and without FCCP pre-incubation. Value of vehicle group was set to 100%. (F) Representative live NRCMs in vehicle and Dex-treated groups with and without FCCP pre-incubation (Scale bars: 100 μm). (G) Area of NRCMs shown in panel (F). (H) Relative expression of Anp, Bnp, Myh7 mRNAs in vehicle and Dex-treated groups with and without FCCP pre-incubation. Values of vehicle group were set to 1. Data are mean ± SEM (n=10-12 per group in A and B, n=3-4 per group in D, n=6-8 per group in E and H; * P<0.05; ** P<0.01; † p<0.001).
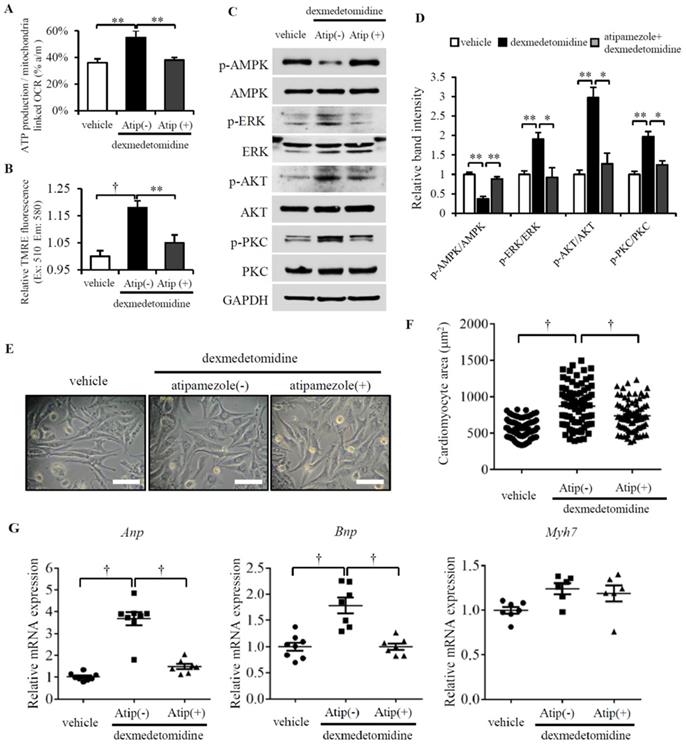
Dex induces α2-adrenergic receptor-mediated cardiac myocyte hypertrophy
Although Dex is an α2-adrenergic receptor agonist, whether Dex-induced cardiomyocyte hypertrophy is mediated by α2-adrenergic receptors is unknown. To resolve this question, we pretreated cardiomyocytes with atipamezole (α2-adrenergic receptor antagonist) for 30 min prior to Dex treatment. After 24 h, we examined mitochondrial coupling efficiency, Δψm and found both are decreased in NRCMs pretreated with atipamezole (Fig. 6A and B). We next further investigated the activation of AMPK, ERK, AKT and PKC. We found Dex increased the phosphorylation of ERK, AKT, and PKC and decreased the phosphorylation of AMPK; however, atipamezole blocked these Dex-induced changes completely (Fig. 6C and D). Next, we quantified the cardiomyocyte area and the involved Anp and Bnp mRNA expression. However, atipamezole-pretreated cardiomyocytes exhibited less hypertrophy, and Anp and Bnp mRNA expression was completely blocked (Fig. 6E-G). These results demonstrate that α2-adrenergic receptors mediate Dex-induced cardiomyocyte hypertrophy.
Atipamezole blocks Dex-induced NRCMs hypertrophy. (A and B) Mitochondrial coupling efficiency determined by ATP production-/mitochondrial-linked OCR (A) and mitochondrial membrane potential (B) in vehicle or Dex-treated NRCMs with or without preconditioning of atipamezole. (C) Representative western blots of expression of ERK, AKT, PKC, and AMPK and their phosphorylated forms in vehicle and Dex-treated groups with and without atipamezole pre-incubation. GAPDH was used as a loading control. (D) Relative quantification of the ratios of p-ERK/ERK, p-AKT/AKT, p-PKC/PKC, and p-AMPK/AMPK as determined from western blot bands shown in panel (C). Values of vehicles were set to 1. (E) Representative live NRCMs in vehicle and Dex-treated groups with and without atipamezole pre-incubation (Scale bars: 100 μm). (F) Area of NRCMs shown in panel (E). (G) Relative expression of Anp, Bnp, Myh7 mRNAs in vehicle and Dex-treated groups with and without atipamezole pre-incubation. Values of vehicle group were set to 1. Data are mean ± SEM (n=6-8 per group in (C), n=3-4 per group in (E). * P<0.05; ** P<0.01; † p<0.001).
Discussion
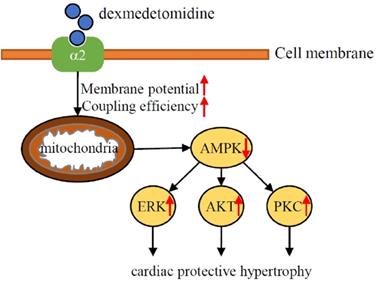
To the best of our knowledge, the results of the present study was the first to demonstrate that Dex induces a protective hypertrophy in mouse heart and NRCMs via α2-adrenergic receptor- and mitochondria- mediated inactivation of AMPK and activation of the ERK, AKT, and PKC pathways. The results of this study found Dex-induced activation of the ERK, AKT, and PKC pathways depends on the inactivation of AMPK and elevated mitochondrial coupling efficiency and membrane potential. When we administered Dex to mice with TAC, although we found that Dex don't affect heart failure related survival, Dex protects against TAC-induced heart failure but had no effect on survival. These results suggest that cardiac preconditioning with Dex in the perioperative setting has a protective effect against cardiac damage (Fig. 7).
Schematic illustration of the results of the present study indicating that Dex preconditioning elevates cardiomyocytes mitochondrial coupling efficiency and membrane potential, which induces the inactivation of AMPK via α2-adrenergic receptor. Down-regulated AMPK further facilitates activation of ERK, AKT, PKC signaling and lead to cardiac hypertrophy, which protects heart against heart failure.
Cardiac hypertrophy is a complex adaptation process involving either extracellular or intracellular signaling circumstance towards to various drugs or physiological requirement. Activation of AMPK inhibits protein synthesis and cardiac hypertrophy [23, 24]; conversely, activation of either ERK, AKT or PKC up-regulates protein synthesis and induces cardiac hypertrophy [20-22]. Furthermore, the effect of AMPK activation was dependent on the down-regulation of ERK, AKT, or PKC [25-27]. These previous reports suggest that suppression of AMPK activation accelerates ERK, AKT, and PKC activation. Although one study suggested that Dex preconditioning induces the expression of pro-survival kinases in heart tissue, including ERK and AKT [14], the results were not significant, and no mechanism leading to up-regulation of these pro-survival kinases was proposed. The present study, however, demonstrated that Dex up-regulates the phosphorylation of ERK, AKT, and PKC in an AMPK dephosphorylation-dependent manner.
Reports indicate that ERK, AKT, and PKC play an important role not only in cardiac hypertrophy, but also cell survival [31, 32]. Previous reports indicated that Dex preconditioning increases the expression of pro-survival kinases and attenuates ischemia/reperfusion injury in rat heart and brain [8, 14]. In these previous studies, the authors only reported the potential pro-survival role of these kinases but ignored their role in morphologic adaptation. We found that administration of Dex for 2 weeks induces cardiac hypertrophy in mice, which has a protective role against TAC-induced heart failure over the short term but does not affect TAC-associated mortality. This result led us to consider that although Dex-induced cardiac hypertrophy is temporary, this short-term effect explains the importance of preconditioning in the perioperative setting.
Exercise is known to induce physiologic cardiac hypertrophy, which protects against heart failure via a very complex mechanism involving the activation of ERK, AKT, and PKC [33-35]. Interestingly, we found that Dex also induces protective cardiac hypertrophy via activation of ERK, AKT, and PKC, which led us to consider that Dex-induced cardiac hypertrophy may be more similar with physiologic hypertrophy than pathologic hypertrophy. In 1984, it was reported that α2-adrenergic receptors are activated during exercise [36]. As the results of our present study demonstrated that Dex preconditioning induces physiologic cardiac hypertrophy involving α2-adrenergic receptors, we hypothesized that exercise-induced α2-adrenergic receptor activation with subsequent activation of ERK, AKT, and PKC is similar with the condition induced by Dex. Moreover, it was reported that failing hearts were accompanied with mitochondria dysfunction and reduced oxygen consumption and exercise enhances mitochondrial function [37, 38]. This similarity could explain why the physiologic-like cardiac hypertrophy induced by Dex protects against heart failure. Contrary to Dex, several studies reported that exercise increased the activation of AMPK in skeletal muscles [39, 40], whereas, Grassi et al. did not support this statement [41] in cardiac functions, hence future studies are needed to compare the protection mechanism between Dex and exercise in heart failure.
Our previous study demonstrated that Dex (100 nM) reduces ROS sensitivity in NRCM mitochondria via downregulation of mitochondrial respiratory complexes I, II, and III and upregulation of Bcl2 protein expression, which protects NRCMs against ROS-induced apoptosis, thus enhancing NRCM survival [11]. However, the mechanism by which Dex induces this mitochondrial phenotype remains to be elucidated. A number of reports have demonstrated that ERK, AKT, and PKC are closely associated with mitochondrial function. For example, phosphorylated ERK translocates to the mitochondria, where it regulates Bcl2 expression and decreases mitochondrial cytochrome C release [42, 43]. AKT accumulation in mitochondria of cardiomyocytes protects the cells by preventing opening of the mitochondrial permeability transition pore [44, 45], whereas PKC activates mitochondrial ATP-sensitive K+ channels, thus protecting against ischemic injury [46]. All of these reports describe ERK, AKT, or PCK activation in mitochondria, which could explain how Dex regulates sensitivity to ROS and Bcl2 expression in NRCM mitochondria. However, very interestingly, we demonstrated mitochondrial coupling efficiency and membrane potential, induced by Dex, are necessary for AMPK inactivation and ERK, AKT, or PCK activation, which are partially consistent with the previous report [28]. This suggests Dex induced preferable mitochondrial change closely linked with the inactivation of AMPK.
For limitation, we hypothesized that AMPK inactivation is essential for the regulation of cardiomyocyte hypertrophy induced by Dex. However, we failed to prove whether AMPK inhibition could induce cardiomyocyte hypertrophy in this study. In addition, we noted that Dex increases Anp in heart but increases Anp, Bnp, Myh7 in cardiomyocytes. This discrepancy may be caused by the complicated atmosphere of tissues or due to species difference, since the cardiomyocytes were isolated from rats and the in vivo analysis were performed on mice. Moreover, clinical data are needed to validate the findings observed in animals and cells.
In summary, the in vitro and in vivo results of the present study demonstrate for the first time that preconditioning with Dex induces cardiomyocyte hypertrophy that protects against heart failure via α2-adrenergic receptor- and mitochondria- mediated AMPK dephosphorylation and ERK, AKT, and PKC phosphorylation. Our demonstration of Dex-induced morphologic cardiac changes in mice provides novel important evidence of the beneficial effects of Dex preconditioning in perioperative settings.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank JinWu (Affiliated Hospital of Jiang Su University, Jiang Su, China) for valuable suggestions and discussion.
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81400895).
Authors' Contributions
Xiaojian Weng and Hua Liu were in charge of performing experiments. Xiaodan Zhang, Qian qian Sun, Cheng Li, Minglu Gu and Yanyifang Xu helped analyze the data. Xiaojian Weng and Hua Liu wrote the manuscript. Shitong Li, Weiwei Li and Jianer Du were the principal investigator of this study and gave final approval for publication of the manuscript. All authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zaugg M, Lucchinetti E, Garcia C, Pasch T, Spahn DR, Schaub MC. Anaesthetics and cardiac preconditioning. Part II. Clinical implications. Br J Anaesth. 2003;91:566-76
2. Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth. 2003;91:551-65
3. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387-407
4. Gerresheim G, Schwemmer U. [Dexmedetomidine]. Anaesthesist. 2013;62:661-74
5. Keating GM. Dexmedetomidine: A Review of Its Use for Sedation in the Intensive Care Setting. Drugs. 2015;75:1119-30
6. Zhang J, Wang Z, Wang Y, Zhou G, Li H. The effect of dexmedetomidine on inflammatory response of septic rats. BMC Anesthesiol. 2015;15:68
7. Chen S, Hua F, Lu J. et al. Effect of dexmedetomidine on myocardial ischemia-reperfusion injury. Int J Clin Exp Med. 2015;8:21166-72
8. Ren X, Ma H, Zuo Z. Dexmedetomidine Postconditioning Reduces Brain Injury after Brain Hypoxia-Ischemia in Neonatal Rats. J Neuroimmune Pharmacol. 2016;11:238-47
9. Liu YE, Tong CC, Zhang YB, Jin HX, Gao Y, Hou MX. Effect of dexmedetomidine on rats with renal ischemia-reperfusion injury and the expression of tight junction protein in kidney. Int J Clin Exp Med. 2015;8:18751-7
10. Willigers HM, Prinzen FW, Roekaerts PM, de Lange S, Durieux ME. Dexmedetomidine decreases perioperative myocardial lactate release in dogs. Anesth Analg. 2003;96:657-64 table of contents
11. Weng X, Zhang X, Lu X, Wu J, Li S. Reduced mitochondrial response sensitivity is involved in the antiapoptotic effect of dexmedetomidine pretreatment in cardiomyocytes. Int J Mol Med. 2018;41:2328-38
12. Ruuskanen JO, Xhaard H, Marjamaki A. et al. Identification of duplicated fourth alpha2-adrenergic receptor subtype by cloning and mapping of five receptor genes in zebrafish. Mol Biol Evol. 2004;21:14-28
13. Kanagy NL. Alpha(2)-adrenergic receptor signalling in hypertension. Clin Sci (Lond). 2005;109:431-7
14. Ibacache M, Sanchez G, Pedrozo Z. et al. Dexmedetomidine preconditioning activates pro-survival kinases and attenuates regional ischemia/reperfusion injury in rat heart. Biochim Biophys Acta. 2012;1822:537-45
15. Clerk A, Michael A, Sugden PH. Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular myocytes by the G protein-coupled receptor agonists, endothelin-1 and phenylephrine: a role in cardiac myocyte hypertrophy? J Cell Biol. 1998;142:523-35
16. Iemitsu M, Miyauchi T, Maeda S. et al. Physiological and pathological cardiac hypertrophy induce different molecular phenotypes in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R2029-36
17. Tian Z, Miyata K, Kadomatsu T. et al. ANGPTL2 activity in cardiac pathologies accelerates heart failure by perturbing cardiac function and energy metabolism. Nat Commun. 2016;7:13016
18. Bello Roufai M, Li H, Sun Z. Heart-specific inhibition of protooncogene c-myc attenuates cold-induced cardiac hypertrophy. Gene Ther. 2007;14:1406-16
19. Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda). 2006;21:362-9
20. Bueno OF, De Windt LJ, Tymitz KM. et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341-50
21. Condorelli G, Drusco A, Stassi G. et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:12333-8
22. Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, Walsh RA. Transgenic overexpression of constitutively active protein kinase C epsilon causes concentric cardiac hypertrophy. Circ Res. 2000;86:1218-23
23. Chan AY, Dyck JR. Activation of AMP-activated protein kinase (AMPK) inhibits protein synthesis: a potential strategy to prevent the development of cardiac hypertrophy. Can J Physiol Pharmacol. 2005;83:24-8
24. Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771-9
25. Meng R, Pei Z, Zhang A. et al. AMPK activation enhances PPARalpha activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway. Arch Biochem Biophys. 2011;511:1-7
26. Kong SS, Liu JJ, Yu XJ, Lu Y, Zang WJ. Protection against ischemia-induced oxidative stress conferred by vagal stimulation in the rat heart: involvement of the AMPK-PKC pathway. Int J Mol Sci. 2012;13:14311-25
27. King TD, Song L, Jope RS. AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem Pharmacol. 2006;71:1637-47
28. Weisova P, Anilkumar U, Ryan C, Concannon CG, Prehn JH, Ward MW. 'Mild mitochondrial uncoupling' induced protection against neuronal excitotoxicity requires AMPK activity. Biochim Biophys Acta. 2012;1817:744-53
29. Maro B, Marty MC, Bornens M. In vivo and in vitro effects of the mitochondrial uncoupler FCCP on microtubules. EMBO J. 1982;1:1347-52
30. Park KS, Jo I, Pak K. et al. FCCP depolarizes plasma membrane potential by activating proton and Na+ currents in bovine aortic endothelial cells. Pflugers Arch. 2002;443:344-52
31. Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776-81
32. Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220-3
33. Kwak HB. Effects of aging and exercise training on apoptosis in the heart. J Exerc Rehabil. 2013;9:212-9
34. Zhang KR, Liu HT, Zhang HF. et al. Long-term aerobic exercise protects the heart against ischemia/reperfusion injury via PI3 kinase-dependent and Akt-mediated mechanism. Apoptosis. 2007;12:1579-88
35. Melling CW, Thorp DB, Milne KJ, Noble EG. Myocardial Hsp70 phosphorylation and PKC-mediated cardioprotection following exercise. Cell Stress Chaperones. 2009;14:141-50
36. Heyndrickx GR, Vilaine JP, Moerman EJ, Leusen I. Role of prejunctional alpha 2-adrenergic receptors in the regulation of myocardial performance during exercise in conscious dogs. Circ Res. 1984;54:683-93
37. Campos JC, Queliconi BB, Bozi LHM. et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy. 2017;13:1304-17
38. Guan Y, Drake JC, Yan Z. Exercise-Induced Mitophagy in Skeletal Muscle and Heart. Exerc Sport Sci Rev. 2019;47:151-6
39. Kjobsted R, Hingst JR, Fentz J. et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018;32:1741-77
40. Laker RC, Drake JC, Wilson RJ. et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8:548
41. Grassi B, Majerczak J, Bardi E. et al. Exercise training in Tgalphaq*44 mice during the progression of chronic heart failure: cardiac vs. peripheral (soleus muscle) impairments to oxidative metabolism. J Appl Physiol (1985). 2017;123:326-36
42. Tamura Y, Simizu S, Osada H. The phosphorylation status and anti-apoptotic activity of Bcl-2 are regulated by ERK and protein phosphatase 2A on the mitochondria. FEBS Lett. 2004;569:249-55
43. Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol. 1999;19:5308-15
44. Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427-35
45. Bhamra GS, Hausenloy DJ, Davidson SM. et al. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol. 2008;103:274-84
46. Wang Y, Hirai K, Ashraf M. Activation of mitochondrial ATP-sensitive K(+) channel for cardiac protection against ischemic injury is dependent on protein kinase C activity. Circ Res. 1999;85:731-41
Author contact
![]() Corresponding authors: Weiwei Li, Department of Anesthesiology and SICU, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Kongjiang Road 1665, Shanghai, 200092, China. E-mail: luckyliweiweicom; Jianer Du, Department of Anesthesiology and SICU, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Kongjiang Road 1665, Shanghai, 200092, China. E-mail: dujianercom.cn.
Corresponding authors: Weiwei Li, Department of Anesthesiology and SICU, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Kongjiang Road 1665, Shanghai, 200092, China. E-mail: luckyliweiweicom; Jianer Du, Department of Anesthesiology and SICU, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Kongjiang Road 1665, Shanghai, 200092, China. E-mail: dujianercom.cn.