Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(5):647-656. doi:10.7150/ijms.41075 This issue Cite
Research Paper
Connective tissue growth factor (CTGF) regulates the fusion of osteoclast precursors by inhibiting Bcl6 in periodontitis
YunJeong Choi1*, Ji Hyun Yoo1*, Jae-Hyung Lee2, Youngkyun Lee3, Moon-Kyoung Bae1, Yong-Deok Kim4 ![]() , Hyung Joon Kim1
, Hyung Joon Kim1 ![]()
1. Department of Oral Physiology, BK21 PLUS Project, Periodontal Diseases Signaling Network Research Center, and Dental and Life Science Institute, School of Dentistry, Pusan National University, Yangsan, Republic of Korea, 50611
2. Department of Maxillofacial Biomedical Engineering, School of Dentistry, Department of Life and Nanopharmaceutical Sciences, Kyung Hee Medical Science Institute, Kyung Hee University, Seoul, Republic of Korea, 02447
3. Department of Biochemistry, School of Dentistry, Kyungpook National University, Daegu, Republic of Korea, 41940
4. Department of Oral and Maxillofacial Surgery, Dental Research Institute, and Dental and Life Science Institute, School of Dentistry, Pusan National University, Yangsan, Republic of Korea, 50611
* These authors contributed equally to this work.
Received 2019-10-11; Accepted 2020-2-11; Published 2020-2-24
Abstract
Connective tissue growth factor (CTGF), an extracellular matrix protein with various biological functions, is known to be upregulated in multiple chronic diseases such as liver fibrosis and congestive heart failure, but the mechanism it undertakes to cause alveolar bone loss in periodontitis remains elusive. The present study therefore investigates the pathways involving CTGF in chronic periodontitis. RNA sequencing revealed a notable increase in the expression of CTGF in chronic periodontitis tissues. Also, TRAP staining, TRAP activity and bone resorption assays showed that osteoclast formation and function is significantly facilitated in CTGF-treated bone marrow-derived macrophages (BMMs). Interestingly, western blotting and immunofluorescence staining results displayed that CTGF had little effect on the osteoclastogenic differentiation mediated by the positive regulators of osteoclastogenesis such as nuclear factor of activated T cells 1 (NFATc1). However, following results showed that both the mRNA and protein expressions of B cell lymphoma 6 (Bcl6), a transcriptional repressor of “osteoclastic” genes, were significantly downregulated by CTGF treatment. Moreover, CTGF upregulated the expressions of v-ATPase V0 subunit d2 (ATP6v0d2) and Dendritic cell-specific transmembrane protein (DC-STAMP) which are osteoclastic genes specifically required for osteoclast cell-cell fusion in pre-osteoclasts. Findings from this study suggest that CTGF promotes the fusion of pre-osteoclasts by downregulating Bcl6 and subsequently increasing the expression of DC-STAMP in periodontitis. Understanding this novel mechanism that leads to increased osteoclastogenesis in periodontitis may be employed for the development of new therapeutic targets for preventing periodontitis-associated alveolar bone resorption.
Keywords: CTGF, DC-STAMP, Bcl6, osteoclast fusion, periodontitis
Introduction
Periodontitis is a chronic inflammatory disease initiated by the colonization of complex subgingival bacterial plaque biofilms [1], and there have been a wide range of host and microbial factors reported to contribute to alveolar bone loss, the hallmark of the disease. Although the complex mechanisms regulating bone resorption in periodontitis is not fully understood, previous studies have postulated that the prolonged and persistent osteoclastic activation in the periodontium is one of the main factors responsible for the alveolar bone loss in periodontitis [2, 3].
Bone resorption is a basic physiologic process mediated by osteoclasts that differentiate from monocyte/macrophage precursors under the regulation of critical cytokines such as macrophage colony stimulating factor (M-CSF), receptor activator of nuclear factor kappa-Β ligand (RANKL), and osteoprotegerin [4]. As osteoclasts are the principal bone resorptive cells, local stimulation of their activity is an essential requirement for alveolar bone loss [5]. In the context of periodontitis, it is now generally accepted that once the biologically active substances within bacterial plaque induce a local inflammatory response in the gingival soft tissues and periodontium [6], T- and B-cell-mediated host immune responses against bacterial components elicit the aberrant activation of osteoclasts based on the production of RANKL by activated lymphocytes [7].
Osteoclasts are multinucleated cells that arise as a result of cell fusion [8]. Studies have shown that fusion is required for the maturation of osteoclasts and for gaining functional ability to resorb bone [9, 10]. The essential cell-cell fusion of mononuclear macrophages to multinuclear osteoclasts is regulated by dendritic cell-specific transmembrane protein (DC-STAMP), a seven-transmembrane protein [10]. Connective tissue growth factor (CTGF) is the second member of the CCN family of proteins (CCN2), and CCN family proteins are involved in a number of biological processes in development, tissue repair, and tumor suppression. In the skeletal system, mounting evidence have indicated that CTGF promotes the proliferation and maturation of chondrocytes and osteoblasts and induces excess osteoclastic functions [11]. Results from a study using MDA231, a human breast cancer cell line, revealed that the osteolysis metastasis was decreased by treating CTGF-neutralizing antibody [12]. CTGF was highly expressed in MDA231 cells, and the bone destruction induced by MDA231 cells may be due to the increased activity of the osteoclasts activated by CTGF [11]. Moreover, another study reported that CTGF induced by tumor necrosis factor α (TNF- α) upregulates osteoclastogenesis in patients with rheumatoid arthritis [13].
In the present study, we hypothesized that CTGF may be enhancing osteoclastogenesis in periodontitis, consequently leading to the alveolar bone loss. Indeed, CTGF is evidenced to potentiate RANKL- induced osteoclastogenesis in its late stage in osteoclast precursor cell line RAW 264.7 cells [14] and in its early stage by binding to RANK in MC3T3‐E1, a mouse osteoblastic cell line [15]. Here, we showed that CTGF causes Bcl6 downregulation, an effect that leads to increased DC-STAMP expression. B cell lymphoma 6 (Bcl6) was originally identified as a proto-oncogene because its chromosomal translocation and constitutive expression promotes lymphomagenesis [16]. Previously, Miyauchi et al. have demonstrated that Bcl6 inhibits osteoclast differentiation by attenuation transcription of osteoclastic genes in vitro [17], but its mechanism involving CTGF that leads to enhanced osteoclastogenesis in periodontitis was unexplored. Therefore, we examined the effects of CTGF on the regulators of osteoclastogenesis.
Materials and methods
Patient recruitment and RNA sequencing of gingival tissue samples
Gingival tissue samples were collected from healthy patients or chronic periodontitis patients at Kyungpook National University Dental Hospital. Periodontitis-affected site had a probing depth of ≥4 mm, clinical attachment level of ≥4 mm, and displayed bleeding upon probing. Ten samples were obtained from 9 healthy patients and 10 from 4 chronic periodontitis patients. All patients were non-smoking, not associated with infection or autoimmune diseases at the time of sample collection, and did not have untreated metabolic or systemic diseases. The gingival biopsies were approximately 3 mm2 in size and collected from gingival margins. They were washed immediately with phosphate-buffered saline (PBS) and stored in RNAlater solution (Thermo Fisher Scientific, Waltham, MA, USA) at -70°C.
Total RNAs were extracted using mirVana RNA isolation kits (Thermo Fisher Scientific, Waltham, MA, USA) after lysing frozen tissues using a disposable pestle grinder system (Thermo Fisher Scientific, Waltham, MA, USA). mRNAs were purified using poly-T oligo-attached magnetic beads and RNAs of ~300 bp were isolated by gel electrophoresis. cDNA libraries were synthesized using Truseq RNA sample preparation kits (Illumina, San Diego, CA, USA) and amplified cDNAs were loaded and sequenced in paired-end (PE) sequencing mode using the HiSeq 2000 sequencing system (Illumina). To determine the significance of differences between groups, the DESeq package was used as previously described [18].
Mice
All animal procedures were approved by the Pusan National University Institutional Animal Care and Use Committee (PNU-IACUC) and carried out according to the guidelines issued by the animal care committee of the Institute of Laboratory Animal Resources of Pusan National University (PNU-2019- 2200).
Reagents
M-CSF and RANKL were purchased from PeproTech (Rocky Hill, NJ, USA). Primary antibodies against p-AKT, AKT, p-ERK, ERK, p-JNK, JNK, p-p38, and p38 were purchased from Cell Signaling (Beverly, MA, USA). HRP-conjugated secondary antibodies were acquired from (GenDEPOT, Barker, TX, USA). Anti-lamin B and anti-NFATc1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). DAPI and Cy3-conjugated secondary antibodies were acquired from Sigma-Aldrich (St. Louis, MO, USA). Keyhole-Limpet-Hemocyanin (KLH)-conjugated anti-DC-STAMP antibody and IgGκ light chain binding protein (m-IgGκ BP-PE) were from Santa Cruz Biotechnology (Millipore, Temecula, CA, USA).
Osteoclast generation
Bone marrow was extracted from the femora and tibiae of 5-week-old female ICR mice and flushed with α-minimum essential medium (α-MEM; Welgene Inc., Deagu, Republic of Korea) using a syringe. Bone marrow cells were collected by centrifugation and incubated with red blood cell lysis buffer for 10 seconds at room temperature. After purification, cells were seeded in 48-well plates at a density of 4 × 104 cells/well and cultured in α-MEM containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin with M-CSF (30 ng/ml) for 2 days. After 2 days, cells were treated with RANKL (100 ng/ml) and different concentrations of CTGF. The adherent cells were used as osteoclast precursors (bone marrow-derived macrophages, BMMs). BMMs were cultured for 4 days after RANKL treatment and the media were changed every 2 days.
Tartrate-resistant acid phosphatase (TRAP) staining and activity assay
Osteoclastic differentiation of BMMs was evaluated by TRAP staining using the Leukocyte Acid Phosphatase Kit (Sigma-Aldrich, St. Louis, MO, USA) and TRAP activity assay (TRAP Assay Kit; Takara, Shiga, Japan). Cultured cells were fixed in 3.7% paraformaldehyde for 10 minutes, treated with 0.1%Triton X-100 in PBS at room temperature for 5 minutes, and rinsed three times with deionized water. Finally, cells were incubated with 0.01% naphthol AS-MX phosphate and 0.05% fast red violet LB salt in 50mM of sodium tartrate and 90mM of sodium acetate (pH 5.0) for 1 hour at 37 ℃ and rinsed 3 times with deionized water. TRAP activity was measured by using the cell culture supernatant generated after staining.
In vitro bone resorption assay
BMMs were seeded on sterilized dentin slices (Immunodiagnostic Systems Inc., Boldon, UK) placed in 48-well plates as previously described [19]. Cells were cultured under three different conditions: the negative control was cultured with α-MEM containing 10 % FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin and 30 ng/ml of M-CSF, the positive control was treated with 100 ng/ml RANKL in addition to the negative control, and the CTGF group was treated with 100 ng/ml of CTGF in addition to the positive control. After 7 days, each well was washed with deionized water. To measure the depth and area of resorption pits, each disc was observed and analyzed using Zeiss LSM 5 PASCAL laser-scanning microscope (Carl Zeiss Inc., Thornwood, NY, USA).
Western blotting
For Western blot analysis, BMMs were grown in 6-well plates at a density of 3 × 105 cells/well and treated with 100 ng/ml RANKL and different concentrations of CTGF. Cells were harvested after lysis in cold RIPA buffer (40 mM Tris-Cl, 10 mM EDTA, 120 mM NaCl, and 0.1 % NP-40) containing protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA), homogenized by sonication, and centrifuged at 4 °C for 10 minutes at 14,000 rpm. The supernatant was collected and the protein concentrations of the samples were determined using the Smith assay. The membranes were denatured using 5× SDS-PAGE sample buffer (TransLab, Daejeon, Republic of Korea) and 50 μg/lane were loaded onto 10% SDS-PAGE gels. Resolved proteins were transferred to nitrocellulose membranes and non-specific sites were blocked by 5% skim milk in TBS-T for 1 hour. Nitrocellulose membranes were immunoblotted overnight at 4 °C with primary antibodies diluted 1:1000 in Tris buffer saline with Tween-20 (TBST; Sigma-Aldrich, St. Louis, MO, USA). After washing 4 times with TBST for 10 minutes, blots were incubated for 1 hour at room temperature with HRP-conjugated secondary antibodies diluted 1:5000. Proteins were visualized by means of an enhanced chemiluminescent detection reagents (SuperSignal West Pico PLUS Chemiluminescent Substrate; Thermo Fisher, Waltham, MA, USA).
Immunofluorescence staining
BMMs were seeded in 48-well plates at a density of 4 × 104 cells/well, plated on sterile glass coverslips, treated with RANL and different concentrations of CTGF for 2 days, fixed for 10 minutes with 3.7 % formaldehyde, permeabilized in 0.1 % Triton X-100, and blocked in 1% BSA in PBS. Cells were then incubated for 1 hour at room temperature with antibodies against lamin B and NFATc1 (1:1000) in 1% BSA solution. After washing with TBS, cells were incubated for 1 hour at room temperature with DAPI or Cy3-conjugated secondary antibodies, washed three times with TBS, and mounted on glass slides. Images were acquired by confocal microscopy (FV300; Olympus, Tokyo, Japan).
RT-PCR
Total RNA was harvested from cultured BMMs using TRIsure™ (Bioline, London, UK) and cDNAs were synthesized from 3 μg of isolated RNAs using Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). Amplification was performed with StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies, Carlsbad, CA, USA).
Real-time PCR
Real-time PCR was performed using SYBR Green Master Mix reagents (Kapa Biosystems, Woburn, MA, USA) and ABI 7500 unit (Applied Biosystems, Carlsbad, CA, USA). HPRT mRNA expression was used as an endogenous control. The sequences of the primers used are as follows: HPRT forward: 5′-CCTAAGATGATCGCAAGTTG-3′; HPRT reverse: 5′-CCACAGGGACTAGAACACCTGCTAA -3′; V-ATPaseV0d2 forward: 5′AGTGCAGTGTGAGACCTTGG-3′; V-ATPaseV0d2 reverse: 5′-CAAAGCAACAGACTCCCAAA-3′; DC-STAMP forward: 5′-GACCTTGGGCACCAGTATTT-3′; DC-STAMP reverse: 5′-CAAAGCAACAGACTCCCAAA-3′; Bcl6 forward: 5′-ATGAGATTGCCCTGCATTTC-3′; Bcl6 reverse: 5′-TTCTTCCAGTTGCAGGCTTT-3′. All reactions were run in triplicate.
Surface antigen expression
BMMs were harvested for flow cytometry analysis of surface DC-STAMP expression. Cells were washed twice with ice cold PBS, stained with KLH-conjugated anti-DC-STAMP antibody for 30 minutes in the dark on ice, washed 3 times, and stained with m-IgGκ BP-PE for 30 min in the dark on ice. Cells were analyzed by BD FACS Cell Analyzer (BD Bioscience, Heidelberg, Germany) equipped with FACSuite software, v1.0.5.3841 (BD Bioscience).
Statistical analysis
The results are presented as means ± standard deviations (SDs). Statistical significance was determined using the two-tailed Student's t-test and p values of < 0.05 or 0.01 -denoted as * and **, respectively -were regarded significant. All experiments were repeated three times.
Results
CTGF gene expression is upregulated in human periodontitis tissues
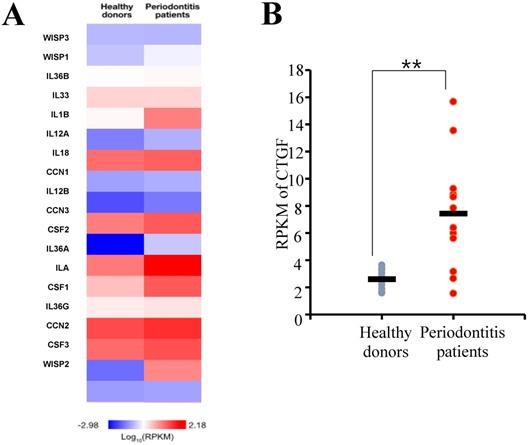
The differential gene expression between the periodontitis-affected and healthy gingiva was evaluated by DESeq package as previously described [20]. From the RNA sequencing assay, genes that showed differences in expressions greater than 2-fold were selected and a false discovery rate (FDR) of < 0.05 were applied. First, the expression levels of gene associated with inflammation responses were evaluated, and the result showed that a number of inflammatory cytokine genes were highly expressed in gingiva samples of periodontitis patient (Fig. 1A). Among the genes, CTGF was found to be upregulated significantly in the tissue samples of periodontitis patients (Fig. 1B).
RNA sequencing analysis of periodontal tissues. (A) A Heat map of genes associated with inflammatory responses. The median gene expression of each gene from the samples was normalized by log10RPKM. RPKM, Reads per kilobase of exon per million mapped reads. (B) Connective tissue growth factor (CTGF) was found to be differentially expressed between the healthy and periodontitis gingival tissues. RPKM, Reads per kilobase of exon per million mapped reads. **p < 0.01.
CTGF enhances osteoclast formation
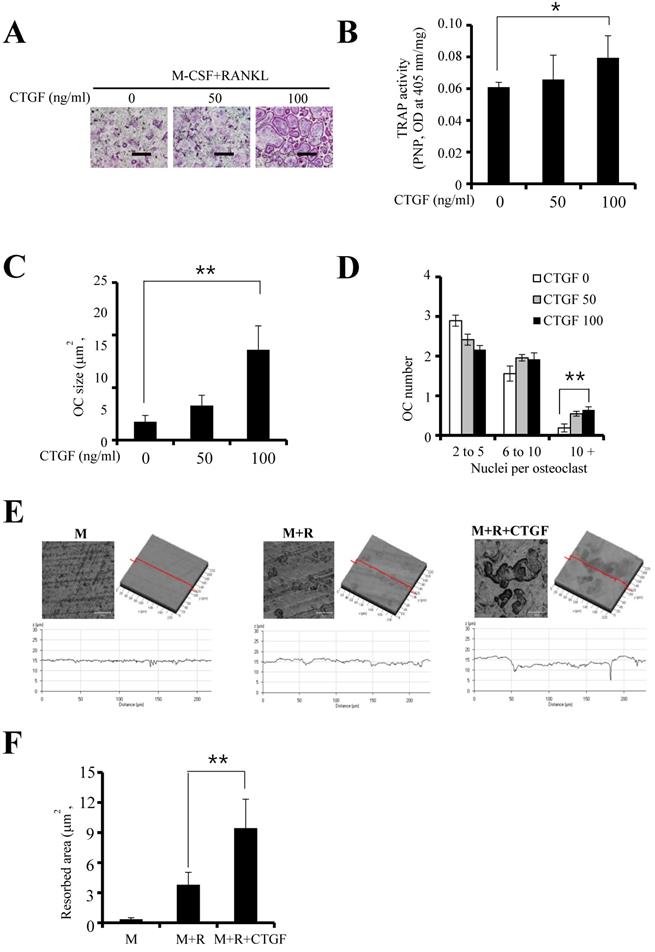
To investigate the effect of CTGF on RANKL- stimulated osteoclastogenesis, TRAP staining and TRAP activity assay were performed. Results showed that CTGF enhanced osteoclastogenesis as well as the expression of its cytochemical marker in a concentration-dependent manner (Fig. 2A and B). Moreover, increasing CTGF concentrations resulted in increased size and number of osteoclasts containing more than 10 nuclei (Fig. 2C and D). When the BMMs were treated with 100 ng/ml CTGF, the depth (Fig. 2E) and area (Fig. 2F) of the dentin samples resorbed by the differentiated osteoclasts increased significantly. These results indicate that CTGF enhances osteoclastogenesis and osteoclast function induced by M-CSF and RANKL.
CTGF enhances osteoclast formation and function. (A) Bone marrow macrophages (BMMs) were treated with different concentrations of CTGF in the presence of 30 ng/ml M-CSF and 100 ng/ml RANKL. Cultured BMMs were stained with tartrate-resistant acid phosphatase (TRAP) and examined under a light microscope. Scale bar, 200 μm. (B) Relative TRAP intensities were measured after 4 days of culture. (C, D) The size and number of OCs were also measured by TRAP staining. (E) Resorption pits on dentin discs were visualized using a confocal laser scanning microscope. Dark areas indicate the resorbed surfaces. Scale bar, 200 μm. (F) Relative resorption areas were measured in four randomly selected images from each bone resorption assay (M: M-CSF, R: RANKL). All experiments were repeated three times. *p < 0.05, ** p < 0.05.
CTGF does not affect the expression of pro-osteoclastogenic factors
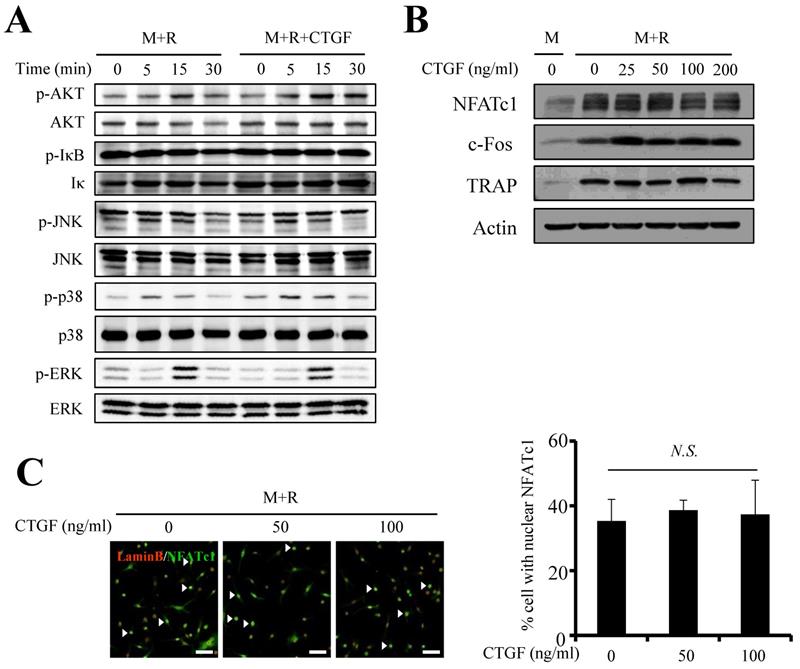
Next, the impact of CTGF treatment on RANKL-induced osteoclast differentiation was assessed by examining expression levels of key modulators in mitogen-activated kinase (MAPK) and Nuclear factor-κB (NF-κB) pathways. The protein expressions of p-IκB, p-JNK, p-ERK, well-known differentiation factors of osteoclastogenesis, were unchanged whether CTGF was treated or not (Fig. 3A). However, the phosphorylations of AKT and p38 showed slight increases upon 100 ng/ml of CTGF treatment. Interestingly, CTGF treatment did not affect the expressions of c-fos, an essential pro-osteoclastogenesis factor expressed during the early stages of osteoclastogenesis, and nuclear factor of activated T cells 1 (NFATc1), the master regulator of osteoclastogenesis (Fig. 3B). Expression levels of TRAP, an important differentiation marker of osteoclastogenesis, was uninfluenced as well. In support of the Western blotting results, immunofluorescence results showed that CTGF had no effect on the nuclear translocation of NFATc1 (Fig. 3C). These data reveal that CTGF does not directly induce osteoclastogenesis by upregulating the positive regulators of RANKL-induced osteoclast differentiation.
CTGF has little effect on the pro-osteoclastogenic signaling pathways. (A) MAPK and NF-κB pathways were examined by western blotting using the indicated antibodies. (B) Protein levels of pro-osteoclastogenic-transcription factors (NFATc1 and c-Fos) and osteoclastogenic differentiation marker (TRAP) were examined. (C) Nuclear translocation of NFATc1 was evaluated by immunofluorescence staining with anti-NFATc1 (FITC-labeled, green) and anti-lamin B (Cy3-labeled, red) antibodies. Numbers of nuclei stained for NFATc1 (arrows) were counted (M: M-CSF, R: RANKL). All experiments were repeated three times. Scale bar, 50 μm. N.S., not significant.
CTGF indirectly induces osteoclastogenesis by suppressing anti-osteoclastogenic factor expression
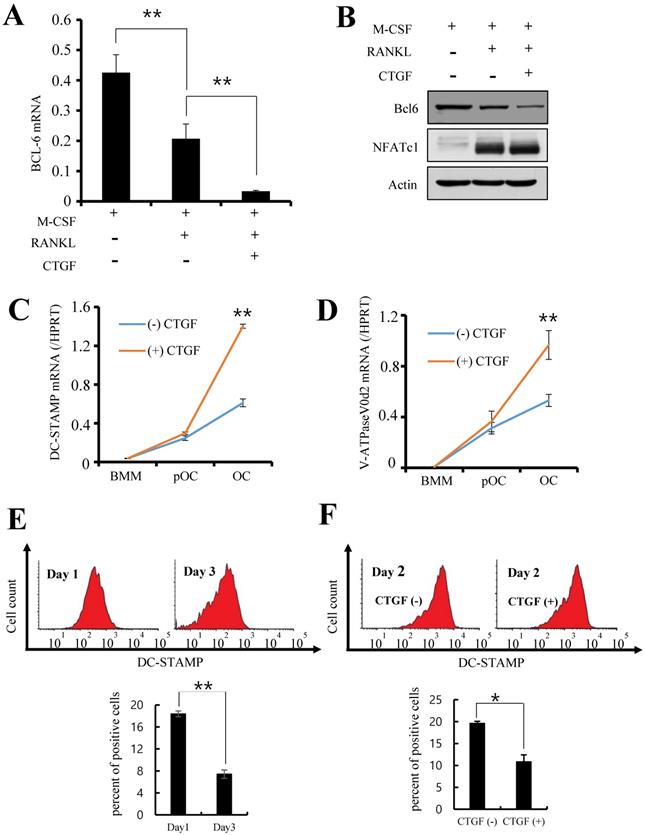
Since CTGF did not have an effect on key inducers of osteoclastogenesis, suppressors of the process were investigated. BMMs were cultured with RANKL and 100 ng/ml of CTGF and the expression levels of B cell lymphoma 6 (Bcl6), a transcriptional repressor of osteoclast differentiation, were observed. As shown in Fig. 4A, the mRNA expression of Bcl6 was significantly suppressed by CTGF treatment. Moreover, its protein expression was markedly downregulated in BMMs treated with CTGF compared to that in the control (Fig. 4B). Thus, CTGF treatment has no significant effect on NFATc1 expression but downregulates Bcl6 in order to facilitate osteoclastogenesis under RANKL stimulation.
CTGF facilitates the multinucleation and maturation of osteoclasts by inhibiting Bcl6 expression and inducing the expressions of DC-STAMP and ATP6v0d2. (A) The mRNA expression of Bcl-6 in osteoclasts was evaluated by real-time PCR, and (B) the protein expressions of Bcl-6 and NFATc1 in osteoclasts treated with 100 ng/ml CTGF were analyzed by western blotting. ** p < 0.01 (C, D) mRNA expressions of DC-STAMP and ATP6v0d2 in BMMs treated with or without 100 ng/ml CTGF determined by real-time PCR. BMMs were cultured for different days: day 0 (BMM), day 2 (pOC), and day 3 (OC). BMM, bone marrow macrophage; pOC; pre-osteoclast; OC; osteoclast. (E) Expressions of DC-STAMP on the surface of BMMs treated with or without 100 ng/ml CTGF were examined on day 1 and 3 by flow cytometry. (F) The ratio of pre-osteoclasts with a fluorescence intensity exceeding the threshold value (103) was calculated. All experiments were repeated three times. ** p < 0.01.
CTGF accelerates cell-to-cell fusion of osteoclast precursors
Next, the downstream factors of the Bcl6 pathway were examined to identify the mechanism underlying the accelerated osteoclastogenesis by CTGF. BMMs were treated with RANKL and 100 ng/ml of CTGF for 0, 2, or 3 days and the mRNA levels of DC-STAMP and v-ATPase V0 subunit d2 (ATP6v0d2) were measured. The promoter region of DC-STAMP is a direct binding target of Bcl6 [21], and both DC-STAMP and ATP6v0d2 are known to regulate the cell-cell fusion of osteoclast precursors [22, 23]. The results show that the expression levels of the two genes were significantly increased in the CTGF-treated pre-osteoclasts compared to the control (Fig. 4C and D).
An earlier study has reported that DC-STAMP migrates from the extracellular surface to the cytoplasm during the fusion and maturation of osteoclasts and therefore its surface expression decreases gradually as mature, multinucleated osteoclasts form during osteoclastogenesis [24]. To show that CTGF is involved in this migration process, the surface expression of DC-STAMP during osteoclastogenesis was observed by flow cytometry. In support of previous data by Chiu et al. [24], DC-STAMP surface expression on culture day 3 was significantly lower than that on day 1 (Fig. 4E). In addition, BMMs treated with CTGF showed markedly lower surface expressions of DC-STAMP compared to the control on day 2 when BMMs are at the pre-osteoclast stage and DC-STAMP expression is supposedly at its peak (Fig. 4F). These results suggest that CTGF may be enhancing osteoclast fusion and maturation by accelerating the DC-STAMP-mediated fusion of pre-osteoclasts (Fig. 5).
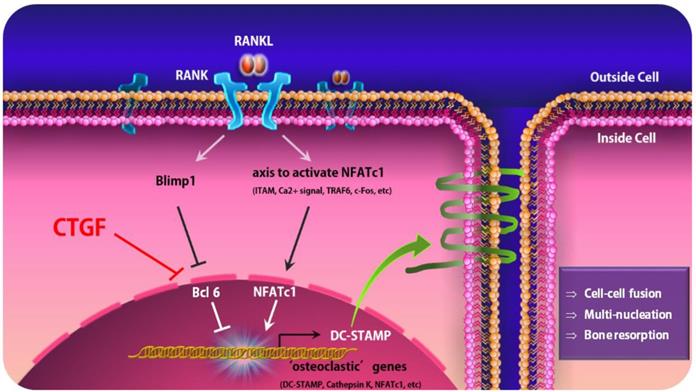
A schematic model of osteoclastogenesis regulated by CTGF that leads to the activation of DC-STAMP. Under normal circumstances, RANKL-RANK interaction leads the activation of NFATc1, a crucial positive regulator for osteoclastogenesis, and Blimp1-Bcl-6, the negative regulators, to regulate osteoclastogenesis and bone homeostasis. When CTGF is upregulated, it suppresses the expression of Bcl6 and induces the dissociation of Bcl-6 from osteoclastic gene promoters, leading to the overexpression of DC-STAMP.
Discussion
CTGF is a cysteine-rich secretory protein containing four conservative modules which are IGFBP module, VWC type C module, TSP type 1 repeat, and C-terminal module [25]. Early studies have focused on its involvement in fibrosis because CTGF had been shown to promote the proliferation, migration, and adhesion of fibroblasts by inducing TGF-beta [26-31]. However, CTGF has recently attracted interest due to its role in oral diseases. For example, increased expressions of CTGF have been documented in lesions caused by phenytoin, an anti-seizure medication, nifedipine, an anti-hypertensive drug, and ciclosporin, an immune suppressor [32]. These drugs from medical therapy ultimately cause tissue-specific gingival overgrowth which presents major problems for maintaining oral hygiene due to increased risk for infection and inflammatory complications, difficulty with mastication, and a disfigured appearance accompanied by the swelling of gingiva. Interestingly, CTGF upregulation in the fibroblasts of gingival tissues have altered signal transduction pathways that confer unexpected resistance to effects of some inflammatory mediators, which are considered to contribute to the tissue specificity and fibrosis [32].
Unfortunately, little information is available on the precise role of CTGF in regard to periodontitis and bone destruction. Early diagnostic study with 21 individuals with or without periodontitis found for the first time the expressions of CTGF along with TGFβ1 mRNA are increased and correlated in patients with periodontitis [33]. Authors from this study suggested from this result that both tissue destruction and tissue regeneration must co-exist in periodontitis. It is compelling to note that although CTGF is involved, the tissue-regenerative process is dominant in drug-induced gingival overgrowth while gingival tissue inflammation and destruction are predominant in periodontitis. In addition, our colleagues recently suggested that CTGF is significantly overexpressed in periodontitis tissues via RNA sequencing analysis [20]. In the present study, our intention was to expand our understanding of the mechanism behind the upregulation of CTGF expression in periodontitis which leads to alveolar bone loss in the end.
Controlling osteoclastogenesis is critical for maintaining physiological bone homeostasis and preventing skeletal disorders, and there are several well-known regulators of osteoclastogenesis. When RANKL binds to its receptor RANK on osteoclast precursors, a broad range of signaling cascades including the canonical and non-canonical NF-κB pathways and MAPK pathways are facilitated, leading to the activation of activator protein-1 (AP-1; c-Fos and c-Jun) and cyclic-AMP response element binding (CREB) transcription factors. In addition, calcium signaling is induced, followed by the expression of key transcription factors such as Blimp1 and NFATc1 for osteoclast differentiation [34-37].
NFATc1 is the master regulator of osteoclastogenesis under RANKL stimulation, responsible for the regulation of genes related to osteoclast function as well as numerous genes non-essential to osteoclast function [38, 39]. Positive regulators such as NFATc1 induce the expression of “osteoclastic” molecules essential for the differentiation and function of osteoclasts. Few of the key osteoclastic genes are: DC-STAMP, which is known to facilitate cell-cell fusion, cathepsin K (Ctsk) which promotes bone matrix proteolysis [40, 41], and NFATc1 which drives differentiation. On the other hand, the Blimp1-Bcl6 axis is a crucial negative regulator of osteoclast differentiation [10]. B lymphocyte-induced maturation protein 1 (Blimp1) is a transcriptional repressor that inhibits the expression of Bcl6, likely by binding directly to the Bcl6 promoter, and Bcl6 is also a transcriptional repressor that antagonizes NFATc1 function [21]. The expression of Bcl6 and Blimp1 is reciprocal in normal osteoclast formation, and the suppression of Bcl6 during osteoclast differentiation is required for the appropriate osteoclastogenesis and regulation of bone homeostasis [21].
In the present study, we discovered that CTGF treatment on BMMs did not affect the protein expressions of key inducers of osteoclastogenesis. Akt and ERK are signaling pathways activated by the binding of M-CSF to its receptor c-Fms, and p-JNK, p-p38, and p-IκB are downstream signaling pathways activated by c-Fos and NFATc1 expression in osteoclast precursors [42]. However, our immunoblotting results demonstrated a slight increase in the expressions of p -AKT and p-ERK, and we predict that CTGF exerts a partial effect on osteoclast differentiation via a mechanism yet to be discovered. Next, we asked if the upstream signaling pathway that regulates NFATc1 expression was affected, and there was no change in the levels of c-Fos in cell cultures with CTGF. Finally, we shed light on the Bcl6-osteoclastic gene axis which is recognized as a force of negative regulation of osteoclast differentiation to study the pathway CTGF undertakes in periodontitis. Balanced osteoclast differentiation is precisely controlled and maintained by complex mechanisms at various levels, and accumulating evidence propose that RANK needs to overcome the transcriptional repressors expressed constitutively in osteoclast precursors in addition to activating positive signaling pathways for osteoclastogenesis [21]. Therefore, it is becoming clearer that negative regulation of osteoclastogenesis and bone resorption plays a key role in bone homeostasis fine tuning bone remodeling and restraining excessive bone resorption in inflammatory settings. Our results revealed that CTGF significantly downregulated the mRNA and protein expressions of Bcl6 in osteoclasts. Taken together, our findings propose a model whereby the overexpression of CTGF in chronic periodontitis tissues induces the suppression of Bcl6, which results in hyper-osteoclast differentiation and subsequent loss of bone mass.
Conclusions
In summary, it has been established that CTGF promotes osteoclastogenesis by inducing DC-STAMP expression in the late stage of osteoclastogenesis [11], but the processes leading to such events were elusive. We report for the first time that CTGF treatment does not affect the expression of NFATc1 but dramatically decreases Bcl-6 expression at the mRNA and protein levels, which in turn leads to the upregulation of DC-STAMP. Understanding these mechanisms provides new molecular targets since inhibition of NFATc1 has failed to increase bone mass but rather reduced bone mass [10]. CTGF as well as the Blimp1-Bcl6 axis may become new therapeutics targets for preventing periodontitis-associated bone resorption.
Acknowledgements
This study was supported by Dental Research Institute (PNUDH DRI-2017-02), Pusan National University Dental Hospital.
Competing Interests
The authors have declared that no competing interest exists.
References
1. D'Aiuto F, Nibali L, Parkar M, Patel K, Suvan J, Donos N. Oxidative stress, systemic inflammation, and severe periodontitis. J Dent Res. 2010;89:1241-1246
2. Kayal RA. The role of osteoimmunology in periodontal disease. Biomed Res Int. 2013;2013:639368
3. Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014;35:3-11
4. Hienz SA, Paliwal S, Ivanovski S. Mechanisms of bone resorption in periodontitis. J Immunol Res. 2015;2015:615486
5. Saffar JL, Lasfargues JJ, Cherruau M. Alveolar bone and the alveolar process: the socket that is never stable. Periodontology 2000. 1997;14:76-90
6. Nanci A, Bosshardt DD. Structure of periodontal tissues in health and disease. Periodontology 2000. 2006;40:11-28
7. Kajiya M, Giro G, Taubman MA, Han X, Mayer MP, Kawai T. Role of periodontal pathogenic bacteria in RANKL-mediated bone destruction in periodontal disease. J Oral Microbiol. 2010;8:2
8. Wisitrasameewong W, Kajiya M, Movila A. et al. DC-STAMP is an osteoclast fusogen engaged in periodontal bone resorption. J Dent Res. 2017;96:685-693
9. Vignery A. Osteoclasts and giant cells: macrophage-macrophage fusion mechanism. Int J Exp Pathol. 2000;81:291-304
10. Miyamoto T. Regulators of osteoclast differentiation and cell-cell fusion. Keio J Med. 2011;60(4):101-105
11. Nishida T, Emura K, Kubota S, Lyons KM, Takigawa M. CCN family2/ connective tissue growth factor (CCN2/CTGF) promotes osteoclastogenesis via induction of and interaction with dendritic cell-specific transmembrane protein (DC-STAMP). J Bone Miner Res. 2011;26:351-363
12. Shimo T, Kubota S, Yoshioka N. et al. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J Bone Miner Res. 2006;21:1045-1059
13. Nozawa K, Fujishiro M, Kawasaki M. et al. Connective tissue growth factor promotes articular damage by increased osteoclastogenesis in patients with rheumatoid arthritis. Arthritis Res Ther. 2009;11:R174
14. Shui C, Riggs BL, Khosla S. The immunosuppressant rapamycin, alone or with transforming growth factor-beta, enhances osteoclast differentiation of RAW 264.7 monocyte macrophage cells in the presence of RANK-ligand. Calcif Tissue Int. 2002;71:437-446
15. Aoyama E, Kubota S, Takigawa M. CCN2/CTGF binds to fibroblast growth factor receptor 2 and modulates its signaling. FEBS Lett. 2012;586:4270-4275
16. Ohno H. Pathogenetic and clinical implications of non-immunoglobulin; BCL6 translocations in B-cell non-Hodgkin's lymphoma. J Clin Exp Hematop. 2006;46:43-53
17. Miyauchi Y, Ninomiya K, Miyamoto H. et al. The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med. 2010;207:751-762
18. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106
19. Kwon JO, Lee YD, Kim H. et al. Tetraspanin 7 regulates sealing zone formation and the bone-resorbing activity of osteoclasts. Biochem Biophys Res Commun. 2016;477:1078-1084
20. Kim YG, Kim M, Kang JH. et al. Transcriptome sequencing of gingival biopsies from chronic periodontitis patients reveals novel gene expression and splicing patterns. Hum Genomics. 2016;10(28):1-10
21. Zhao B, Ivashkiv LB. Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Res Ther. 2011;13:234
22. Kukita T, Wada N, Kukita A. et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200:941-946
23. Lee SH, Rho J, Jung D. et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med. 2006;12:1403-1409
24. Chiu YH, Mensah KA, Schwarz EM. et al. Regulation of human osteoclast development by dendritic cell-specific transmembrane protein (DC-STAMP). J Bone Miner Res. 2012;27:79-92
25. Takigawa M. CCN2: a master regulator of the genesis of bone and cartilage. J Cell Commun Signal. 2013;7:191-201
26. Perbal B, Takigawa M. et al. CCN Proteins: A New Family of Cell Growth and Differentiation Regulators. London, UK: Imperial College Press. 2005:61-79
27. Takigawa M. CTGF/Hcs24 as a multifunctional growth factor for fibroblasts, chondrocytes, and vascular endothelial cells. Drug News Perspect. 2003;16:11-21
28. Mori T, Kawara S, Shinozaki M. et al. Role and interaction of connective tissue growth factor with transforming growth factor-β in persistent fibrosis: a mouse fibrosis model. J Cell Physiol. 1999;181:153-159
29. Sato S, Nagaoka T, Hasegawa M. et al. Serum levels of connective tissue growth factor are elevated in patients with systemic sclerosis: association with the extent of skin sclerosis and the severity of pulmonary fibrosis. J Rheumatol. 2000;27:149-154
30. Brigstock DR. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr Rev. 1999;20:189-206
31. Moussad EE, Brigstock DR. Connective tissue growth factor: what's in a name? Mol Genet Metab. 2000;71:276-292
32. Trackman PC, Kantarci A. Molecular and clinical aspects of drug-induced gingival overgrowth. J Dent Res. 2015;94:540-546
33. Mize TW, Sundararaj KP, Leite RS, Huang Y. Increased and correlated expression of connective tissue growth factor and transforming growth factor beta 1 in surgically removed periodontal tissues with chronic periodontitis. J Periodontal Res. 2015;50:315-319
34. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251-264
35. Humphrey MB, Lanier LL, Nakamura MC. Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev. 2005;208:50-65
36. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res. 2013;92:860-867
37. Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457-484
38. Aliprantis AO, Ueki Y, Sulyanto R. et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. The Journal of clinical investigation. 2008;118:3775-3783
39. Charles JF, Coury F, Sulyanto R. et al. The collection of NFATc1-dependent transcripts in the osteoclast includes numerous genes non-essential to physiologic bone resorption. Bone. 2012;51:902-912
40. Yagi M, Miyamoto T, Sawatani Y. et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345-351
41. Li CY, Jepsen KJ, Majeska RJ. et al. Mice lacking cathepsin K maintain bone remodeling but develop bone fragility despite high bone mass. J Bone Miner Res. 2006;21:865-875
42. Abbas S, Clohisy JC, Abu-Amer Y. Mitogen-activated protein (MAP) kinases mediate PMMA-induction of osteoclasts. J Orthop Res. 2003;21:1041-1048
Author contact
![]() Corresponding authors: Yong-Deok Kim, Address: Department of Oral Physiology, Pusan National University School of Dentistry, 49 Busandaehak-ro, Yangsan, Republic of Korea, 50611. E-mail: ydkimddsac.kr; Tel: +82-55-360-5100; Fax: +82-55-510-8208 or Hyung Joon Kim, Address: Department of Oral Physiology, Pusan National University School of Dentistry, 49 Busandaehak-ro, Yangsan, Republic of Korea, 50611. E-mail: hjoonkimac.kr; Tel: +82-55-510-8278; Fax: +82-55-510-8208
Corresponding authors: Yong-Deok Kim, Address: Department of Oral Physiology, Pusan National University School of Dentistry, 49 Busandaehak-ro, Yangsan, Republic of Korea, 50611. E-mail: ydkimddsac.kr; Tel: +82-55-360-5100; Fax: +82-55-510-8208 or Hyung Joon Kim, Address: Department of Oral Physiology, Pusan National University School of Dentistry, 49 Busandaehak-ro, Yangsan, Republic of Korea, 50611. E-mail: hjoonkimac.kr; Tel: +82-55-510-8278; Fax: +82-55-510-8208