Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Statistical analysis
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(8):1149-1156. doi:10.7150/ijms.34749 This issue Cite
Research Paper
The phenotype of vascular smooth muscle cells co-cultured with endothelial cells is modulated by PDGFR-β/IQGAP1 signaling in LPS-induced intravascular injury
Xia Zheng1 ![]() *, Xiaotong Hu2*, Wang Zhang1
*, Xiaotong Hu2*, Wang Zhang1
1. Department of Critical Care Medicine, The First Affiliated Hospital of Zhejiang University, Hangzhou, Zhejiang, 310003, P.R. China.
2. Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases; The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, 310003, P.R. China.
*Co-first author
Received 2019-3-10; Accepted 2019-7-9; Published 2019-8-6
Abstract
Background Sepsis, a leading cause of death in intensive care units, is generally associated with vascular dysfunction. However, its pathophysiological process has not been fully clarified, lacking in-depth knowledge of its pathophysiological process may hinder the improvement of diagnosis and therapy for sepsis. Hence, as the key parts of the vascular wall, the interaction between endothelial cells (ECs) and smooth muscle cells (SMCs) under septic situation need to be further studied.
Methods ECs and SMCs were co-cultured using Transwell plates. Lipopolysaccharide (LPS) was used to induce sepsis. A scratch-wound assay was used to assess cell migration, and western blotting was used to assess the level of redifferentiation of SMCs as well as the expression of PDGFR-β and IQGAP1.
Results Co-culture with ECs reduced the redifferentiation of SMCs induced by LPS (10 μg/ml), which was characterized by increased migration ability and decreased expression of contractile proteins (e.g., SM22 and α-SMA). The production of TNF-α could decrease the level of PDGFR-β in SMCs. Treatment of SMCs with the PDGFR-β inhibitor imatinib (5 μM) was able to counteract LPS-induced SMC redifferentiation and reduce IQGAP1 protein expression, especially when SMCs were co-cultured with ECs.
Conclusion The phenotype of vascular SMCs co-cultured with ECs was modulated by IQGAP1 through the PDGFR-β pathway, which may lead to vascular remodeling and homeostasis in LPS-induced intravascular injury. This pathway could be a novel target for the treatment of vascular damage.
Keywords: sepsis, endothelial cells, smooth muscle cells, phenotype, Intravascular Injury.
Introduction
Sepsis is a major challenge in public health care. Considerable resources have been invested in developing potential therapies [1]. The current treatment for sepsis is primarily symptomatic support [2]. This is likely a result of the lack of in-depth knowledge of the underlying pathophysiological processes of sepsis. It is well known that vascular dysfunction is a decisive factor in the development of several inflammatory diseases. The mechanisms underlying septic induction of oxidative and nitrosative stresses, the functional consequences of these stresses, and potential adjunct therapies for microvascular dysfunction have been identified [3]. As demonstrated in the literature, the interaction between endothelial cells (ECs) and smooth muscle cells (SMCs) is an essential maturation process in physiological conditions [4, 5]. Generally, in vitro studies are based on single cell cultures, which exclude interactions between different cell types. Previous studies have suggested that ECs could regulate vascular tone through the release of vasoactive molecules such as platelet-derived growth factor (PDGF)-BB [6, 7] and tumor necrosis factor-α (TNF-α) [8]. There are two types of SMCs: contractile and synthetic SMCs. Synthetic SMCs have stronger migratory ability compared with contractile SMCs [9]. Contractile and synthetic SMCs can be distinguished by differences in the expression levels of marked proteins, such as smooth muscle 22 (SM22) and α-smooth muscle actin (α-SMA), which are known as contractile SMC proteins. Studies have shown that the PDGF receptor (PDGFR)-β pathway plays a key role in SMC phenotypic modulation by suppressing the expression of SM22 and α-SMA [10, 11], resulting in a synthetic phenotype that can facilitate the infiltration of inflammatory cells [12].
IQ-domain GTPase-activating protein 1 (IQGAP1) plays a key role in regulating cell migration [13-15]. A previous study demonstrated that IQGAP1 expression was markedly increased in vascular diseases caused by complete removal of the endothelium [16], and that IQGAP1 played a critical role in SMC migration at least in part by increasing PDGFR in focal adhesions, as well as by increasing focal adhesion formation at the leading edge [16]. However, the effects of IQGAP1 on SMC phenotypic transformation and migration following vascular damage caused by sepsis remain unknown. The present study investigated the role of the PDGFRβ/IQGAP1 pathway in EC-mediated SMC phenotypic transformation and migration during sepsis in a co-culture cell model.
Methods
Reagents
Lipopolysaccharide (LPS, Escherichia coli 055: B5, Cat. No. L2880; Sigma-Aldrich) was used to mimic a septic condition. LPS was diluted by phosphate-buffered saline (PBS); The chemical inhibitor imatinib mesylate (Cat. No. S1026; Selleck) was used to inhibit the PDGFR; The first antibodies included anti-IQGAP1 (1:1000; # ab86064, Abcam), anti-α-SMA (1:1000; #A5228; Sigma-Aldrich), anti-SM22 (1:1000; #ab137453; Abcam), anti-GAPDH (1:1000; #5174; Cell Signaling Technology); A horseradish peroxidase (HRP)-conjugated secondary antibody was purchased from Cell Signaling Technology (1:5000, #7074).
Cell Culture and intervention
Human umbilical vein SMCs (Cat. No. 8020) and ECs (Cat. No. 8000) were purchased from ScienCell. SMCs were cultured in basal medium (SMCM, Cat. No.1101; ScienCell), supplemented with 2% fetal bovine serum (FBS, Cat. No. 0010; ScienCell), 1% smooth muscle cell growth supplement (SCGS, Cat.No.1152; ScienCell). ECs were cultured in basal medium (ECM, Cat. No.1001; ScienCell) containing 5% fetal bovine serum (FBS, Cat. No. 0025; ScienCell), and 1% endothelial cell growth supplement (ECGS, Cat. No. 1052; ScienCell). After 1% penicillin/streptomycin (P/S, Cat. No. 0503; ScienCell) was added, they were maintained at 37 °C in a humidified 5% CO2 incubator. Passages 3-8 were used for the experiments.
Co-culture of ECs and SMCs
The co-culture system was established by using the Transwell plates (Cat. No. 3470; Corning)[17], with SMCs were seeded in the lower wells and the ECs were planted in the transwell inserts. Before ECs were co-cultured with SMCs for 24 hours, they both were separately pretreated as follows: culturing with control vehicle (Control), culturing with LPS (LPS), culturing with LPS and imatinib (LPS + imatinib) according to different experiment design, imatinib was given before LPS for 90 min, then SMCs and ECs were extensively washed with PBS to remove excess LPS and/or imatinib which were not taken up. Serum-free mediums were added, in order to exclude any confounding factors contained in the serum.
Enzyme-Linked immunosorbnent assay
Levels of PDGF-BB and TNF-α were determined in the supernatants of different groups using commercial high-sensitivity ELISAs, according to the manufacturer's instructions (Cat. No.EK91372, MultiSciences Biotech, Co., Ltd).
Wound healing assay
SMCs were seeded in six-well plates according to different groups. The cell monolayer was scratched using a 200μl pipette tip before washing three times with PBS to clear cell debris and floating cells. One thousand microliters of serum-free SMCM was then added, and the cells were incubated for 24 h at 37 °C in 5% CO2. Images were captured under a microscope before and after the 24 h incubation at the same position. Migration ability was measured by calculating the rate of scratch wound confluence after 24 h using Adobe Photoshop 2016 software (Adobe Systems Inc.,).
Western Blot Analysis
According to general procedure, western Blot Analysis was performed, briefly, Equal amounts of lysates of cells were applied to 4-12% SDS-PAGE precast gels (Cat. No. NP0335, Invitrogen; Thermo Fisher Scientific), Resolved proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Cat. No. IPVH00010; Merck Millipore), blocked, and then incubated with the primary and second antibody, then the protein bands were visualized by enhanced chemiluminescence kit (Cat. No. 70-P1421; MultiSciences Biotech, Co., Ltd.,) and exposed to X-ray film. The expression of the protein was analyzed by Image J software.
CCK-8 assay
Cells (1 × 105 cells/ml) were grown in 96-well plates and then starved for 24 h before being subjected to treatment according to the experimental requirement, cells were then harvested and washed with PBS and cell counting kit-8 (CCK-8; Dojindo) mixed with FBS-free medium was used for cell viability assay.
Statistical analysis
All results were shown as mean ± SD. Statistical significance was assessed by unpaired Student's t-test or ANOVA, P-values of 0.01 and 0.05 were considered significant. *p < 0.05, **p < 0.01.
Results
Co-culture of ECs and SMCs resulted in higher SMC TNF-α expression
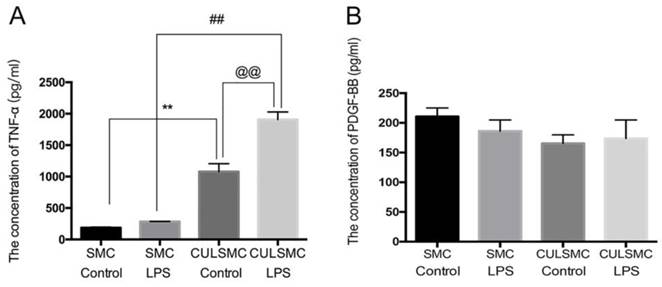
SMCs and ECs were treated according to the experimental design shown in Figure 1. The supernatants were collected from SMCs for an enzyme-linked immunosorbent assay (ELISA), which indicated a slight upward trend in the level of TNF-α in single-culture SMCs treated with lipopolysaccharide (LPS); there was no significant difference between the LPS-treated and control groups. However, in the co-culture system, ECs induced higher TNF-α expression in SMCs compared with single-culture SMCs with or without LPS treatment (mean ± standard deviation, 1077.37 ± 127.90 pg/ml vs. 187.47 ± 10.45 pg/ml; P < 0.01 without LPS treatment; 1907.69 ± 119.79 pg/ml vs. 284.17 ± 1.60 pg/ml; P < 0.01 with LPS treatment). When the co-culture system was stimulated with LPS, TNF-α expression reached the highest level of the four subgroups (Figure 1A). There were no statistical differences in the levels of PDGF-BB in the above-mentioned groups (Figure 1B).
ECs induced higher TNF-α expression in SMCs. (A), The quantification of TNF-α expression in SMCs under single-culture or co-culture system. **: P<0.01, SMCs were treated with control vehicle in single-culture system (SMC Control) vs SMCs were treated with control vehicle in co-culture system (CULSMC Control), ##: P<0.01, SMCs were treated with LPS in single-culture (SMC LPS) vs SMCs were treated with LPS in co-culture (CULSMC LPS), @@: P<0.01, CULSMC Control vs CULSMC LPS; (B), No significant difference in the level of PDGF-BB was shown in the above-mentioned groups.
ECs affect the SMC phenotype in a paracrine manner
To ascertain whether ECs can affect the SMC phenotype in a paracrine manner, single-culture SMCs were treated with control vehicle or LPS for 24 h. After the cell monolayer was scratched, serum-free medium was added to the culture and the cells were incubated for another 24 h. In the co-culture system, SMCs and ECs were treated with or without LPS separately for 24 h prior to co-culture, and the subsequent steps were performed as described above for the single-culture SMCs. The scratch-wound assay revealed that LPS increased the migration ability of SMCs compared with the control group in both the single- and co-culture systems. When SMCs were co-cultured with ECs, the increased migration induced by LPS was alleviated. ECs had no influence on the migration ability of SMCs in the absence of LPS (Figure 2A and 2A1).
SMCs Shift phenotypic transition to synthetic type from contractile type orchestrated by ECs. (A), LPS increased the migration ability of SMCs. When SMCs were co-cultured with ECs, the increased migration induced by LPS was alleviated. (A1), Quantification of A is shown in A1, [% width of injury line = (a - b) × 100%/a; a = Initial scratch wound area at 0 h, b = Scratch wound area at 24 h] **: P<0.01, SMC Control vs SMC LPS, ##: P<0.01, CULSMC Control vs CULSMC LPS, @@: P<0.01, SMC LPS vs CULSMC LPS; (B), LPS induced the reduction of SM22 and α-SMA in SMCs, however, when SMCs were co-cultured with ECs, the reduction of SM22 and α-SMA protein induced by LPS were partly alleviated. Meanwhile, ECs could enhance the expression of SM22 and α-SMA in SMCs in normal condition. (B1 and B2). Quantification of B is shown in B1 and B2, **: P<0.01, SMC Control vs SMC LPS, ##: P<0.01, #: P<0.05, SMC LPS vs CULSMC LPS; CULSMC Control vs CULSMC LPS, @@: P<0.01, SMC LPS vs CULSMC LPS; &: P<0.05, SMC Control vs CULSMC Control.
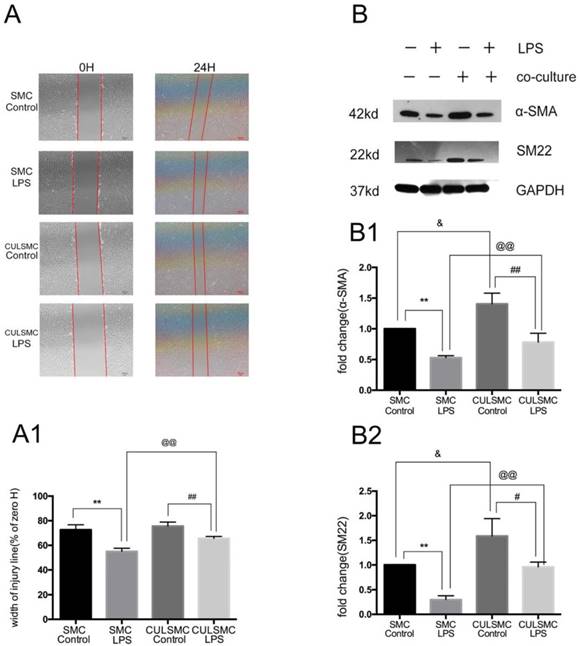
Western blotting demonstrated that LPS treatment for 24 h eventually led to a phenotypic transition characterized by the reduction of SM22 and α-SMA in SMCs. When SMCs were co-cultured with ECs, the protein levels of SM22 and α-SMA were significantly increased; when treated with LPS, the levels of SM22 and α-SMA were somewhat decreased. Additionally, when exposed to LPS, SM22 and α-SMA protein expression in SMCs co-cultured with ECs was markedly higher compared with single-culture SMCs (Figure 2B).
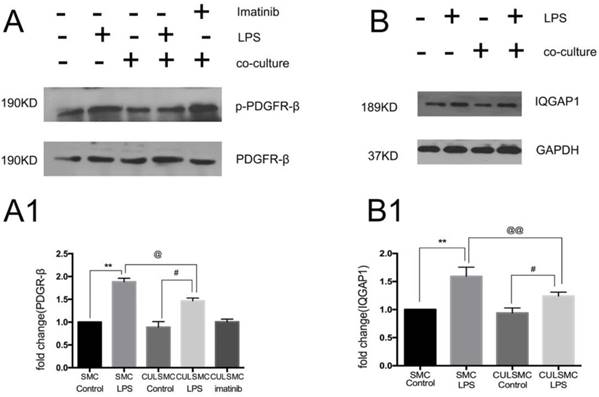
As shown in Figure 3A, when compared to control groups, LPS induced a higher level of phosphorylated PDGFR-β (p-PDGFR-β) in SMCs in both the single and co-culture systems. ECs induced a slight decrease in p-PDGFR-β levels of SMCs in the absence of LPS treatment, but this difference was not statistically significant. With LPS stimulation, p-PDGFR-β levels in co-culture SMCs were remarkably lower than those of single-culture SMCs.
LPS induced a higher higher level of PDGFR-β and IQGAP1 in SMCs in both the single and co-culture systems. (A), LPS induced the level of p-PDGFR-β expression in SMCs in both single and coculture system, but when SMCs were co-cultured with ECs, both control and LPS group experienced the relative lower p-PDGFR-β protein expression than that in single culture system. With LPS stimulation, p-PDGFR-β levels in co-culture SMCs were remarkably lower than those of single-culture SMCs. Quantification of A is shown in A1; **: P<0.01, SMC Control vs SMC LPS, #: P<0.05, CULSMC Control vs CULSMC LPS, @: P≤0.05, SMC LPS vs CULSMC LPS; (B), the change of IQGAP1 expression was mediated by LPS, Quantification of B is shown in (B1); **: P<0.01, SMC Control vs SMC LPS, #: P<0.05, CULSMC Control vs CULSMC LPS, @@: P<0.01, SMC LPS vs CULSMC LPS.
IQGAP1 is involved in SMC differentiation in the co-culture system
As illustrated by western blotting, increased expression of IQGAP1 was observed in LPS-treated SMCs in both the single and co-culture systems. In the co-culture system, the increased expression of IQGAP1 in LPS-treated SMCs was partly alleviated (Figure 3B).
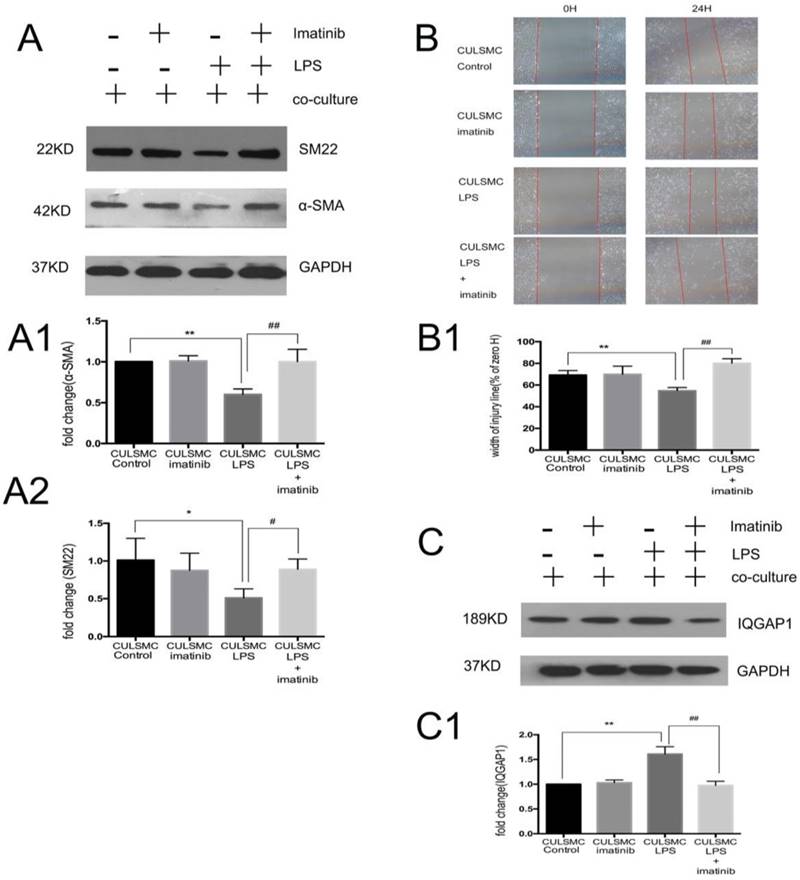
SMCs co-cultured in the absence of LPS were treated with imatinib (Figure 3A). Imatinib had no effect on the levels of p-PDGFR-β; however, as a PDGFR antagonist, the effects of imatinib on LPS induced migratory ability in SMCs. The phenotypic transition of SMCs and expression of IQGAP1 in the co-culture system were measured. We used imatinib at 5 μM for the following experiments; this concentration was confirmed by CCK8 assay (Figure S1). In Figure 4, the scratch-wound assay and western blotting showed that LPS induced a decrease in the expression of α-SMA and SM22, and an increase in the migration ability of SMCs in the co-culture system; these effects were attenuated by the PDGFR-β antagonist (Figure 4A and 4B). Next, IQGAP1 protein expression was determined in the absence and presence of imatinib. The results showed that LPS induced increased expression of IQGAP1 in SMCs, and imatinib prevented this increase in expression (Figure 4C).
Imatinib attenuated vascular smooth muscle cell phenotype switching to a migration state by IQGAP1. (A), The changes of α-SMA and SM22 expression were mediated by imatinib. Quantification of α-SMA is shown in A1; **: P<0.01, CULSMC Control vs CULSMC LPS, ##: P<0.01, CULSMC LPS vs SMCs were treated with LPS and imatinib in co-culture system (CULSMC LPS + imatinib). Quantification of SM22 expression is shown in A2; *: P<0.05, CULSMC Control vs CULSMC LPS, #: P<0.05, CULSMC LPS vs CULSMC LPS + imatinib; (B) LPS induced an increase in the migration ability of SMCs in the co-culture system were attenuated by imatinib. Quantification of B is shown in B1, **: P<0.01, CULSMC Control vs CULSMC LPS, ##: P<0.01, CULSMC LPS vs CULSMC LPS + imatinib; (C) The change of IQGAP1 expression was mediated by imatinib. Quantification of IQGAP1 is shown in C1; **: P<0.01, CULSMC Control vs CULSMC LPS, ##: P<0.01, CULSMC LPS vs CULSMC LPS + imatinib.
Discussion
The most characteristic aspects of sepsis may be its complex pathophysiological processes, in which vascular events are considered prominent; the role of ECs in vascular events has been expanded immensely [18-20]. However, SMCs are another component of blood vessel with a less well-defined role during sepsis, and the relationship between ECs and SMCs has not yet been characterized.
A growing body of literature is indicating that SMCs exhibit considerable phenotypic plasticity [21-23]. When vascular damage occurs, constricted SMCs are responsible for vascular contraction and expansion, and can be transformed into synthetic SMCs, which are characterized by decreased expression of SM22 and α-SMA [21, 24, 25] and increased migration ability. To date, few studies have reported on the involvement of ECs in transformation of the SMC phenotype during sepsis in a co-culture system.
In the present study, LPS treatment resulted in a decrease in the expression of contractile proteins and an increased migration ability in single-culture SMCs, and co-culture of ECs and SMCs resulted in alleviation of the expression of contractile proteins and suppression of migration ability in the presence or absence of LPS. Together, our results provide evidence that LPS induces SMC injury, and that ECs could partly reverse the change in the phenotypic transition and migration of SMCs.
PDGF is a potent mitogen for cells of mesenchymal origin, including fibroblasts, smooth muscle cells, and glial cells [26, 27]. In both mouse and human, the PDGF signaling network consists of five ligands, PDGF-AA through -DD (including -AB), and two receptors, PDGFR-α and PDGFR-β. In general, expression of PDGFRs is low in vivo, but increases dramatically during inflammation. In human pulmonary alveolar epithelial cells, LPS has been shown to induce p42/p44 MAPK activation via the PDGFR/PI3K/Akt pathway [28]. Previous research has revealed that PDGFR-β engages several well-characterized signaling pathways known to be involved in multiple LPS-induced cellular and developmental responses [29-31]. Moreover, Oison et al. found that PDGFR-β signaling is involved in the differentiation of vascular smooth muscle [21]. Similarly, in the present study, when compared to control groups, LPS induced a higher level of p-PDGFR-β in SMCs in both the single and co-culture systems. In co-culture SMCs, imatinib had no effect on p-PDGFR-β levels and could reverse the LPS-mediated higher level of p-PDGFR-β in SMCs.
A previous study found PDGFR-β could be suppressed by TNF-α [32], which is often involved in sepsis, and that TNF-α could reduce cell proliferation in response to PDGF-BB [33]. In the present study, TNF-α only exhibited a slight upward trend, while the expression of PDGFR-β was significantly increased following treatment with LPS, which may be affected by multiple factors related to sepsis. In the absence of LPS stimulation, ECs promoted an increase in TNF-α expression in SMCs and tended to slightly decrease PDGFR-β expression; with LPS stimulation, there was an obvious increase in TNF-α expression and a decrease in PDGFR-β expression. Thus, TNF-α/PDGFR-β may be involved in the interaction between ECs and SMCs in co-culture, especially with LPS stimulation.
PDGF-BB is the highest affinity ligand for PDGFR-β. In a recent study involving a co-culture system, LPS-activated microglia stimulated PDGF-BB expression, enhanced angiogenesis, migration, proliferation, and permeability, and altered the phenotype of co-cultured renal microvascular endothelial cells [34]. Previous studies have demonstrated that PDGF can be induced in ECs in response to injury or stimulus and play a vital role in SMCs and vessel remodeling [35, 36]. However, no statistical differences were observed in PDGF-BB levels between the different groups in this study. We can speculate that not all PDGF family members were detected, only PDGF-BB. A similar hypothesis was proposed by Kim and colleagues; they determined that PDGF-BB levels did not respond to LPS treatment, but that PDGF-AA increased in a dose-dependent manner with LPS stimulation [37]. Therefore, we suggest that the change in PDGFR-β was affected by TNF-α rather than PDGF-BB in our experiment.
In fact, a large corpus of data has shown that PDGFR-β participates in SMC migration [38, 39]. Kohno and colleagues suggested that IQGAP1 might contribute to SMC migration through interaction with PDGFR-β [16]. IQGAP1 mediates protein-protein interactions with a myriad of binding sites that regulate numerous signaling pathways, contributing to its multiple domains. For example, IQGAP1 forms scaffolds with several components of the Akt [15] and ERK [40, 41] pathways to facilitate diverse cellular functions. Further evidence has suggested that Akt signaling may regulate the SMC phenotype [23], and that IQGAP1 operates at both the leading and trailing edge of migrating cells via the ERK pathway [16]. These results prompted us to further explore the regulation of IQGAP1 in response in injury and to determine whether IQGAP1 was involved in SMC differentiation. Consistent with the findings of previous studies [21, 42], increased expression of IQGAP1 was observed in LPS-treated SMCs in both the single and co-culture systems. Expression of IQGAP1 was higher in single culture compared with co-culture SMCs. In the co-culture system, the increased expression of IQGAP1 in LPS-treated SMCs was partly alleviated by the PDGFR-β inhibitor imatinib. When SMCs were treated with imatinib, the decreased expression of α-SMA and SM22, and LPS-induced increased migration of co-cultured SMCs, were attenuated. Thus, we conclude that LPS could induce the phenotypic transition of SMCs to migratory SMCs in the co-culture system via the TNF-α/PDGFR-β/IQGAP1 pathway, and that the different levels of these parameters in the single and co-culture systems may be due to the interaction between ECs and SMCs.
Conclusions
Our findings indicate that the PDGFR-β/IQGAP1 pathway is involved in the interaction between ECs and SMCs, and that TNF-α may regulate PDGFR-β, especially in vascular injury with LPS stimulation. Thus, the interaction between ECs and SMCs serves an important role in vascular homeostasis and remodeling during sepsis. Indeed, this pathway may be a new target for the treatment of vascular damage that occurs with sepsis. Further study of additional signaling pathways involved in EC-mediated SMC phenotypic transformation will be necessary.
Supplementary Material
Supplementary figure S1.
Acknowledgements
The work was supported by Young Science Foundation of National Natural Science Foundation of China (81101445) and Zhejiang Provincial Natural Science Foundation (LY16H150002).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Critical care medicine. 2001;29:1303-10
2. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. Bmj. 2016;353:i1585
3. Cepinskas G, Wilson JX. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. J Clin Biochem Nutr. 2008;42:175-84
4. Jain RK. Molecular regulation of vessel maturation. Nature medicine. 2003;9:685-93
5. Lao KH, Zeng L, Xu Q. Endothelial and smooth muscle cell transformation in atherosclerosis. Curr Opin Lipidol. 2015;26:449-56
6. Labrecque L, Lamy S, Chapus A, Mihoubi S, Durocher Y, Cass B. et al. Combined inhibition of PDGF and VEGF receptors by ellagic acid, a dietary-derived phenolic compound. Carcinogenesis. 2005;26:821-6
7. Rolny C, Nilsson I, Magnusson P, Armulik A, Jakobsson L, Wentzel P. et al. Platelet-derived growth factor receptor-beta promotes early endothelial cell differentiation. Blood. 2006;108:1877-86
8. Zhaowei J, D'Souza MJ, Oettinger CW. Reversal of LPS induced endothelial cell TNF synthesis and increased permeability with microencapsulated antisense oligomers to NF-kappaB. J Microencapsul. 2007;24:596-607
9. Hao H, Gabbiani G, Bochaton-Piallat ML. Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:1510-20
10. McDonald OG, Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100:1428-41
11. Millette E, Rauch BH, Kenagy RD, Daum G, Clowes AW. Platelet-derived growth factor-BB transactivates the fibroblast growth factor receptor to induce proliferation in human smooth muscle cells. Trends Cardiovasc Med. 2006;16:25-8
12. Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15:100-8
13. Brown MD, Sacks DB. IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 2006;16:242-9
14. Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. IQGAP1: a key regulator of adhesion and migration. Journal of cell science. 2005;118:2085-92
15. Yamaoka-Tojo M, Ushio-Fukai M, Hilenski L, Dikalov SI, Chen YE, Tojo T. et al. IQGAP1, a novel vascular endothelial growth factor receptor binding protein, is involved in reactive oxygen species-dependent endothelial migration and proliferation. Circ Res. 2004;95:276-83
16. Kohno T, Urao N, Ashino T, Sudhahar V, Inomata H, Yamaoka-Tojo M. et al. IQGAP1 links PDGF receptor-beta signal to focal adhesions involved in vascular smooth muscle cell migration: role in neointimal formation after vascular injury. American journal of physiology Cell physiology. 2013;305:C591-600
17. Zhao L, Luo H, Li X, Li T, He J, Qi Q. et al. Exosomes Derived from Human Pulmonary Artery Endothelial Cells Shift the Balance between Proliferation and Apoptosis of Smooth Muscle Cells. Cardiology. 2017;137:43-53
18. De Backer D, Ortiz JA, Salgado D. Coupling microcirculation to systemic hemodynamics. Curr Opin Crit Care. 2010;16:250-4
19. Koh IH, Menchaca-Diaz JL, Koh TH, Souza RL, Shu CM, Rogerio VE. et al. Microcirculatory evaluation in sepsis: a difficult task. Shock. 2010;34(Suppl 1):27-33
20. Levi M. Pathogenesis and treatment of disseminated intravascular coagulation in the septic patient. Journal of critical care. 2001;16:167-77
21. Olson LE, Soriano P. PDGFRbeta signaling regulates mural cell plasticity and inhibits fat development. Dev Cell. 2011;20:815-26
22. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN. et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705-10
23. Tang R, Zhang G, Chen SY. Smooth Muscle Cell Proangiogenic Phenotype Induced by Cyclopentenyl Cytosine Promotes Endothelial Cell Proliferation and Migration. J Biol Chem. 2016;291:26913-21
24. Sandbo N, Taurin S, Yau DM, Kregel S, Mitchell R, Dulin NO. Downregulation of smooth muscle alpha-actin expression by bacterial lipopolysaccharide. Cardiovasc Res. 2007;74:262-9
25. Leimgruber C, Quintar AA, Sosa LD, Garcia LN, Figueredo M, Maldonado CA. Dedifferentiation of prostate smooth muscle cells in response to bacterial LPS. Prostate. 2011;71:1097-107
26. Hannink M, Donoghue DJ. Structure and function of platelet-derived growth factor (PDGF) and related proteins. Biochim Biophys Acta. 1989;989:1-10
27. Heldin CH. Structural and functional studies on platelet-derived growth factor. The EMBO journal. 1992;11:4251-9
28. Cho RL, Yang CC, Lee IT, Lin CC, Chi PL, Hsiao LD. et al. Lipopolysaccharide induces ICAM-1 expression via a c-Src/NADPH oxidase/ROS-dependent NF-kappaB pathway in human pulmonary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2016;310:L639-57
29. Ha T, Hua F, Liu X, Ma J, McMullen JR, Shioi T. et al. Lipopolysaccharide-induced myocardial protection against ischaemia/reperfusion injury is mediated through a PI3K/Akt-dependent mechanism. Cardiovasc Res. 2008;78:546-53
30. Zhang L, Li HY, Li H, Zhao J, Su L, Zhang Y. et al. Lipopolysaccharide activated phosphatidylcholine-specific phospholipase C and induced IL-8 and MCP-1 production in vascular endothelial cells. J Cell Physiol. 2011;226:1694-701
31. Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW. et al. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1282-92
32. Molander C, Kallin A, Izumi H, Ronnstrand L, Funa K. TNF-alpha suppresses the PDGF beta-receptor kinase. Exp Cell Res. 2000;258:65-71
33. Battegay EJ, Raines EW, Colbert T, Ross R. TNF-alpha stimulation of fibroblast proliferation. Dependence on platelet-derived growth factor (PDGF) secretion and alteration of PDGF receptor expression. J Immunol. 1995;154:6040-7
34. Ding X, Gu R, Zhang M, Ren H, Shu Q, Xu G. et al. Microglia enhanced the angiogenesis, migration and proliferation of co-cultured RMECs. BMC Ophthalmol. 2018;18:249
35. Drusbosky L, Gars E, Trujillo A, McGee C, Meacham A, Wise E. et al. Endothelial cell derived angiocrine support of acute myeloid leukemia targeted by receptor tyrosine kinase inhibition. Leuk Res. 2015;39:984-9
36. Lin CM, Chiu JH, Wu IH, Wang BW, Pan CM, Chen YH. Ferulic acid augments angiogenesis via VEGF, PDGF and HIF-1 alpha. J Nutr Biochem. 2010;21:627-33
37. Kim TH, Moon JH, Savard CE, Kuver R, Lee SP. Effects of lipopolysaccharide on platelet-derived growth factor isoform and receptor expression in cultured rat common bile duct fibroblasts and cholangiocytes. J Gastroenterol Hepatol. 2009;24:1218-25
38. Tan M, Yan HB, Li JN, Li WK, Fu YY, Chen W. et al. Thrombin Stimulated Platelet-Derived Exosomes Inhibit Platelet-Derived Growth Factor Receptor-Beta Expression in Vascular Smooth Muscle Cells. Cell Physiol Biochem. 2016;38:2348-65
39. Cai Y, Nagel DJ, Zhou Q, Cygnar KD, Zhao H, Li F. et al. Role of cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ Res. 2015;116:1120-32
40. Bardwell AJ, Lagunes L, Zebarjedi R, Bardwell L. The WW domain of the scaffolding protein IQGAP1 is neither necessary nor sufficient for binding to the MAPKs ERK1 and ERK2. J Biol Chem. 2017;292:8750-61
41. Sbroggio M, Bertero A, Velasco S, Fusella F, De Blasio E, Bahou WF. et al. ERK1/2 activation in heart is controlled by melusin, focal adhesion kinase and the scaffold protein IQGAP1. Journal of cell science. 2011;124:3515-24
42. Yang P, Wu J, Miao L, Manaenko A, Matei N, Zhang Y. et al. Platelet-Derived Growth Factor Receptor-beta Regulates Vascular Smooth Muscle Cell Phenotypic Transformation and Neuroinflammation After Intracerebral Hemorrhage in Mice. Critical care medicine. 2016;44:e390-402
Author contact
![]() Corresponding author: X.Z. email: zxicuedu.cn; Postal address: 79 QingChun Road, Hangzhou, Zhejiang, 310003, P.R. China.
Corresponding author: X.Z. email: zxicuedu.cn; Postal address: 79 QingChun Road, Hangzhou, Zhejiang, 310003, P.R. China.