Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(11):1412-1423. doi:10.7150/ijms.34398 This issue Cite
Research Paper
Synergistic apoptosis of human gastric cancer cells by bortezomib and TRAIL
Hang Thi Thuy Bui1,2†, Nhu Huynh Le1,2†, Qui Anh Le1,2, Sung Eun Kim3, Sooho Lee1, Dongchul Kang1,2 ![]()
1. Ilsong Institute of Life Science, Hallym University, Anyang, Kyonggi-do, 14066, Republic of Korea.
2. Department of Biomedical Gerontology, Hallym University Graduate School, Chuncheon, Kangwon-do, 24252, Republic of Korea.
3. Department of Internal Medicine, Hallym University Sacred Heart Hospital, College of Medicine, Hallym University, Anyang, Kyonggi-do, 14068, Republic of Korea.
† These authors contributed equally to this work.
Received 2019-2-25; Accepted 2019-8-8; Published 2019-9-20
Abstract
Resistance against tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced cell death of cancer cells is a major obstacle in clinical application of TRAIL. Variable response to TRAIL of gastric cancer cells, synergy of TRAIL with bortezomib and potential mechanisms behind the phenomena were investigated in this study. The response to TRAIL varied among six gastric cancer cell lines, which correlated with the expression of apoptotic TRAIL receptors. Analysis of TCGA gene expression data showed that DR4 expression correlated with DR5 in gastric cancer. Although higher expression of DR4 was significantly associated with lower T, N and TNM stages, neither DR4 nor DR5 expression meaningfully influenced overall survival rate. Combined treatment of TRAIL with bortezomib resulted in strong synergistic response with enhanced activation of caspases-8, -9 and -3, and increased Annexin V-binding cell fractions in TRAIL-resistant SNU-216 cells. Bortezomib increased the expression of p21cip1/waf1, but p21cip1/waf1 silencing did not restore cell viability significantly. Bortezomib also increased DR5 expression and knockdown of DR5 expression significantly recovered cell viability reduced by the combination treatment. Bortezomib decreased phosphorylation of ERK1/2, but increased that of JNK. Treatment with either an ERK1/2 inhibitor U0126 or a JNK inhibitor SP600125 rescued SNU-216 from dying of bortezomib or combined treatment. However, upregulation of DR5 by bortezomib was knocked down only by inhibition of ERK1/2 activation significantly, but not by JNK activity inhibition. In summary, upregulation of DR5 by bortezomib is of critical significance in the synergy of bortezomib with TRAIL in apoptosis of TRAIL-resistant SNU-216 and that activity of ERK1/2 is required in the bortezomib-induced DR5 overexpression.
Keywords: Gastric cancer, TRAIL, Bortezomib, DR4, DR5, ERK, p21cip1/waf1
Introduction
Gastric cancer is the third leading cause of cancer death in the world and half of the total cases occur in East Asia, particularly in China, Japan and Korea [1]. Almost one million new cases of gastric cancer were diagnosed each year (6.8% of the total), making it the fifth common malignancy in the world according to GLOBOCAN 2012 [2]. Mortality by gastric cancer has been decreased by advances in diagnosis, surgery and novel treatment regimens, but the prognosis of the patients with advanced gastric cancer still remains poor [3].
The defect in apoptosis is a causative factor of tumorigenesis, tumor metastasis and anticancer drug resistance [4]. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induces apoptotic cell death in various cancer cell types including breast, bladder, lung, liver and stomach cancers, whilst generally sparing non-malignant cells [5, 6]. The selective cytotoxicity of TRAIL against cancer cells has gained an intense interest in exploring the potential utility of TRAIL as an anticancer therapeutics [5]. However, resistance to TRAIL-induced apoptosis of various cancer cells has been reported in numerous cases [6, 7]. TRAIL-induced apoptosis can be inhibited by diverse mechanisms such as blocking death inducing signalling complex (DISC) formation, overexpression of anti-apoptotic proteins such as Bcl-2 and Bcl-XL and inhibition of active caspases by IAPs. The exact mechanism of the resistance to TRAIL-induced apoptosis varies depending on cell types and has not been fully understood, yet. Drug combination with conventional and targeted cancer therapeutics is most widely attempted to overcome the TRAIL resistance [7, 8].
In this study, we attempted to determine differential response to TRAIL-induced apoptosis of human gastric cancer cells and to identify potential indicators of the TRAIL response. An association of the expression of DR4 and DR5 with clinicopathological phenotypes of gastric cancer patients was also analyzed with TCGA (The Cancer Genome Atlas) data. TRAIL-induced apoptosis of gastric cancer cells was enhanced by combined treatment with various reagents including 5-fluorouracil, cisplatin, paclitaxel and bortezomib [9, 10]. Therefore, we have examined whether combined treatment of TRAIL with conventional chemotherapeutics can overcome the TRAIL resistance of the gastric cancer cells and what would be a potential mechanism underlying the synergistic induction of apoptosis.
Materials and methods
Gastric cancer cell lines and cell culture
The human gastric cancer cell lines, SNU-216, SNU-484, SNU-601, SNU-638, SNU-668 and SNU-719 were purchased from Korea Cell Line Bank (Seoul, Korea). They were cultured in RPMI-1640 (Gibco, Grand Islands, NY, USA) supplemented with 10% fetal bovine serum (Welgene, Daegu, Korea), 5% L-Glutamine (Gibco), 5% penicillin/streptomycin (Gibco) and maintained at 37°C in a humidified 5% CO2 atmosphere.
MTT assay
The viability of cells was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded in 96-well plates at density of 8 x 103 cells per well in 100 µl culture medium (RPMI-1640) one day prior to the treatment. Cells were treated with reagents as specified in figure legends. MTT (5 mg/ml MTT in PBS, Sigma-Aldrich, St. Louis, MO, USA) was treated and left for 3 h, and solubilized as previously described [11]. The absorbance at 570 nm with reference absorbance at 650 nm was measured using a MultiskanTM GO spectrophotometer (Thermo Scientific, Rockland, IL, USA). Results were calculated by subtracting blank readings, in which cells were not seeded.
Western blot analysis
Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 0.1% SDS, 1% Triton X-100, 0.1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM DTT and protease inhibitors). Protein amount was estimated by BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). Equal amounts of protein samples (25 µg/lane) were resolved by 10% or 12% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane (PALL Corporation, Port Washington, NY, USA). Immunodetection except visualization was performed as previously described [11]. ECL-treated blots (Advansta, Menlo Park, CA, USA) were exposed to a ChemiDocTM MP System (BioRad, Hercules, CA, USA) to visualize specific proteins. Intensity of detected bands was analyzed with Image J software (ij152-win-java8 downloaded from https://imagej.nih.gov/ij/) and quantification results are provided in Supplemental Figures. Antibodies used in the western blotting were anti-DR4 (Abnova Corporation, Taipei, Taiwan), anti-DR5, anti-p21cip1/waf1, anti-caspase-8, anti-caspase-9, anti-caspase-3, cleaved anti-caspase-8, cleaved anti-caspase-9, cleaved anti-caspase-3, anti-FLIP, anti-XIAP, anti-Bid, anti-Puma, (Cell Signaling Tech., Danvers, MA, USA), anti-BAX, anti-p53, anti-ERK, anti-phospho-ERK, anti-JNK, anti-phospho-JNK, anti-cIAP2, anti-β-actin (Santa Cruz Biotech., Dallas, Texas, USA), anti-p38 MAPK (ABM, Richmond, BC, Canada), and anti-phospho-p38 MAPK (Chemicon, Temecula, CA, USA).
Flow cytometric analysis
Cells were seeded in 6-well plates at density of 5 x 105 cells per well one day prior to treatment with indicated drugs. At specific time point, cells were harvested by trypsinization. The collected cells were washed once with 1X Annexin V binding buffer (eBioscience, San Diego, CA, USA) and then incubated in the buffer containing FITC-conjugated Annexin V (eBioscience). After incubated for 30 min at room temperature in dark, the cells were washed once with binding buffer and resuspended in 500 µl binding buffer containing propidium iodide solution (PI, 0.5 μg/ml). Annexin V binding and PI infiltration were evaluated by flow cytometry using a FACSCalibur™ (BD Bioscience, Sparks, MD, USA) and analyzed with CellQuest Pro™ software (BD Bioscience). To measure receptor expression on cell surface, cells were harvested by trypsinization at time points as specified in figure legends. The collected cells were incubated in 100 µl phycoerythrin (PE)-conjugated anti-DR4 or anti-DR5 antibodies (eBioscience) at RT for 30 min in dark. A PE-conjugated mouse IgG isotype control (eBioscience) was used as negative control. Fluorescence signals were then acquired on a FACSCalibur™ and analyzed as described above.
Silencing gene expression with siRNAs and shRNA lentiviruses
SNU-216 was transiently transfected with the following siRNAs procured from Genolution Pharmaceuticals, Inc. (Seoul, Korea): DR5 siRNA (5′-AAGACCCTTGTGCTCGTTGTC-3′) and scramble siRNA as a control. Cells were transfected at 40 µM siRNAs using the Lipofectamine 2000® transfection reagent (Life Technologies, Carlsbad, CA, USA) as instructed in the manufacturer's protocol. Cells were treated with TRAIL and/or bortezomib as specified in each experiment at 48 h after transfection. The pLKO.1 lentiviral vector with a scramble sequence, p21cip1/waf1 shRNA or DR5 shRNA (all from Sigma-Aldrich) were transfected into HEK293 cells by calcium phosphate precipitation. After 48h of transfection, viral supernatant was collected and filtered through 0.45 µm strain and stored at -80°C. SNU-216 was transduced with corresponding lentivirus in the presence of 8 µg/ml polybrene (Sigma-Aldrich). Infected cells were then selected by using medium containing 2.5 µg/ml of puromycin for 7 days before performing further experiments.
Gene expression analysis
Gene expression data in median z scores from RNASeq V2 RSEM of 478 gastric cancer patients with clinical information was obtained from TCGA via cBioPortal for Cancer Genomics (http://www.cbioportal.org/, [12, 13]). Kernel density plot, Shapiro-Wilk normality test, Wilcoxon rank sum test, Kruskal-Wallis rank sum test, Kaplan-Meyer survival analysis and log rank test were carried out with R statistical computing software (https://www.r-project.org/). Correlation coefficients among expression of TRAIL receptors and TRAIL were calculated by CORREL implemented in Microsoft Excel.
Statistical analysis
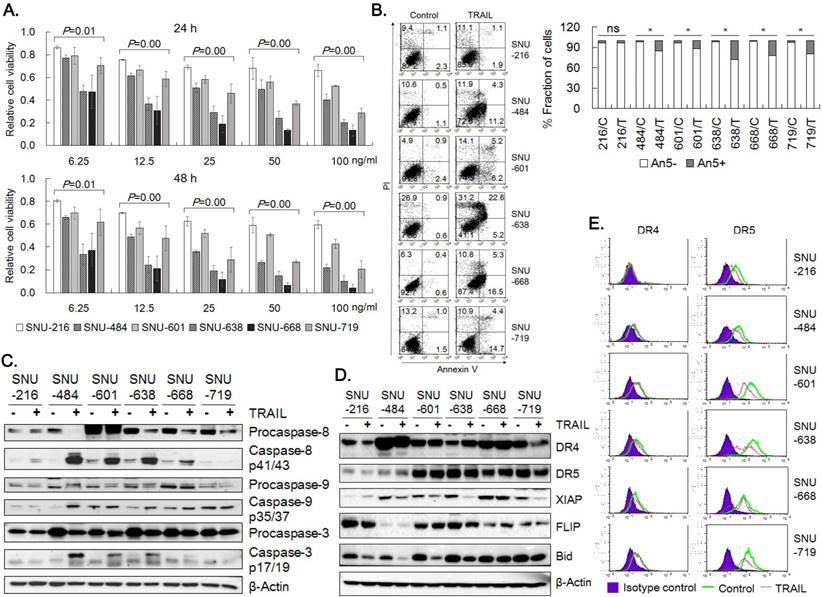
All of the data are shown either as the mean 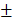 standard error of deviation (SE) or as the mean
standard error of deviation (SE) or as the mean 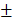 standard deviation (SD) from at least three independent experiments. Statistical comparison was performed with two-tailed Student's t-test or one-way ANOVA with post hoc Turkey's test. The p value smaller than 0.05 was considered statistically significant. The combination index (CI) was determined by using Compusyn software (http://www.combosyn.com/) in order to analyze the cooperation between TRAIL and bortezomib regarding to synergism, additivity, and antagonism [14, 15].
standard deviation (SD) from at least three independent experiments. Statistical comparison was performed with two-tailed Student's t-test or one-way ANOVA with post hoc Turkey's test. The p value smaller than 0.05 was considered statistically significant. The combination index (CI) was determined by using Compusyn software (http://www.combosyn.com/) in order to analyze the cooperation between TRAIL and bortezomib regarding to synergism, additivity, and antagonism [14, 15].
Results
Sensitivity of six gastric cancer cell lines to TRAIL
Six gastric cancer cell lines were treated with increasing concentration of TRAIL and cell viability was measured by MTT assay at 24 h and 48 h (Fig. 1A). Sensitivity to TRAIL ranked in the order of SNU-668 and SNU-638 > SNU-719, SNU-484 and SNU-601 > SNU-216. Cell viability of the SNU cells except SNU-216 was decreased in a dose-dependent manner up to 100 ng/ml TRAIL, while the viability of SNU-216 was decreased up to 25 ng/ml TRAIL, but not significantly reduced further in 50~100 ng/ml TRAIL range. TRAIL-induced apoptosis of the gastric cancer cells was further validated by analyzing Annexin V binding with flow cytometry and caspase activation with western blotting. TRAIL treatment significantly increased Annexin V-positive cell fractions in the gastric cancer cells except SNU-216 (Fig 1B). The cleaved caspases-8, -9, and -3 were evidently detected in all cell lines, again except SNU-216 (Fig 1C). Obviously, TRAIL induced apoptotic cell death at varying extent in the six gastric cancer cells, and SNU-216 was found the most resistant among them.
TRAIL-induced apoptosis of six gastric cancer cell lines. (A) TRAIL-induced reduction of cell viability of six gastric cancer cell lines. Cells were seeded in 96-well plates one day prior to TRAIL treatment at 0, 6.25, 12.5, 25, 50 and 100 ng/ml for 24 h and 48 h. Cell viability was assessed by MTT assay and relative cell viability was calculated against untreated control. Data shown is the mean ± standard error (SE) of three independent experiments. P values were calculated from one-way ANOVA. (B) Six gastric cancer cell lines were treated with TRAIL at 25 ng/ml for 24 h, and then TRAIL-induced apoptosis was determined by flow cytometric analysis of Annexin V/PI binding. Right panel shows fractions of Annexin V positive and negative cells, respectively. * for P<0.05 calculated by Student's t-test. (C) Activation of caspases-3, -8 and -9 was examined by detection of procaspases and active caspase fragments in western blot analysis with whole-cell lysate preparation from indicated cell lines treated with TRAIL at 25 ng/ml for 24 h. (D) Expression of death receptors and apoptosis modulators in untreated and treated cells with TRAIL (25 ng/ml) for 24 h were examined by western blot analysis. (E) Surface expression of DR4 and DR5 in untreated and treated cells with TRAIL (25 ng/ml) for 24 h were analyzed by flow cytometry. Results shown are representatives of three independent experiments (B, C, D and E).
In order to identify molecular determinants of the differential sensitivity, we analyzed the expression of molecules in the apoptotic signaling pathway of TRAIL by western blotting (Fig. 1D). DR4 expression was detected in all six cell lines, but lowest in SNU-216. DR5 expression was also evident, but the expression level was lower in SNU-484 and SNU-216. Basal XIAP level was lowest in SNU-216 and decreased upon TRAIL treatment in all cells, whereas FLIP expression was highest in SNU-216 and lowest in SNU-484. Bid expression varied among the cells, but decrease in full length Bid upon TRAIL treatment was observed in all six cells. The expression of DR4 and DR5 was evaluated in the six gastric cancer cells by flow cytometry (Fig. 1E). Flow cytometry verified comparable DR4 expression in all cells except SNU-216. DR5 expression was detected in all six cell lines by flow cytometry, but low in SNU-216 and SNU-484 as was shown in the western blotting (Fig. 1D). Reduction in surface DR5 expression upon TRAIL treatment was obvious in all six cells, while surface DR4 level noticeably decreased only in SNU-638 and SNU-668 upon TRAIL treatment. Collectively, expression levels of DR4, DR5 and FLIP might be associated with observed high resistance of SNU-216 against TRAIL.
Gene expression analysis of DR4 and DR5 in gastric cancer tissue
Resistance to TRAIL-induced apoptosis of gastric cancer cells appeared to be associated with the expression of apoptotic TRAIL receptors. Therefore, we analyzed the expression of DR4 and DR5 in gastric cancer tissue using RNASeq results in TCGA database. Median z-values were distributed between -2.19~9.90 for DR4 and -2.07~6.57 for DR5, respectively (Fig. 2A). Expression of DR4 correlated with that of DR5 with correlation coefficient of 0.657, which was highest among all 18,447 genes included in the analysis (Fig. 2B). Correlation coefficients of DR4 or DR5 expression with TRAIL and TRAIL decoy receptors were less than 0.5. DR4 expression was significantly higher in early stage tumor with low infiltration and no nodal involvement (p=0.014, p=0.015 and p=0.018 for T stage, N stage and TNM stage, respectively, Table 1). In contrast, expression of DR5 was marginally associated with N stage and grade (p=0.064 and p=0.065, respectively) and was significantly higher in intestinal type than diffused type (p=0.018). However, survival analysis with Kaplan-Meier estimation showed that either DR4 or DR5 expression did not significantly affect overall survival (p=0.37 for DR4 and p=0.37 for DR5, respectively (Fig. 2C). Expression of DR4 and DR5, especially DR4, appears to be decreased with progression of gastric cancer at early stage, but not with development of metastatic capacity, which might explain the observed irrelevance of patient survival with DR4 and DR5 expression.
Association of clinicopathological parameters and death receptor expression in gastric cancer.
| TNFRSF10A | TNFRSF10B | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No | 1st Q | Median | 3rd Q | p value | 1st Q | Median | 3rd Q | p value | ||
| Sex | Female | 147 | -0.713 | -0.064 | 1.074 | 0.047 | -0.748 | 0.002 | 0.815 | 0.096 |
| Male | 267 | -0.892 | -0.325 | 0.655 | -0.803 | -0.261 | 0.527 | |||
| Age | <65 | 171 | -0.801 | -0.163 | 0.871 | 0.466 | -0.807 | -0.155 | 0.643 | 0.685 |
| >=65 | 234 | -0.853 | -0.211 | 0.805 | -0.796 | -0.115 | 0.688 | |||
| Histology | Intestinal | 176 | -0.849 | -0.205 | 0.727 | 0.581 | -0.613 | -0.089 | 0.671 | 0.018 |
| Diffused | 69 | -0.851 | -0.209 | 0.532 | -0.932 | -0.311 | 0.301 | |||
| Grade | G1/G2 | 159 | -0.756 | -0.097 | 0.763 | 0.358 | -0.552 | -0.119 | 0.859 | 0.065 |
| G3 | 246 | -0.873 | -0.321 | 0.822 | -0.863 | -0.158 | 0.568 | |||
| T status | T1/T2 | 109 | -0.678 | 0.152 | 1.115 | 0.014 | -0.803 | -0.033 | 1.232 | 0.237 |
| T3/T4 | 296 | -0.892 | -0.291 | 0.665 | -0.801 | -0.158 | 0.534 | |||
| N status | N0 | 122 | -0.693 | 0.068 | 1.115 | 0.015 | -0.772 | -0.011 | 0.998 | 0.064 |
| N1/N2/N3 | 273 | -0.921 | -0.341 | 0.668 | -0.836 | -0.195 | 0.534 | |||
| M status | M0 | 367 | -0.871 | -0.221 | 0.846 | 0.827 | -0.801 | -0.154 | 0.671 | 0.834 |
| M1 | 27 | -0.660 | -0.176 | 0.259 | -0.686 | 0.010 | 0.615 | |||
| Tumor stage | Stage1/2 | 179 | -0.790 | 0.096 | 1.047 | 0.018 | -0.769 | -0.061 | 0.882 | 0.262 |
| Stage3/4 | 210 | -0.921 | -0.333 | 0.572 | -0.844 | -0.156 | 0.477 | |||
Analysis of DR4 and DR5 expression in gastric cancer tissue with RNASeq results in TCGA database. (A) A Kernel density plot represents distribution of median z-score of DR4 (TNFRSF10A) and DR5 (TNFRSF10B) expression in gastric cancer tissue. (B) A table of correlation coefficients among DRs, DcRs (TNFRSF10C and TNFRSF10D) and TRAIL (TNFSF10). (C) Kaplan-Meier plots for overall survival according to expression of DR4 (upper panel) and DR5 (lower panel). Solid lines represent for expression lower than the first quartile, dotted lines for expression between the first and third quartile and dashed lines for expression higher than the third quartile, respectively.
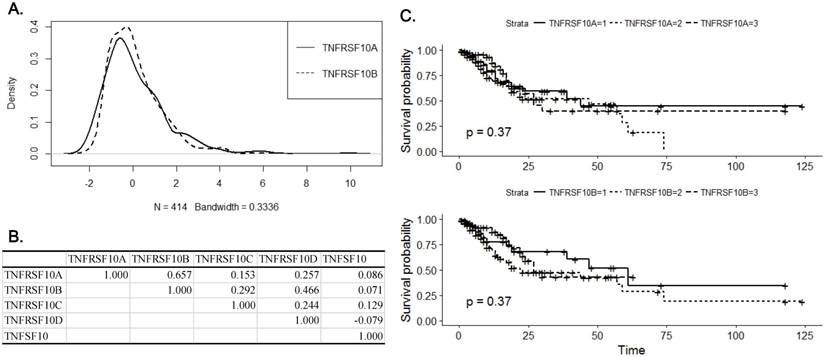
Combined treatment of TRAIL with bortezomib
In order to potentiate the efficacy of TRAIL, we treated the gastric cancer cells with 20 different known and putative cancer therapeutics plus TRAIL at single concentration combinations and evaluated their ability to enhance TRAIL cytotoxicity. Four reagents including proteasome inhibitors (bortezomib and MG132) and anthracyclines (doxorubicin and daunorubicin) were found to potentiate cytotoxicity of TRAIL more than expected by simple multiplication of individual drug effect. Since bortezomib enhanced TRAIL cytotoxicity, the effect of bortezomib on TRAIL was further characterized in TRAIL-resistant SNU-216 cells. Viable cell amount was significantly decreased when combined treatment of TRAIL and bortezomib was compared with that of TRAIL or bortezomib alone (Fig. 3A). IC50-equivalent amount of TRAIL at various TRAIL/bortezomib concentration combinations in SNU-216 were located well below the line of additivity in isobolograms, indicating strong synergistic interaction at all combinations (Fig. 3B). TRAIL-induced apoptosis of SNU-216 was further analyzed by Annexin V binding after 24 h and 48 h treatment of TRAIL alone or in combination with bortezomib. In accordance with the MTT results, combined treatment of TRAIL and bortezomib significantly increased Annexin V positive fractions over TRAIL treatment alone (Fig. 3C). In addition, TRAIL/bortezomib enhanced the activation of caspases-8, -9 and -3 (Fig. 3D). Collectively, these results clearly demonstrated that bortezomib could synergize TRAIL-mediated apoptosis of the gastric cancer cells.
Synergistic effects of TRAIL and bortezomib in SNU-216. (A) SNU-216 was treated with TRAIL (12.5 ng/ml) in combination with bortezomib (22.75 nM) for 24 h and 48 h. Cell viability was analyzed by MTT assay and relative cell viability was calculated against untreated control. Results are presented as the mean ± SE of three independent experiments. P values were calculated from one-way ANOVA. (B) The cells were co-treated with TRAIL (0.0, 12.5, 25.0 and 50 ng/ml) and bortezomib (0.0, 5.7, 11.4, 22.8, 45.5 and 91.0 nM) in a range of concentration for 24 h and 48 h. Cell viability was measured by MTT assay and synergism of drug combinations was analyzed with isobologram. (C) Apoptotic cells were determined by flow cytometry of Annexin V/PI staining. Lower panel shows fractions of Annexin V positive and negative cells, respectively. P values were calculated from one-way ANOVA. (D) Western blot was used to detect the activation of caspases-3,-8 and -9. The data shown are representatives of triple experiments (C and D).
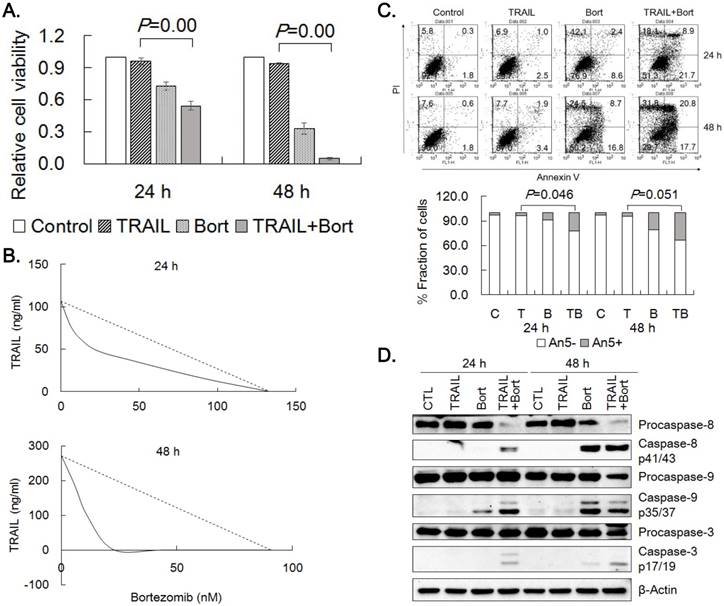
Bortezomib upregulates p21cip1/waf1
To understand the mechanism underlying the synergistic effect of bortezomib on TRAIL, we examined the expression of proteins modulating cell proliferation and apoptosis. p21cip1/waf1 expression was markedly increased by 24 h and 48 h treatment of bortezomib and bortezomib plus TRAIL, whereas expression of p53, Bid and Puma was decreased by bortezomib plus TRAIL treatment at 48 h noticeably (Fig. 4A, Supplemental Fig. 1). However, bortezomib did not significantly alter protein levels of Bax, FLIP, cIAP2 and XIAP. To determine whether bortezomib-enhanced TRAIL sensitivity was ascribed to increased expression of p21cip1/waf1, expression of p21cip1/waf1 was knocked down with p21cip1/waf1 shRNA and susceptibility of cells to the combined treatment was examined. Cell viability of the p21cip1/waf1-knockdown cells were insignificantly increased at 24 h and 48 h after treatment of bortezomib alone or TRAIL/bortezomib, compared to that of scramble shRNA expressing cells (Fig. 4B). However, activation of caspases-3, -8, and -9 upon combined treatment of TRAIL and bortezomib was mitigated in the p21cip1/waf1-knockdown cells than in scramble shRNA expressing ones (Fig. 4C), which appears to reflect marginal increase in viability of p21cip1/waf1-knockdown cells treated with TRAIL plus bortezomib for 24 h (Fig. 4B). These results suggested that accumulation of p21cip1/waf1 by bortezomib contributes to the enhanced TRAIL-induced apoptosis of SNU-216 cells insignificantly.
Bortezomib upregulates p21cip1/waf1. (A) SNU-216 cells were treated with TRAIL and/or bortezomib for 24 h and 48 h. The expression of indicated proteins including p21cip1/waf1 were visualized by western blot analysis. (B) SNU-216 cells were transduced with scramble or p21cip1/waf1 shRNA expressing lentiviruses, then were selected by puromycin (2.5 µg/ml) for one week. The p21cip1/waf1 knockdown cells were treated with TRAIL in the presence or absence of bortezomib for 24 h and 48 h. Cell viability of SNU-216 after 24 h and 48 h treatment was measured by MTT assay and relative cell viability was calculated against untreated control. MTT results shown are the means ± SE of three independent experiments. 'ns' for P>0.05 in Student's t-test. (C) Expression of p21cip1/waf1 and activation of caspases in the p21cip1/waf1 knockdown cells were visualized by western blot analysis. All cells (A~C) were treated with TRAIL at 12.5 ng/ml and/or bortezomib (22.75 nM) for indicated time period. The images shown are representatives of triple experiments (A and C).
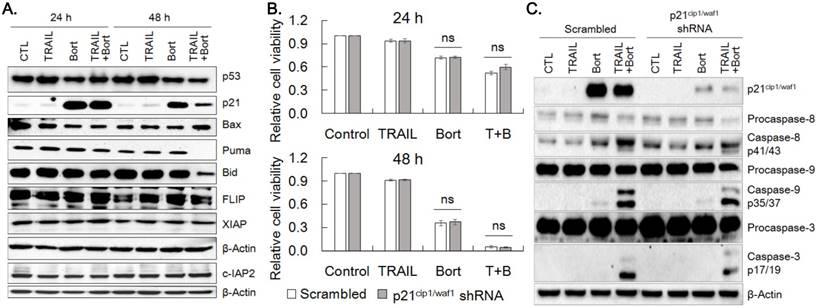
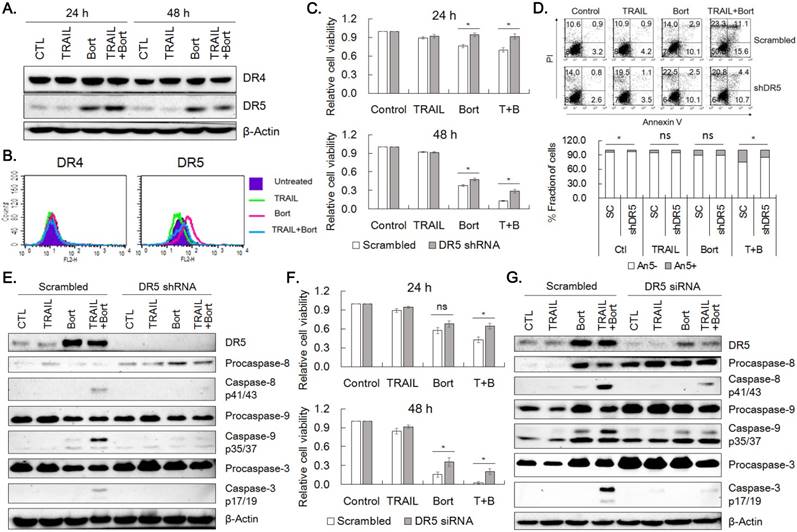
Overexpression of DR5 by bortezomib
Since accumulation of p21cip1/waf1 was not enough to explain viability reduction observed in the combined treatment, the effect of combined treatment of TRAIL and bortezomib on the expression of DR4 and DR5 was examined in SNU-216 cells. Bortezomib increased expression of DR5 obviously and DR4 to a lesser extent at 24 h and 48 h of the drug treatment (Fig. 5A). Consistently, flow cytometric analysis for the receptors confirmed that the expression of DR5 was increased in SNU-216 treated with bortezomib alone or in combination with TRAIL (Fig. 5B). These observations implied that upregulation of DR5 might play a critical role in sensitization of TRAIL-induced apoptosis of SNU-216 by bortezomib. In order to assess the involvement of DR5 in sensitization of TRAIL-induced apoptosis by bortezomib, we knocked down DR5 expression by shRNA. Knockdown of DR5 significantly increased cell viability when compared with scramble control in both 24 h and 48 h of TRAIL and bortezomib treatment (Fig. 5C). Of notice, cell viability of DR5-silenced cells treated with TRAIL plus bortezomib was comparable with that of scramble control treated with bortezomib alone. In addition, Annexin V-positive fraction of the DR5-silenced cells was reduced by 38% in average when compared with that of scramble control upon combined treatment of TRAIL and bortezomib for 24 h (p=0.03, Fig. 5D). The activation of caspases-8, -9 and -3 was also reduced in DR5-silenced cells when compared with scramble control (Fig. 5E). Furthermore, silencing DR5 expression by siRNA transfection also resulted in comparable viability increase shown in DR5 shRNA expressing cells and decreased activation of caspases -8, -9 and -3 (Fig. 5F and 5G, respectively). Taken together, upregulation of DR5 by bortezomib was supposed to be a critical factor for the TRAIL-bortezomib synergy.
DR5 contributes to bortezomib-induced TRAIL sensitization. (A) Effects of bortezomib on the expression of DR4 and DR5 in SNU-216. Cells were treated with TRAIL and/or bortezomib for 24 h and 48 h, and expression of DR4 and DR5 was examined by western blot analysis. (B) Surface expression of DR4 and DR5 in SNU-216 upon 24 h expose to TRAIL in the presence or absence of bortezomib, was analyzed by flow cytometry. (C) Lentivirus of scramble control or DR5 shRNA was transduced into SNU-216, and then selected with puromycin (2.5 µg/ml) for a week. Cell viability of scramble and DR5-silenced cells upon TRAIL and/or bortezomib treatment for 24 h and 48 h was measured by MTT assay and relative cell viability was calculated against untreated control. Results shown are means ± SE of three independent experiments. (D) Reduced Annexin V binding in the DR5-silenced cells treated with TRAIL and/or bortezomib was analyzed by flow cytometry. Lower panel shows fractions of Annexin V positive and negative cells, respectively. (E) Expression of DR5 and activation of caspases in the DR5-silenced cells treated with TRAIL and/or bortezomib were analyzed by western blot. (F) Expression of DR5 in SNU-216 was knocked down by transfection of DR5 siRNA. DR5-silenced cells by siRNA transfection were treated with TRAIL and/or bortezomib for 24 h and 48 h and cell viability was measured by MTT assay. Relative cell viability was calculated against untreated control and shown are the means ± SE of three independent experiments. (G) Expression of DR5 and activation of caspases in the DR5-silenced cells were examined by western blot analysis. The siRNA transfected cells were treated with TRAIL and/or bortezomib for 24 h. All cells (A~G) were treated with TRAIL at 12.5 ng/ml and/or bortezomib (22.75 nM) for indicated time period. * for P< 0.05 and 'ns' for P>0.05 calculated from Student's t-test (C, D and F). Results shown are representatives of triple experiments (A, B D, E and G).
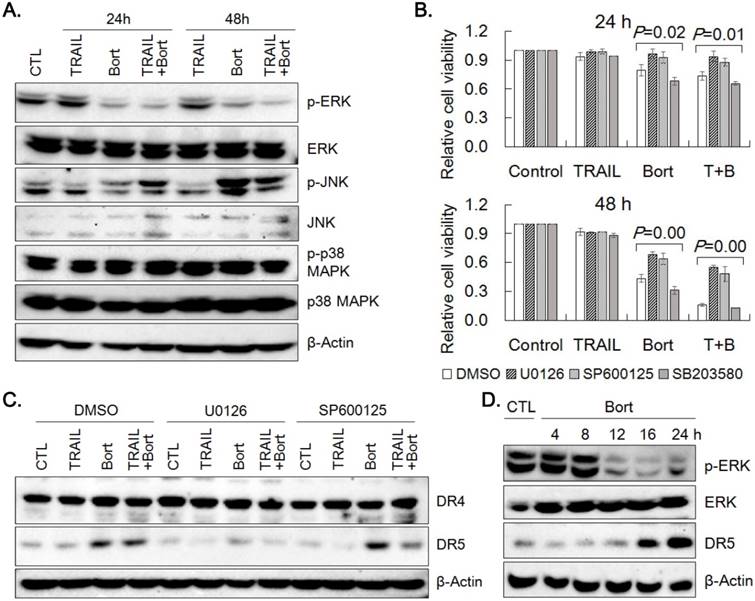
ERK1/2 activity is required in the upregulation of DR5 by bortezomib
Since the overexpression of DR5 was found critical in TRAIL and bortezomib synergism, we attempted to explore mechanism by which bortezomib could upregulate DR5 expression. DR5 expression is known to be modulated by ERK, JNK and p38 MAPK [16, 17]. Hence, the effect of TRAIL and bortezomib on expression and phosphorylation of the MAPKs was examined by western blotting (Fig. 6A). Bortezomib reduced phosphorylation of ERK1/2, but increased that of JNK, and did not alter that of p38 MAPK without significant changes in the expression level of the kinases. Reduction of cell viability caused by treatment of bortezomib and bortezomib plus TRAIL was significantly recovered by pretreatment of SNU-216 with U0126 (an inhibitor of ERK1/2 activation) and SP600125 (a JNK inhibitor), but not with SB203580 (a p38MAPK inhibitor) (Fig. 6B). The effects of U0126 and SP600125 on the expression of DR4 and DR5 was determined by western blotting. Whereas DR4 expression remained unchanged upon pretreatment of U0126 and SP600125, upregulation of DR5 level by bortezomib was dampened significantly only by U0126 pretreatment, but not by SP600125 (Fig. 6C, Supplemental Fig. 2). Since DR5 upregulation by bortezomib appeared to be associated with ERK activation, time-dependent expression of phospho-ERKs and DR5 was examined by western blotting. The level of phospho-ERKs was maintained until 8 h, but dropped in 12 h, whilst DR5 level was increased after 16 h of bortezomib treatment (Fig. 6D). These results suggest that upregulation of DR5 expression by bortezomib might be dependent on ERK1/2 activity at early time point.
Activation of MAPK signaling pathway by bortezomib. (A) Activation of ERK1/2, JNK and p38 MAPK in SNU-216 treated with TRAIL and/or bortezomib for 24 h and 48 h was analyzed by western blotting with antibodies against activation-associated phosphorylation of the kinases. (B) Cells were pretreated with U0126 (20 µM), SP600125 (20 µM) or SB203580 (20 µM) for 1 h followed by TRAIL and/or bortezomib treatment for 24 h and 48 h. Cell viability was measured by MTT assay and relative cell viability was calculated against untreated control. Results shown are the means ± SE of three independent experiments. P values were calculated from one-way ANOVA. (C) Expression of DR4 and DR5 in SNU-216 pretreated with U0126 (20 µM) or SP600125 (20 µM) for 1 h followed by TRAIL and/or bortezomib treatment for 24 h was visualized by western blot analysis. (D) Time-dependent expression of DR5, phospho-ERKs and ERKs in cells treated with bortezomib was examined by western blot analysis. All cells (A~D) were treated with TRAIL at 12.5 ng/ml and/or bortezomib (22.75 nM) for indicated time period. The data shown are representatives of triple experiments (A, C and D).
Discussion
The correlation of TRAIL sensitivity with the expression of TRAIL receptors and their immediate signal modulators was examined in six human gastric cancer cell lines. SNU-216 in which level of DR4 and DR5 was lowest, but that of FLIPL was highest among them, was found the most resistant to TRAIL. Correlation of death receptor expression with TRAIL sensitivity was also found in SNU-1 gastric cancer cells that showed negligible DR4 expression and manifested strong resistance against TRAIL [11]. Although a critical role of various other modulators in TRAIL-induced apoptosis cannot be ruled out, therefore, surface expression level of DRs should be considered as a critical factor in TRAIL response of the gastric cancer cells. TRAIL resistance of SNU-216 might also be ascribed to high expression level of FLIP through constitutive activation of PI3K/AKT pathway [18], which suggests considerable significance of other apoptosis regulators in modulation of TRAIL response of the gastric cancer cells.
Association of death receptor expression with pathology of gastric cancer tissue at mRNA level revealed that DR4 expression was significantly higher in early stage tumor without distant metastasis. DR5 expression was also associated with nodal status with marginal significance. Although clinical association of DR expression at mRNA level in gastric cancer has not been established yet, negative DR4 protein expression was found to correlate with lower nodal status with marginal significance in gastric cancer [19]. Aside from gastric cancer, DR4 expression correlated with more differentiated tumors and negative nodal status in an immunohistochemical study of breast cancer, while DR5 expression correlated with higher tumor grade, proliferative index, positive nodal status and reduced overall survival rate [20]. On the contrary, DR5 expression was reported to be reduced in higher grade prostate cancer [21]. Therefore, DR4 expression appeared to be negatively associated with phenotypes of progressed tumor, albeit correlation of DR5 expression with tumor grade and survival is controversial, yet.
In order to overcome TRAIL resistance observed in the gastric cancer cells, TRAIL cytotoxicity was examined in combination with various chemotherapeutics. Strong synergy in induction of apoptosis of the gastric cancer cells was observed by combined treatment of TRAIL with proteasome inhibitors (bortezomib and MG132) and anthracyclines (doxorubicin and daunorubicin). Bortezomib (VELCADE®) that was approved by the Food and Drug Administration (FDA) for the treatment of multiple myeloma is a selective 26S proteasome inhibitor [22, 23]. Bortezomib elicits G2/M arrest and induces apoptosis of diverse cancer cells by itself and in combined treatment with various known and potential cancer therapeutics. Bortezomib-induced growth arrest and apoptosis are mediated by inhibition of NF-κB activation, increased expression of growth arresting and apoptotic proteins as well as induction of ER stress [24-26]. Bortezomib also enhances the efficacy of TRAIL in several cancer cells including gastric cancer cells [27]. Depending on the cellular context, bortezomib modulates the expression of TRAIL receptors, c-FLIP, Bik, Bim, IAPs, p21cip1/waf1 and p27kip, and activation of NF-κB, Akt and MAPKs, which has been suggested for potential mechanisms behind the synergy [22].
Treatment of bortezomib on TRAIL-resistant SNU-216 cells elicited G2/M arrest and apoptosis of the cancer cells. Bortezomib treatment significantly increased expression of p21cip1/waf1, DR5 and DR4 which could directly modulate cell cycle progression and apoptosis. Bortezomib is a proteasome inhibitor that can stabilize and increase p53 level [28]. However, bortezomib by itself did not increase level of p53 and its targets including Bax and Puma in SNU-216 cells. Instead, expression of p53 and Puma was significantly decreased by combined treatment of TRAIL plus bortezomib for 48 h, suggesting that downregulation of p53 and Puma might result from massive cell death at late time point. On the other hand, expression of p21cip1/waf1 and DR5 was significantly increased in bortezomib-treated cells even without noticeable change in p53 at early time point. Oridonin, a herbal diterpenoid, increased expression of p53 and Bax in SNU-216 cells, demonstrating its inducibility and functionality in the cells [29]. Therefore, it is conceivable that bortezomib might activate an alternative signaling pathway that obviates p53 induction to upregulate DR5 and p21cip1/waf1 expression. Activation of ATF4-ATF3/CHOP axis via PKCdelta [30] or change in ERK1/2 activity profile (Fig. 6) by bortezomib could be an alternative signaling pathway for induction of DR5. Indeed, bortezomib-induced DR5 expression is regulated by CHOP, an ER-stress mediator in several cells including human non-small cell lung cancer cells [31]. Insignificant change in Bax and decrease in p53 and Puma in conjuction with seemingly p53-independent induction of DR5 and p21cip1/waf1 cast doubt on the role of p53 in the synergistic death of SNU-216 cells by combined treatment of bortezomib and TRAIL.
Upregulation of p21cip1/waf1 by bortezomib has been reported in many cancer cells, which might be associated with cell cycle arrest upon bortezomib treatment [32]. Increase in p21cip1/waf1 and concomitant decrease in CDK activity by bortezomib are found responsible for sensitization of bladder and prostate cancer cells to TRAIL-induced apoptosis [33]. However, silencing p21cip1/waf1 in SNU-216 cells marginally increased cell viability and decreased level of cleaved caspases upon treatment of bortezomib alone or TRAIL/bortezomib for 24 h. The rescuing effect of p21cip1/waf1 knockdown was too modest to support a determinative role of p21cip1/waf1 in the TRAIL/bortezomib synergy. Hence, although p21cip1/waf1 level is strongly increased by bortezomib treatment, the role of p21cip1/waf1 in the TRAIL/bortezomib synergy remains to be doubtful.
Liu et al. reported synergistic apoptosis of different gastric cancer cells by co-treatment of bortezomib and TRAIL [27]. Increase in both DR4 and DR5 and decrease in c-IAP1 were observed in the SGC7901 gastric cancer cells. Increase in DR5 only or in both DR4 and DR5 was also reported in other cancer cells, which was considered as a critical component in TRAIL/bortezomib synergy [27, 31]. In accord, silencing DR5 expression in SNU-216 by shRNA expression or siRNA transfection increased cell viability of combined treatment of TRAIL and bortezomib up to ~80% of bortezomib single treatment (Fig. 5). Thus, upregulation of DR5 by bortezomib was mainly responsible for bortezomib synergy in TRAIL-induced apoptosis, as was reported in many other cancer cells [31, 34].
How bortezomib modulates the expression of DR5 has not been fully understood. Activation of the MAPKs including ERKs, JNK and p38 MAPK was known to stimulate DR5 expression in many cancer cells [17, 35, 36]. In SNU-216, phospho-ERK1/2 was decreased, but phospho-JNK was increased by 24 h treatment of bortezomib. Both ERK and JNK inhibitors were able to counteract the cytotoxicity of bortezomib only and TRAIL/bortezomib treatment. However, inhibition of ERK1/2 activation, not JNK inhibition attenuated bortezomib-induced DR5 upregulation. Collectively, although both ERK and JNK pathway in parallel contribute to synergistic apoptosis by TRAIL and bortezomib, DR5 expression was regulated in ERK1/2-dependent manner.
Upregulation of DR5 was associated with increased phosphorylation of ERK1/2 in bortezomib-treated lung cancer cells [31]. However, bortezomib was also known to decrease phosphorylation of ERK1/2 via a MAPK phosphatase-3-dependent pathway in transformed endothelial cells [37]. In SNU-216, phospho-ERK level was significantly reduced after 8 h treatment of bortezomib, whereas increased DR5 expression was detected after 16 h of treatment (Fig. 6D). Apparently, these results suggest that upregulation of DR5 could result from reduction of phospho-ERKs. Surprisingly, however, inhibition of ERK1/2 by U0126 prevented either basal or induced level of DR5 expression from increasing and partially reverted cell viability reduction upon bortezomib treatment, which argues for the role of ERKs in the process. In reconciliation, presence of active ERKs at the early stage of bortezomib treatment appears to be required for bortezomib-induced cytotoxicity and DR5 upregulation. However, mode and mechanism of action of ERKs in the bortezomib-induced changes remains to be elucidated in detail.
Conclusion
Gastric cancer cells varied in response to TRAIL, which might be explained by differential expression of DR4 and FLIP. Although expression of DR4 and DR5 did not influence overall survival rate of gastric cancer patients, higher DR4 expression was found to correlate with lower T, N and TNM stages. TRAIL resistance of SNU-216 could be efficiently overcome by combined treatment of TRAIL and bortezomib. Upregulation of DR5 by bortezomib was found to contribute significantly to the TRAIL/bortezomib synergy. Both JNK and ERK1/2 were involved in cytotoxic effect of bortezomib and TRAIL/bortezomib treatments. However, only ERK1/2 activity at early time point of bortezomib treatment was required for the upregulation of DR5. Bortezomib also enhances the efficacy of TRAIL against tumor xenografts and endogenous cancers in animal studies [38]. In addition, TRAIL is implicated in antitumor effect of NK cells which can be potentiated by bortezomib treatment [39]. Thus, these results would provide important information on the utility of TRAIL as a therapeutic measure of gastric cancer with synergistic agents including bortezomib.
Supplementary Material
Supplementary figures.
Abbreviations
TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; DR: death receptor; FLIP: FADD-like IL-1β-converting enzyme(FLICE)-inhibitory protein; IAP: inhibitor of apoptosis; XIAP: X-linked inhibitor of apoptosis; MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; JNK: Jun N-terminal kinase; CDK: cyclin-dependent kinase; NK: natural killer; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; PI-3K: phosphoinositide 3-kinase; TCGA: The Cancer Genome Atlas; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT); PI: propidium iodide.
Acknowledgements
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01058830).
Author's contributions
HTTB: acquisition, analysis and interpretation of data, drafting the manuscript; NHL and QAL: acquisition, analysis and interpretation of data; SL: design of experiments, acquisition, analysis and interpretation of data; SEK: clinical data analysis; DK: conception and design of experiments, analysis and interpretation of data, drafting and revising the manuscript; All authors approved the final version of manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rahman R, Asombang AW, Ibdah JA. Characteristics of gastric cancer in Asia. World J Gastroenterol. 2014;20:4483-4490
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
3. Shin A, Kim J, Park S. Gastric cancer epidemiology in Korea. J Gastric Cancer. 2011;11:135-140
4. Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950-2966
5. Kruyt FA. TRAIL and cancer therapy. Cancer Lett. 2008;263:14-25
6. Refaat A, Abd-Rabou A, Reda A. TRAIL combinations: The new 'trail' for cancer therapy (Review). Oncol Lett. 2014;7:1327-1332
7. Thorburn A, Behbakht K, Ford H. TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Updat. 2008;11:17-24
8. Shankar S, Srivastava RK. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: mechanisms and clinical implications. Drug Resist Updat. 2004;7:139-156
9. Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21:1350-1364
10. Newsom-Davis T, Prieske S, Walczak H. Is TRAIL the holy grail of cancer therapy? Apoptosis. 2009;14:607-623
11. Song NM, Jun S, Zang DY, Kim SG, Park HR, Kang D. Differential susceptibility of gastric cancer cells to TRAIL-induced apoptosis. Oncol Rep. 2013;29:1224-1230
12. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
13. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-404
14. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-681
15. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440-446
16. Do MT, Na M, Kim HG, Khanal T, Choi JH, Jin SW, Oh SH, Hwang IH, Chung YC, Kim HS. et al. Ilimaquinone induces death receptor expression and sensitizes human colon cancer cells to TRAIL-induced apoptosis through activation of ROS-ERK/p38 MAPK-CHOP signaling pathways. Food Chem Toxicol. 2014;71:51-59
17. Pennati M, Sbarra S, De Cesare M, Lopergolo A, Locatelli SL, Campi E, Daidone MG, Carlo-Stella C, Gianni AM, Zaffaroni N. YM155 sensitizes triple-negative breast cancer to membrane-bound TRAIL through p38 MAPK- and CHOP-mediated DR5 upregulation. Int J Cancer. 2015;136:299-309
18. Nam SY, Jung GA, Hur GC, Chung HY, Kim WH, Seol DW, Lee BL. Upregulation of FLIP(S) by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Sci. 2003;94:1066-1073
19. Hu JK, Yang K, Li CM, Zhang B, Chen ZX, Chen XZ, Chen JP. The expression of TRAIL and its receptors in gastric cancer and the apoptotic effect of rh-TRAIL on SGC7901 cells. Oncol Rep. 2009;21:681-688
20. Ganten TM, Sykora J, Koschny R, Batke E, Aulmann S, Mansmann U, Stremmel W, Sinn HP, Walczak H. Prognostic significance of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in patients with breast cancer. J Mol Med (Berl). 2009;87:995-1007
21. Hernandez-Cueto A, Hernandez-Cueto D, Antonio-Andres G, Mendoza-Marin M, Jimenez-Gutierrez C, Sandoval-Mejia AL, Mora-Campos R, Gonzalez-Bonilla C, Vega MI, Bonavida B. et al. Death receptor 5 expression is inversely correlated with prostate cancer progression. Mol Med Rep. 2014;10:2279-2286
22. de Wilt LH, Kroon J, Jansen G, de Jong S, Peters GJ, Kruyt FA. Bortezomib and TRAIL: a perfect match for apoptotic elimination of tumour cells? Crit Rev Oncol Hematol. 2013;85:363-372
23. Mujtaba T, Dou QP. Advances in the understanding of mechanisms and therapeutic use of bortezomib. Discov Med. 2011;12:471-480
24. Brooks AD, Jacobsen KM, Li W, Shanker A, Sayers TJ. Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol Cancer Res. 2010;8:729-738
25. Vaziri SA, Grabowski DR, Hill J, Rybicki LR, Burk R, Bukowski RM, Ganapathi MK, Ganapathi R. Inhibition of proteasome activity by bortezomib in renal cancer cells is p53 dependent and VHL independent. Anticancer Res. 2009;29:2961-2969
26. Williams SA, McConkey DJ. The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 prostate cancer cells. Cancer Res. 2003;63:7338-7344
27. Liu J, Qu XJ, Xu L, Zang Y, Qu JL, Hou KZ, Liu YP. Bortezomib synergizes TRAIL-induced apoptosis in gastric cancer cells. Dig Dis Sci. 2010;55:3361-3368
28. Xue Y, Barker N, Hoon S, He P, Thakur T, Abdeen SR, Maruthappan P, Ghadessy FJ, Lane DP. Bortezomib Stabilizes and Activates p53 in Proliferative Compartments of Both Normal and Tumor Tissues. Cancer Res. 2019;79:3595-3607
29. Bi E, Liu D, Li Y, Mao X, Wang A, Wang J. Oridonin induces growth inhibition and apoptosis in human gastric carcinoma cells by enhancement of p53 expression and function. Braz J Med Biol Res. 2018;51:e7599
30. Seol DW. p53-Independent up-regulation of a TRAIL receptor DR5 by proteasome inhibitors: a mechanism for proteasome inhibitor-enhanced TRAIL-induced apoptosis. Biochem Biophys Res Commun. 2011;416:222-225
31. Xu L, Su L, Liu X. PKCdelta regulates death receptor 5 expression induced by PS-341 through ATF4-ATF3/CHOP axis in human lung cancer cells. Mol Cancer Ther. 2012;11:2174-2182
32. Maynadier M, Shi J, Vaillant O, Gary-Bobo M, Basile I, Gleizes M, Cathiard AM, Wah JL, Sheikh MS, Garcia M. Roles of estrogen receptor and p21(Waf1) in bortezomib-induced growth inhibition in human breast cancer cells. Mol Cancer Res. 2012;10:1473-1481
33. Lashinger LM, Zhu K, Williams SA, Shrader M, Dinney CP, McConkey DJ. Bortezomib abolishes tumor necrosis factor-related apoptosis-inducing ligand resistance via a p21-dependent mechanism in human bladder and prostate cancer cells. Cancer Res. 2005;65:4902-4908
34. Bullenkamp J, Raulf N, Ayaz B, Walczak H, Kulms D, Odell E, Thavaraj S, Tavassoli M. Bortezomib sensitises TRAIL-resistant HPV-positive head and neck cancer cells to TRAIL through a caspase-dependent, E6-independent mechanism. Cell Death Dis. 2014;5:e1489
35. Kim EY, Yu JS, Yang M, Kim AK. Sub-toxic dose of apigenin sensitizes HepG2 cells to TRAIL through ERK-dependent up-regulation of TRAIL receptor DR5. Mol Cells. 2013;35:32-40
36. Trivedi R, Maurya R, Mishra DP. Medicarpin, a legume phytoalexin sensitizes myeloid leukemia cells to TRAIL-induced apoptosis through the induction of DR5 and activation of the ROS-JNK-CHOP pathway. Cell Death Dis. 2014;5:e1465
37. Suares A, Mori Sequeiros Garcia M, Paz C, Gonzalez-Pardo V. Antiproliferative effects of Bortezomib in endothelial cells transformed by viral G protein-coupled receptor associated to Kaposi's sarcoma. Cell Signal. 2017;32:124-132
38. Christian PA, Thorpe JA, Schwarze SR. Velcade sensitizes prostate cancer cells to TRAIL induced apoptosis and suppresses tumor growth. Cancer Biol Ther. 2009;8:73-80
39. Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180:163-170
Author contact
![]() Corresponding author: Dongchul Kang, dckangac.kr
Corresponding author: Dongchul Kang, dckangac.kr