Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(6):872-881. doi:10.7150/ijms.29322 This issue Cite
Research Paper
Differential Analysis of Hypertension-Associated Intestinal Microbiota
Xie Dan1, Zhang Mushi1, Wang Baili2, Lin Han1, Wu Enqi1, Zhao Huanhu1, Li Shuchun1, ![]()
1. School of Pharmacy, Minzu University of China, Key Laboratory of Ethnomedicine (Minzu University of China), Ministry of education, Beijing 100081, P.R. China
2. School Hospital, Minzu University of China, Beijing 100081, China
Received 2018-8-18; Accepted 2019-3-27; Published 2019-6-2
Abstract
Hypertension is the main risk factor for cerebral stroke and death resulting from cerebral stroke. Current association studies on hypertension and intestinal microbiota focus on patients with hypertension (HTN); however, no investigations involving patients with isolated diastolic hypertension (IDH) or systolic hypertension (SH) have been conducted to date. In this study, fecal samples from 62 cases with normal blood pressure (BP) and 67 cases with high BP were used for 16S amplicon sequencing. Sixty-one cases of HTN and 61 corresponding cases with normal BP were obtained by propensity score matching (PSM), and differential analysis was conducted using the DEseq2 package. PSM was also used to match six IDH patients with six controls and to match 35 cases of SH with 35 controls. There were 54 differential genera between the HTN and normal BP groups, and there were five differential genera between the IDH and normal BP groups. There were 38 differential genera between the SH and normal BP groups, including Christensenella. Bayesian network analysis showed that variations in BP influenced microbial abundance. Pearson's correlation analysis showed that bacterial abundance is correlated with BP. Significant differences between the intestinal microbiota of high and normal BP groups were observed. Gut microbiota dysbiosis differed among HTN, IDH, and SH patients. In particular, diastolic blood pressure (DBP) and systolic blood pressure (SBP) were related to different intestinal microbiota.
Keywords: Hypertension, intestinal microbiota, differential species, systolic pressure, diastolic pressure
Background
Hypertension (HTN) is the most common chronic disease and is the main risk factor for cardiovascular and cerebrovascular diseases. Its etiology includes both genetic and environmental factors [1,2]. Environmental factors include lack of exercise, sedentary lifestyle, obesity, tobacco smoking, alcohol drinking, high stress environment, and poor diet. Currently, as the number of patients with HTN has increased, antihypertensive therapy has become a popular topic and a challenge in medical research. The intestinal microbiota is considered the “second genome” of the human body, influencing human health from birth to old age. Intestinal microbes are mainly composed of bacteria, archaea, fungi, and viruses, of which more than 99% are bacteria, mainly including Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria. Studies have shown that intestinal microbes are associated with several diseases, including diabetes [3], cardiovascular disease [4], inflammatory bowel disease [5-7], liver cirrhosis [8], arthritis [9], obesity [10,11], atherosclerosis [12], depression [13], and asthma [14].
It has been proposed that changes in intestinal microbiota may be associated with HTN. Relevant resources include animal experiments and clinical case studies. The aim of these studies was to explore the correlation between microbes and their metabolites and blood pressure (BP). Hypertensive models were used in animal experiments, and fecal samples were collected from clinical hypertension cases or fecal transplantation experiments. Subsequently, the intestinal microbiota was studied using 16S amplicon sequencing and metagenome sequencing, and microbial sequencing data were analyzed using bioinformatics tools. Changes in the composition and abundance of intestinal microbiota in hypertensive patients were observed in all those studies. Studies involving patients with clinical HTN and healthy controls showed that Bifidobacterium, Coprococcus, Roseburia, Clostridium, Faecalibacterium, Butyrivibrio, Enterococcus, Blautia, Oscillibacter, and Synergistetes were present at lower levels in the HTN group than in the control group, whereas Actinomyces, Porphyromonas, Streptococcus, Clostridium, Prevotella, Klebsiella, Desulfovibrio, Parabacteroides, Eggerthella, and Salmonella occurred at higher levels [15-17]. In animal experiments, compared to controls, Bifidobacterium, Rothia, Roseburia, Coprococcus, Pseudobutyrivibrio, Clostridiales (order), Akkermansia, Roseburia, Lactobacillus, Clostridium, Blautia, Anaerotruncus, Ruminococcus, Bifidobasterium, Pseudoflavonifractor, Oscillibacter, Allobaculum, Microtuberculosis, and Verrucomicrobiaceae occurred at lower levels in the HTN model group, whereas Bacteroidetes (phylum), Veillonellaceae (family), Rothia, Veilaceae (family), Streptococcus, Turicibacter, Streptococcaceae (family), Coprobacillus, Lactococcus, Turicibacter, S24-7 (family), Prevotella, and Parasutterella were distributed at higher levels [17-22]. Studies have also shown that microbial populations in the cecum of hypertensive donors could induce hypertension in recipient animals without pre-existing HTN [15,23].
The diagnostic criteria for HTN are SBP > 140 mmHg and/or DBP> 90 mmHg in the absence of antihypertensive medications. Existing studies have only compared the intestinal microbiota of HTN and normotensive people, but lacked differential intestinal microbiota analysis between SH (SBP > 140 mmHg, DBP < 90 mmHg) or IDH (SBP < 140 mmHg, DBP > 90 mmHg) and the normal BP population. In this study, the intestinal microbiota of 67 HTN patients and 62 normal BP individuals were compared. Six IDH patients and six matching normal cases and 35 SH patients and 35 matching normal cases were matched with PSM [24], and the differential microbiotas were analyzed.
Methods
Subjects
All subjects were teachers and staff from the Hospital of Minzu University of China (Beijing, China) who participated in the physical examination and screening of diabetes, HTN, and colorectal cancer. Fecal and blood samples were collected. They completed informed consent forms and were numbered according to the standard code of human genetic resources. Approximately 5 g of stool sample were collected from each participant and placed in a stool stock solution. Height, weight, age, and ethnicity were recorded during sample collection, and these were used in the subsequent screening and analysis of subjects.
Inclusion criteria: Each patient's BP was evaluated based on the diagnostic criteria of the Chinese Guidelines for the Prevention and Treatment of Hypertension 2010. For the normal BP population, 90 mmHg ≤ SBP ≤ 140 mmHg and 60 mmHg ≤ DBP ≤ 90 mmHg. For hypertensive patients, SBP ≥ 140 mmHg or DBP ≥ 90 mmHg. Exclusion criteria: Patients with mental illness, history of drug abuse, cancer, heart failure, renal failure, stroke, or peripheral arterial disease were excluded, as were patients who had received antihypertensive therapy. If they had received antibiotics in the past eight weeks, patients using probiotics or other medications were excluded.
Ethics Statement
This study received approval from the Ethics Committee of Minzu University of China, and written informed consent was obtained from each participant.
DNA Extraction and Sequencing
Total DNA was extracted from the 129 fecal samples. Primers were designed according to the V3+V4 regions of bacteria (the upstream primer was 5'-ACTCCTACGGGAGCAGCA-3', and the downstream primer was 5'-GGACTACHVGGTWTCTAAT-3'). The primers were linked to adapters and used in PCR amplification; the PCR products were purified, quantified, and then normalized to form a sequencing library. These libraries first underwent quality control, and qualified libraries were sequenced by Illumina HiSeq 2500. The raw image data files obtained from the high-throughput sequencing platform (Illumina HiSeq) were analyzed and transformed into raw sequence reads by base calling, and the results were stored as FASTQ files, containing sequence information of reads and their corresponding sequencing qualities.
Bioinformatics Analysis
The data were acquired by pair-end sequencing and were saved as FASTQ files. The sequencing quality of the 129 sequencing samples was evaluated by fastQC, and the correct rate of each base was evaluated based on quality score (Q-score). Because the number of reads per sample differed significantly, to avoid the deviation caused by the different sizes of sample data, each sample with sufficient sequencing depth was subsampled to the minimum number of reads.
After quality control, sequences from the 129 samples were assembled. Usearch (version 9.0) was installed in Quantitative Insights Into Microbial Ecology (QIIME) [25]. Raw data were assembled, filtered, deduplicated, combined, re-deduplicated, and then clustered using the default similarity of 97%. Then, an evolutionary tree was constructed for the representative sequences of operational taxonomic units (OTUs), and a table of OTUs was generated. Based on the valid data, OTU phylogenetic analysis was conducted, and the representative sequence of each OTU was annotated using RDP Classifier [26].
Statistical Analysis
After the table of OTUs with annotation was produced, alpha diversity analysis was performed. A differential analysis of intergroup alpha diversity index was conducted using the R language, and the Wilcoxon rank-sum test was used. Box plots were generated based on the alpha diversity indices, using the ggplot2 package [27] of the R language. Beta diversity analysis was conducted, and a distance matrix was generated using the vegan package [28] of the R language. The distance between the samples was visualized on a principal component analysis (PCoA) plot. After correcting other confounding factors by using PSM with the language R, significantly different genera were mined using the DEseq2 package of the language R and then visualized with a volcano plot using a ggplot2 package. Figtree (version 1.4.3) was used to construct a dendrogram, and a Bayesian network was used to show the association between the differential microbiotas and disease. Pearson's correlation test was used to analyze the correlation between the differential microbiotas and BP. The data were expressed as the mean ± standard deviation (SD); P < 0.05 and q < 0.05 indicated statistical significance.
Results
Physical and biochemical indices of the included groups
A total of 62 cases of HTN and 67 cases of normal BP were included in this study. Clinical data, including age, SBP, DBP, body mass index (BMI), waist-to-hip ratio (WHR), glucosuria (GLU), high density lipoprotein (HDL), low density lipoprotein (LDL), total triglyceride (TG), total cholesterol (CHO), blood urea nitrogen (BUN), uric acid (UA), and homocysteine (HCY). Other than SBP and DBP, the other clinical parameters showed no significant differences between groups (Table 1).
Characteristics of subjects
| Hypertension | Normal blood pressure | P-value | |
|---|---|---|---|
| Sample | 62 | 67 | |
| Gender (% Female) | 62 (53.23%) | 67 (61.19%) | 0.360 |
| Age, year | 69.322 ± 10.613 | 69.492 ± 9.630 | 0.509 |
| SBP, mmHg | 122.935 ± 6.902 | 153.298 ± 14.917 | 0.000* |
| DBP, mmHg | 76.209 ± 6.902 | 84.313 ± 10.739 | 0.000* |
| BMI, kg/m2 | 26.089 ± 3.112 | 25.051 ± 4.436 | 0.169 |
| WHR | 0.864 ± 0.065 | 0.853 ± 0.130 | 0.645 |
| GLU, mmol/L | 6.729 ± 1.956 | 6.030 ± 1.176 | 0.059 |
| CHO, mmol/L | 5.249 ± 1.022 | 5.093 ± 1.062 | 0.443 |
| HDL, mmol/L | 1.403 ± 50.827 | 1.441 ± 0.346 | 0.496 |
| LDL, mmol/L | 1.986 ± 1.250 | 3.182 ± 0.865 | 0.322 |
| TG, mmol/L | 1.986 ± 1.250 | 1.812 ± 1.206 | 0.336 |
| BUN, mmol/L | 5.373 ± 1.285 | 5.739 ± 1.418 | 0.181 |
| UA, μmol/L | 344.709 ± 89.964 | 346.224 ± 76.830 | 0.386 |
| HCY, μmol/L | 13.940 ± 6.080 | 13.150 ± 3.620 | 0.607 |
The data on age, SBP, DBP, BMI, WHR, GLU, CHO, HDL, LDL, TG, BUN, UA, and HCY are expressed as the mean ± SD. P-values for gender, age, SBP, DBP, BMI, WHR, GLU, CHO, HDL, LDL, TG, BUN, UA, and HCY were calculated using Student's t-test. *P < 0.05. SBP: systolic blood pressure, DBP: diastolic blood pressure, BMI: body mass index, WHR: waist-to-hip ratio; GLU: glucosuria; HDL: high density lipoprotein; LDL: low density lipoprotein; TG: total triglyceride, CHO: total cholesterol; BUN blood urea nitrogen; UA: uric acid; HCY: homocysteine. *P < 0.05.
Diversity Analysis
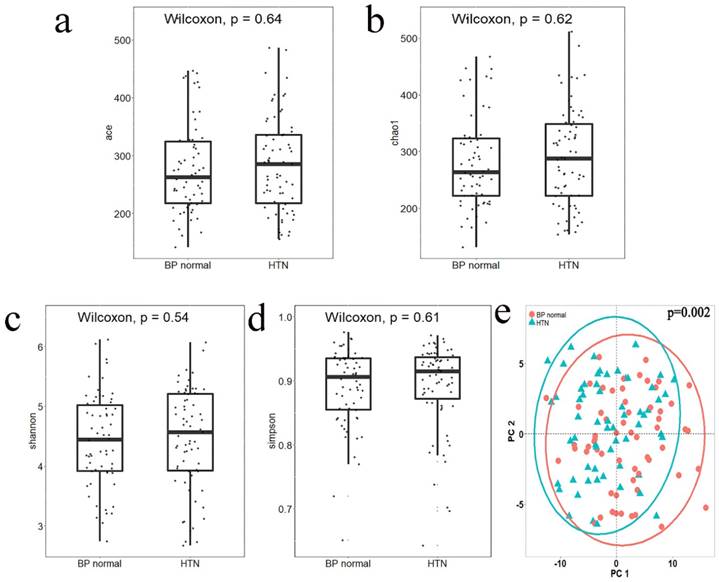
Alpha diversity reflects the species diversity within a single sample. Four indices of alpha diversity were analyzed by QIIME and plotted into box plots of Ace (Figure 1a), Chao1 (Figure 1b), Shannon (Figure 1c), and Simpson (Figure 1d). The results of a Wilcoxon rank-sum test showed no significant differences between the alpha diversity indices of the two groups. Beta diversity analysis was used to compare the differences in species diversity between the two groups. A Jaccard distance matrix was generated using the table of differential OTUs and then plotted into a PCoA graph (Figure 1e). Significant differences between the two groups were detected by the Adonis test, indicating that the difference between groups was greater than that within groups (P = 0.002).
The alpha diversity index box plot and beta diversity PCoA plot of HTN and normal blood pressure (BP normal) groups; significance was determined by Wilcoxon rank-sum test. a. Ace index, box plot generated by R (version 3.5.0). b. Chao1 index, box plot generated by R (version 3.5.0). c. Shannon index, box plot generated by R (version 3.5.0). d. Simpson index, box plot generated by R (version 3.5.0). e. PCoA plot according to the Jaccard distance matrix generated from the table of differential OTUs, plotted by the ggplot2 package of the language R. Groups were compared using Adonis.
Differential microbiota between the HTN and normal BP groups
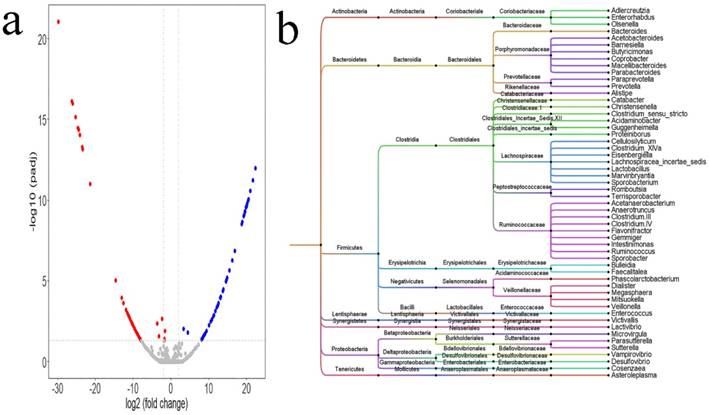
Alternations in intestinal microbiota were influenced by a variety of confounding factors. PSM in the language R was adopted to correct these confounding factors. After PSM was completed, differential analysis was conducted between the two groups using the DEseq2 package of the language R. Based on the table of OTUs, a total of 98 differential OTUs (Figure 2a, Supplemental Table 1) between the two groups were identified. The 98 OTUs were clustered and visualized using Figtree, and the clustering results (Figure 2b) showed that most of the differential genus were clustered to the Firmicutes and Bacteroidetes phyla, and Ruminococcaceae, Prevotellaceae, Porphyromonadaceae, Lachnospiraceae, Veillonellaceae families. At the genus level, there were 54 differential genera between the two groups: 18 genera, namely, Acetobacteroides, Alistipes, Bacteroides, Barnesiella, Butyricimonas, Christensenella, Clostridium sensu stricto, Cosenzaea, Desulfovibrio, Dialister, Eisenbergiella, Faecalitalea, Megasphaera, Microvirgula, Mitsuokella, Parabacteroides, Proteiniborus, and Terrisporobacter showed a higher abundance in the HTN group; and 36 genera, namely, Acetobacteroides, Acidaminobacter, Adlercreutzia, Anaerotruncus, Asteroleplasma, Bulleidia, Cellulosilyticum, Clostridium III, Clostridium IV, Clostridium XlVa, Coprobacter, Enterococcus, Enterorhabdus, Flavonifractor, Gemmiger, Guggenheimella, Intestinimonas, Lachnospiracea_incertae_sedis, Lactivibrio, Lactobacillus, Macellibacteroides, Marvinbryantia, Olsenella, Paraprevotella, Parasutterella, Phascolarctobacterium, Prevotella, Romboutsia, Ruminococcus, Sporobacter, Sporobacterium, Sutterella, Vampirovibrio, Veillonella, and Victivallis showed higher abundance in the normal BP group.
Differential microbiota between HTN group and normal blood pressure (BP) group. a. The differential OTUs between the HTN group and the normal BP group were analyzed using the DEseq2 package of the language R, and OTUs in accordance with corrected P value < 0.05 and fold-change > 2 or fold change < 0.5 were identified. There were 54 OTUs with higher abundances in the HTN group, and 44 OTUs with higher abundances in the normal BP group. A volcano plot was generated by Vegan package. b. The differential OTUs between the HTN and normal BP groups were clustered into 54 genera. Bayesian networks between the SH and normal BP groups, and between the IDH and normal BP groups. a. Analysis of differential microbiota between the HTN and normal BP groups using the DEseq2 package of the language R. The dendrogram was plotted using Figtree (version 1.4.3).
Differential Microbiota between IDH and Normal BP Groups
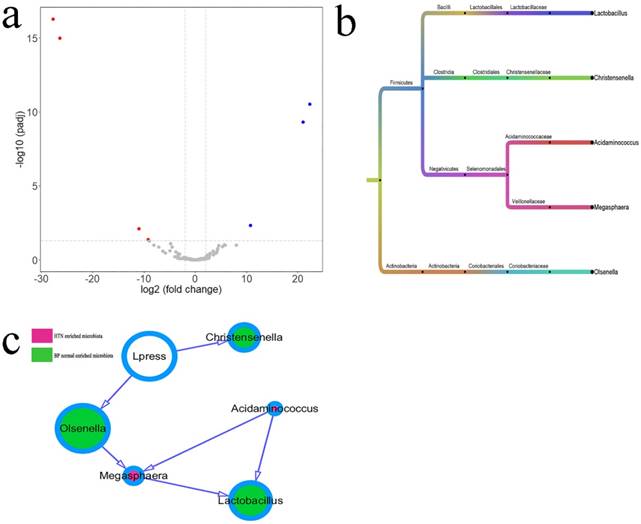
The different intestinal OTUs in six cases of IDH and six matched cases of normal BP were analyzed by the DEseq2 package (Figure 3a, Supplemental Table 2). According to the clustering of differential OTUs, there were five differential microbiotas between the two groups. Acidaminococcus and Megasphaera showed higher abundances in patients with HTN, and Christensenella, Lactobacillus, and Olsenellathose showed higher abundances in individuals with normal BP (Figure 3b). The relationship between microbiota and diastolic pressure was inferred using a Bayesian network, and visualization using Cytoscape showed that variations in diastolic BP could change the abundances of Christensenella and Olsenella (Figure 3c).
Differential microbiotas between the IDH group and normal blood pressure (BP) group and their associations. a. The differential OTUs between the IDH and normal BP groups were analyzed using the DEseq2 package of the language R, and OTUs in accordance with corrected P value < 0.05 and fold-change > 2 or fold change < 0.5 were identified. There were four OTUs with higher abundances in the IDH group, and three OTUs with high abundances in the normal BP group. A volcano plot was generated by Vegan package. b. The differential OTUs between the IDH group and normal BP group were clustered into five genera. c. The associations between microbiotas were deduced by a Bayesian network, and the network diagram was generated using Cytoscape (version 3.6.1). The relative size of the circles indicates relative abundance. c. Differences in the microbiota between the IDH group and normal BP group were analyzed using the DEseq2 package of the language R. The associations between microbiotas were deduced by a Bayesian network, and the network diagram was generated by Cytoscape (version 3.6.1).
Differential Microbiota between the SH and Normal BP Groups
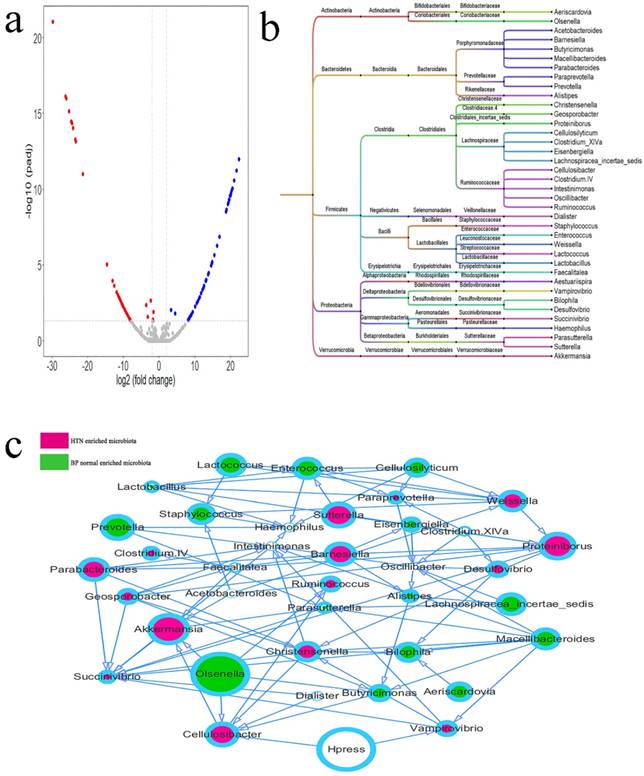
The differential intestinal OTUs in 35 cases of SH and 35 matched cases of normal BP were analyzed using the DEseq2 package (Figure 4a, Supplemental Table 3). According to the clustering of differential OTUs, there were 38 differential microbiotas between the two groups. Acetobacteroides, Aestuariispira, Akkermansia; Barnesiella, Cellulosibacter, Christensenella, Clostridium IV, Clostridium XlVa, Desulfovibrio, Dialister, Faecalitalea, Geosporobacter, Intestinimonas, Parabacteroides, Paraprevotella, Proteiniborus, Ruminococcus, Succinivibrio, Sutterella, Vampirovibrio, and Weissella showed higher abundances in hypertensive patients, and Aeriscardovia, Alistipes, Bilophila, Butyricimonas, Cellulosilyticum, Eisenbergiella, Enterococcus, Haemophilus, Lachnospiracea_incertae_sedis, Lactobacillus, Lactococcus, Macellibacteroides, Olsenella, Oscillibacter, Parasutterella, Prevotella, and Staphylococcus showed higher abundance in the normal BP group (Figure 5b). The relationship between microbiota and systolic pressure was inferred by the Bayesian network, and visualization using Cytoscape showed that variations in SBP could change the abundances of Cellulosibacter and Vampirovibrio (Figure 3c).
Differential microbiotas between the SH group and normal blood pressure (BP) group and their associations. a. The differential OTUs between the SH and normal BP groups were analyzed using the DEseq2 package of the language R, and OTUs in accordance with corrected P value < 0.05 and fold-change > 2 or fold-change < 0.5 were screened. There were 23 OTUs with high abundances in the SH group, and 32 OTUs with high abundances in the normal BP group. A volcano plot was generated by Vegan package. b. The differential OTUs between the SH and normal BP groups were clustered into 38 genera. c. The associations between microbiotas were deduced by a Bayesian network, and the network diagram was generated by Cytoscape (version 3.6.1). The relative size of each circle represents the relative abundance.
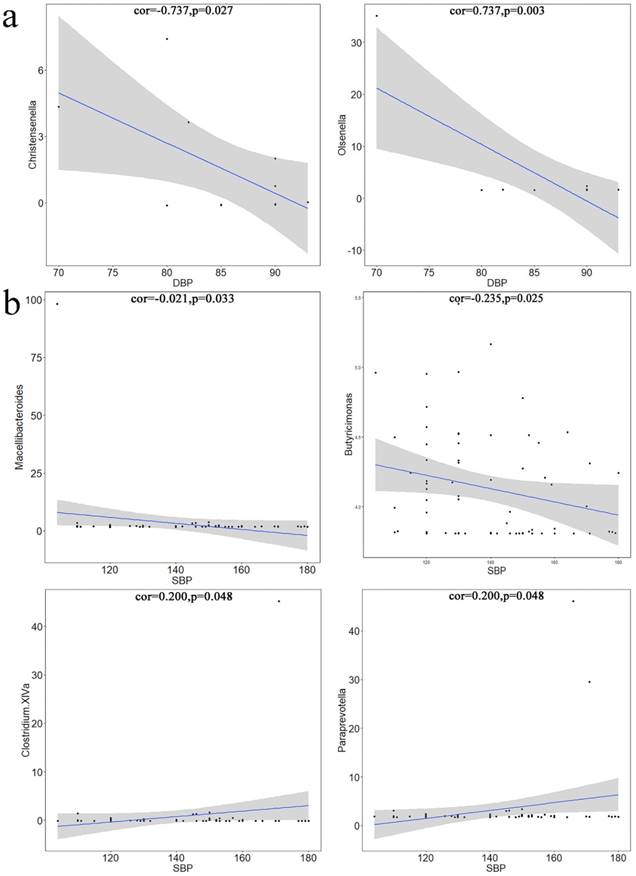
Pearson's correlation coefficients between differential microbiotas and blood pressure (BP). a. Differential microbiota was significantly correlated with DBP. Christensenella and Olsenella were negatively correlated with the systolic pressure. The X-axis represents diastolic pressure, and the Y-axis represents the abundance of bacteria. Data were plotted using the language. b. differential microbiota was significantly correlated with SBP. The X-axis represents the diastolic pressure, and the Y-axis represents the abundance of bacteria. Data were plotted using the language R. Macellibacteroides was significantly negatively correlated with diastolic pressure, whereas Butyricimonas, Clostridium XlVa, and Paraprevotella were significantly positively correlated with diastolic pressure.
Pearson's Correlation Coefficient
Using Pearson's correlation test, microbiotas that were significantly related to BP variation were identified from the differential microbiotas and plotted. Christensenella (Figure 5a1) and Olsenella (Figure 5a2) were significantly correlated with SBP; Macellibacteroides (Figure 5b1) and Butyricimonas (Figure 5b2) were significantly negatively correlated with DBP; Clostridium XIVa (Figure 5b3) and Paraprevotella (Figure 5b4) were significantly positively correlated with DBP.
Discussion
Over the past three years, the potential of intestinal microbiota dysbiosis causing HTN has become a popular research topic, and most studies have shown a correlation between intestinal microbiota and HTN [15-23]. Comparative studies of individuals with HTN or models of HTN against control groups have shown genera with different abundance among groups [15,16,19-21,23]. Transplanting fecal bacteria from a HTN group into germ-free mice increased the BP of the recipient mice [15]). However, due to the differences among models (spontaneously hypertensive rats, SHR) [19], angiotensin II perfusion model (Ang II), and fecal bacteria transplantation to germ-free mice [15,29]), populations, sequencing techniques (16S amplicon sequencing and metagenome sequencing), and analytical methods, these studies have generated conflicting results. For example, Yan et al. [16] showed a higher abundance of Clostridium in a HTN group than in a control group, but Li et al. [15] found that Clostridium was less abundant in their HTN group than in their control group.
At the genus level, Parabacteroides showed a higher abundance in our HTN and SH groups, which was consistent with the results of Yan et al. [16]. Parabacteroides is usually an opportunistic pathogen in infectious diseases and can develop antimicrobial resistance [30], and the mechanism of Parabacteroides in the pathogenesis of HTN is unclear. In the present study, Desulfovibrio showed higher abundance in the HTN and SH groups, which was consistent with the findings of Li et al. [15]. In a study on gestational diabetes, the intestinal microbiota of patients with gestational diabetes was abnormal at the phylum level compared to pregnant women with normal blood glucose, and Desulfovibrio also showed higher abundance in the patients with diabetes [31]. Because Desulfovibrio can reduce sulfate to hydrogen sulfide (H2S), and H2S inhibits the epithelial oxidation of butyrate, which is the main energy source of the epithelium, it has been suggested that the possible mechanism of this bacterium leading to HTN may be related to the vascular epithelial changes caused by its metabolites. Christensenella showed higher abundance in the HTN and SH groups, and was negatively correlated with systolic pressure; however, it was less abundant in the IDH group, indicating that Christensenella had a bidirectional effect. This bacterium has been reported in a previous study of Parkinson's disease, in which the abundance of Christensenella was higher in the disease group [32]. Alistipes has been purported to play a role in ameliorating obesity [10], and it showed higher abundance in the HTN group in our study. Kim et al. [22] found that Alistipes could induce intestinal inflammation, suggesting a strong association between intestinal changes and HTN.
BP alterations are always accompanied by changes in short chain fatty acids (SCFAs) [33,34]. SCFAs interact with G protein-coupled receptors (GPCRs) such as G protein coupled receptor 41 (Gpr41) and olfactory receptor 78 (Olfr78) to influence host cells [35]. The relevance of SCFAs to BP regulation was demonstrated in a previous study by Pluznick et al. [36]. Among bacteria that generate SCFAs, the butyric acid-producing bacteria Anaerotruncus was present at a lower abundance in our HTN group, and Prevotella was present at a lower abundance in the HTN and SH groups, which contradicted the results of previous reports [15]. Interestingly, Butyricimonas showed a higher abundance in the SH group. Butyricimonas was significantly negatively correlated with DBP by Pearson's correlation test, but its abundance was lower in the HTN group. Therefore, the abundance of Butyricimonas might be related to the stages of HTN.
Lactobacillus, which can inhibit the reproduction of pathogenic bacteria, showed higher abundance in the normal BP group, which was consistent with the results reported by Wilck et al. [17]. That group of researchers studied the feeding behavior of mice with Lactobacillus. The Lactobacillus rhamnosus GG strain mitigated the development of obstructive sleep apnea-induced hypertension in the situation of a high salt diet via regulating trimetlylamine oxide (TMAO) level and CD4+ T cell induced-type I inflammation [37].
The lower Oscillibacter abundance in the SH group was consistent with the findings of previous studies [15]. This genus might generate metabolites that directly regulate the integrity of epithelium, or it might have a double effect following changes in the overall composition of the microbiota [38].
One of the important points in this study is the application of PSM. Several previous studies are associated with data deviations and confounding variables. The PSM was designed to reduce the effects of these deviations and confounding variables to conduct a reasonable comparison between groups. The intestinal microbiota was affected by various factors, including diet, age, gender, BMI, and WHR, and the application of PSM eliminated confounding factors between groups.
This study has a number of limitations. The study population consisted of faculty and thus does not represent the general population, and there were fewer IDH cases than SH cases. Metabolites in the intestinal microbiota of hypertensive patients undergo changes compared to those with normal BP [15], suggesting that intestinal microbiota metabolites play a role in the pathogenesis of HTN [39]. However, this study did not measure metabolites, although we plan to investigate this matter in subsequent studies.
Studies have shown a correlation between intestinal microbiota and HTN. In most studies, only bacteria that might be associated with HTN were identified, but none of them has been confirmed to be associated with HTN. One possible solution is to identify bacteria associated with HTN using larger clinical cohorts or animal experiments. In addition, a large cohort of HTN patients should be investigated to study the causal relationship between HTN and intestinal microbiota, which will provide new prospects for the treatment and prevention of HTN and associated diseases.
Supplementary Material
Supplementary tables.
Acknowledgements
The Key Laboratory of Ethnomedicine (Minzu University of China) and the Ministry of Education (No. KLEM-ZZ201801) supported this study.
Authors' Contributions
Xie Dan and Zhang Mushi performed the experiments; Xie Dan, Zhao Huanhu, and Wu Enqi analyzed the data; Li Shuchun obtained funding for the project and planned the experiments; and Xie Dan, Zhang Mushi, Wang Baili, and Lin Han collected samples. All authors have read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. He FJ, Li J, Macgregor GA. Effect of longer-term modest salt reduction on blood pressure. The Cochrane database of systematic reviews. 2013(4):CD004937
2. Kato N, Takeuchi F, Tabara Y. et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nature genetics. 2011;43(6):531-538
3. Forslund K, Hildebrand F, Nielsen T. et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528(7581):262-266
4. Garcia-Rios A, Torres-Pena JD, Perez-Jimenez F. et al. Gut Microbiota: A New Marker of Cardiovascular Disease. Current pharmaceutical design. 2017;23(22):3233-3238
5. Thorburn AN, Macia L, Mackay CR. Diet, metabolites, and "western-lifestyle" inflammatory diseases. Immunity. 2014;40(6):833-842
6. Goto Y, Kurashima Y, Kiyono H. The gut microbiota and inflammatory bowel disease. Current opinion in rheumatology. 2015;27(4):388-396
7. Walujkar SA, Kumbhare SV, Marathe NP. et al. Molecular profiling of mucosal tissue associated microbiota in patients manifesting acute exacerbations and remission stage of ulcerative colitis. World journal of microbiology & biotechnology. 2018;34(6):76
8. Qin N, Yang F, Li A. et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513(7516):59-64
9. Scher JU, Sczesnak A, Longman RS. et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife. 2013;2:e01202
10. Ridaura VK, Faith JJ, Rey FE. et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214
11. Turnbaugh PJ, Ley RE, Mahowald MA. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027-1031
12. Koeth RA, Wang Z, Levison BS. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature medicine. 2013;19(5):576-585
13. Foster JA, McVey Neufeld KA. Gut-brain axis: how the microbiome influences anxiety and depression. Trends in neurosciences. 2013;36(5):305-312
14. Venkatesan P. Gut microbiota and the risk of childhood asthma. The Lancet. Respiratory medicine. 2015;3(11):843
15. Li J, Zhao F, Wang Y. et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5(1):14
16. Yan Q, Gu Y, Li X. et al. Alterations of the Gut Microbiome in Hypertension. Frontiers in cellular and infection microbiology. 2017;7:381
17. Wilck N, Matus MG, Kearney SM. et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551(7682):585-589
18. Santisteban MM, Qi Y, Zubcevic J. et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circulation research. 2017;120(2):312-323
19. Yang T, Santisteban MM, Rodriguez V. et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65(6):1331-1340
20. Mell B, Jala VR, Mathew AV. et al. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiological genomics. 2015;47(6):187-197
21. Durgan DJ, Ganesh BP, Cope JL. et al. Role of the Gut Microbiome in Obstructive Sleep Apnea-Induced Hypertension. Hypertension. 2016;67(2):469-474
22. Kim S, Goel R, Kumar A. et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clinical science. 2018;132(6):701-718
23. Adnan S, Nelson JW, Ajami NJ. et al. Alterations in the gut microbiota can elicit hypertension in rats. Physiological genomics. 2017;49(2):96-104
24. Austin PC. The performance of different propensity-score methods for estimating differences in proportions (risk differences or absolute risk reductions) in observational studies. Statistics in medicine. 2010;29(20):2137-2148
25. Caporaso JG, Kuczynski J, Stombaugh J. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7(5):335-336
26. Wang Q, Garrity GM, Tiedje JM. et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology. 2007;73(16):5261-5267
27. Ito K, Murphy D. Application of ggplot2 to Pharmacometric Graphics. CPT: pharmacometrics & systems pharmacology. 2013;2:e79
28. Philip D. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science. 2003;14(6):927-930
29. Jama HA, Kaye DM, Marques FZ. The gut microbiota and blood pressure in experimental models. Current opinion in nephrology and hypertension. 2019;28(2):97-104
30. Boente RF, Ferreira LQ, Falcao LS. et al. Detection of resistance genes and susceptibility patterns in Bacteroides and Parabacteroides strains. Anaerobe. 2010;16(3):190-194
31. Crusell MKW, Hansen TH, Nielsen T. et al. Gestational diabetes is associated with change in the gut microbiota composition in third trimester of pregnancy and postpartum. Microbiome. 2018;6(1):89
32. Petrov VA, Saltykova IV, Zhukova IA. et al. Analysis of Gut Microbiota in Patients with Parkinson's Disease. Bulletin of experimental biology and medicine. 2017;162(6):734-737
33. Felizardo RJF, Watanabe IKM, Dardi P. et al. The interplay among gut microbiota, hypertension and kidney diseases: The role of short-chain fatty acids. Pharmacological research. 2019;141:366-377
34. de la Cuesta-Zuluaga J, Mueller NT. Higher Fecal Short-Chain Fatty Acid Levels Are Associated with Gut Microbiome Dysbiosis, Obesity, Hypertension and Cardiometabolic Disease Risk Factors. Nutrients. 2018:11 (1)
35. Pluznick JL. Microbial Short-Chain Fatty Acids and Blood Pressure Regulation. Current hypertension reports. 2017;19(4):25
36. Pluznick JL, Protzko RJ, Gevorgyan H. et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110(11):4410-4415
37. Liu J, Li T, Wu H. et al. Lactobacillus rhamnosus GG strain mitigated the development of obstructive sleep apnea-induced hypertension in a high salt diet via regulating TMAO level and CD4(+) T cell induced-type I inflammation. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2019;112:108580
38. Lam YY, Ha CW, Campbell CR. et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PloS one. 2012;7(3):e34233
39. Natarajan N, Hori D, Flavahan S. et al. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiological genomics. 2016;48(11):826-834
Author contact
![]() Corresponding author: ShuChun Li, MinZu University of China, 27 ZhongGuanCun South Street, 100081, Beijing, China. Tel: 86-10-6893-3254; Fax: 86-10-6893-9905; E-mail: jasonedu.cn
Corresponding author: ShuChun Li, MinZu University of China, 27 ZhongGuanCun South Street, 100081, Beijing, China. Tel: 86-10-6893-3254; Fax: 86-10-6893-9905; E-mail: jasonedu.cn