Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(6):854-863. doi:10.7150/ijms.31690 This issue Cite
Research Paper
Hepatocyte growth factor plays a particular role in progression of overall cardiac damage in experimental pulmonary hypertension
Michal Radik, Zuzana Kmecova, Jana Veteskova, Eva Malikova, Gabriel Doka, Peter Krenek ![]() , Jan Klimas
, Jan Klimas
Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University in Bratislava, Odbojarov 10, 83232 Bratislava, Slovak Republic
Received 2018-11-21; Accepted 2019-3-27; Published 2019-6-2
Abstract
Background: HGF/MET pathway may have a role in pulmonary hypertension (PH). However, the link between the pathway and development of target organ damage in PH remains elusive. We aimed to demonstrate the relation between plasma HGF and HGF/MET tissue expressions in affected organs during PH progression.
Methods: 12 weeks old male Wistar rats were injected with monocrotaline (MCT, 60 mg/kg, s.c.) to induce PH and sacrificed after 1, 2 and 4 weeks. Controls received saline. mRNA levels of HGF regulatory complex (Hgf, Met, Hgfa, Hai-1, Hai-2) were determined in right and left ventricles (RV, LV), lungs, pulmonary artery and liver by RT-qPCR. HGF protein levels in plasma were analysed by ELISA.
Results: PH development was associated with a progressive elevation of HGF plasma levels that correlated with relative RV mass. Furthermore, Hgf mRNA expressions at week 4 were upregulated solely in the cardiac ventricles while being downregulated in a. pulmonalis, lungs and liver. Met and Hai-1/Hai-2 followed a similar pattern and were upregulated in cardiac ventricles, where Hgfa remained unchanged, but downregulated in lungs.
Conclusion: We suggest that cardiac overexpression of Hgf might contribute to increased plasma HGF in MCT-induced PH. HGF could be exploited as a cardiospecific biomarker and HGF/MET pathway as a target in drug discovery for PH.
Keywords: HGF, MET receptor, pulmonary hypertension, monocrotaline, biomarker
Introduction
Pulmonary hypertension (PH) is a rare, progressive disease with poor prognosis and limited therapeutic options [1]. It is assessed by functional tests [1], but a reliable and specific prognostic biomarker is lacking. PH pathogenesis involves processes, which progressively increase pulmonary vascular resistance leading to myocardial remodeling and failure of the right ventricle (RV) [2]. This outcome is driven by alterations in cytokines and growth factors [3], including the hepatocyte growth factor (HGF) that via its receptor MET (mesenchymal-epithelial transition) promotes proliferation and morphogenesis as well as anti-apoptotic and anti-fibrotic effects on various types of cells, including cardiomyocytes [4]. HGF activity is managed by its set of endogenous regulators, namely HGF activator [5], and respective inhibitors of HGF activator, HAI-1 [6] and HAI-2 [7]. HGFA is the main factor responsible for splitting pro-HGF form to a mature, biologically active HGF protein [8], while HAI-1/HAI-2 are binding HGFA in an intrinsic inhibitory mechanism [6, 7]. HGF/MET are involved in tissue repair [9]. In diseases like pulmonary arterial hypertension (PAH) [10], lung fibrosis [11], myocardial infarction [12], and heart failure [13], exogenous HGF or gene transfer was protective and attenuated disease progression. HGF activates pathways known to be involved in PH pathophysiology and target organ damage [14].
Since PH is often diagnosed only at an advanced stage of disease [15], the lack of specific biomarker for detection of early stages of PH represents a crucial problem. HGF has been suggested as a potential predictor of mortality in heart failure patients [16], hypertension severity [17] and could also be relevant in diagnostics of PH [18]. HGF plasma levels correlated with mean pulmonary arterial pressure and HGF was detectable already at early stages of the disease [19], making it a potential biomarker candidate. However, its relation to target organ damage during disease progression remains unclear.
We hypothesize, that HGF/MET system dysregulation could reflect development and progression of target organ damage in PH. We also hypothesize, that such a dysregulation would be accompanied by elevated plasma HGF levels when right ventricular pressure (RVP) increases. We aim to evaluate the significance of plasma levels of HGF as a potential PH biomarker and to investigate a possible link of HGF plasma level alteration to Hgf and Met gene expressions in affected tissues in the monocrotaline (MCT)-induced rat model of PH.
Materials and Methods
Animal experiment design
12 weeks old male Wistar rats (Dobra Voda, Slovak Republic) were randomized according to experiment duration: 1, 2, 4 weeks (1W, 2W, 4W) and type of treatment (saline - CON, monocrotaline - MCT). Rats were subcutaneously injected with 60 mg/kg dose of monocrotaline [20], or saline. In experimental model of MCT-induced PH in rat, males are preferred over females as they are more susceptible to MCT toxicity than female rats [21]. Animals were sacrificed 1, 2 and 4 weeks after MCT injection, to study different PH progression stages. Procedures involving the use of animals were approved by the Ethics Committee of the Faculty of Pharmacy, Comenius University in Bratislava, Slovak Republic and the State Veterinary and Food Administration of the Slovak Republic. The investigations were conducted in accordance with NIH Guide for the Care and Use of Laboratory Animals: Eight Edition (2010) published by the US Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council, the EU adopted Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for experimental and other scientific purposes and the Slovak law regulating animal experiments.
Right ventricular pressure measurement
RVP was measured by RV catheterization using a polyethylene catheter, filled with heparinized saline and connected to the pressure transducer (Spel Advanced HaemoSys, Experimetria Ltd., Hungary). The catheter was advanced into the RV via right jugular vein under tribromoethanol anaesthesia [22].
Collection of samples
Rats were sacrificed in CO2 atmosphere. Blood was collected from caudal vena cava using EDTA as an anticoagulant, plasma was separated by centrifugation and stored at -80 °C. Cardiac ventricles, lungs and livers were blotted dry and weighed. Relative organ weights were calculated compared to total body weights and ratios were used as measures of organ damage. Samples of LV, RV, a. pulmonalis, lungs and liver were frozen in liquid nitrogen and stored at -80 °C until further processing.
ELISA
To measure HGF protein plasma concentrations, the Quantikine® ELISA Mouse/Rat HGF Immunoassay MHG00 (R&D Systems, USA) was used according to the manufacturer's instructions. The assay uses a quantitative sandwich enzyme immunoassay technique and detects natural and recombinant HGF, with less than 0.5% cross-reactivity and no significant interference with related molecules.
RT-qPCR
Total RNA was isolated from tissues using Tri Reagent® (Sigma-Aldrich, USA). RNA quality was verified by gel electrophoresis and quantified by spectrophotometry (NanoDrop® ND-1000, Thermo Fisher Scientific, USA). Reverse transcription was performed using High Capacity cDNA Reverse Transcription Kit with RNAse inhibitor (Thermo Fisher Scientific, USA). Quantitative real-time PCR was performed using SYBR™ Select Master Mix (Thermo Fisher Scientific, USA) on StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific, USA). Hgf, Met, Hgfa, Hai-1, Hai-2, Nppa, and Nppb mRNA levels were evaluated using gene-specific primers (Sigma-Aldrich, USA) verified to yield a single PCR product with a correct length (Table 1). Results were normalized to expression of reference genes beta-2-microglobulin (B2m) and hypoxanthine phosphoribosyltransferase 1 (Hprt1) and calibrated to appropriate control groups [23].
Primer sequences for qRT-PCR.
| Gene Name | GenBank Access No. | Primer Sequences (5'→3') | PCR product size (bp) |
|---|---|---|---|
| B2m1 | NM_012512.1 | forward: ATGGAGCTCTGAATCATCTGG reverse: AGAAGATGGTGTGCTCATTGC | 105 |
| Hai-11 | NM_001004265.2 | forward: ACACGCCTGACTGCCCTGAT reverse: TCCAGTGTCTGGCAGCTCTGCA | 128 |
| Hai-21 | NM_199087.1 | forward: CCTAGACGTCCACGAGAACACCAT reverse: CCAGTGACCGCCTTTGGGACA | 150 |
| Hgf1 | NM_017017.2 | forward: TCAGCGCTGGGATCAGCAGACA reverse: TGTAGCACCATGGCCTCGGCTT | 122 |
| Hgfa1 | NM_053320.1 | forward: TGTGCTGGGAGCCTCGTCCA reverse: GTGTCACATCCGTCGTGCGGT | 130 |
| Hprt11 | NM_012583.2 | forward: CAGCTTCCTCCTCAGACCGCTTT reverse: TCACTAATCACGACGCTGGGACTG | 150 |
| Met1 | NM_031517.1 | forward: GTGGCTGGTGCCACAATCGGT reverse: TCCTCCTTCGAGGGGTGCACT | 150 |
| Nppa1 | NM_012612.2 | forward: GGGGGTAGGATTGACAGGAT reverse: GGATCTTTTGCGATCTGCTC | 104 |
| Nppb1 | NM_031545.1 | forward: GACCGGATCGGCGCAGTCAGT reverse: GGAGTCTGCAGCCAGGAGGTCT | 78 |
1 B2m: beta-2-microglobulin, Hai-1: hepatocyte growth factor activator inhibitor 1, Hai-2: hepatocyte growth factor activator inhibitor 2, Hgf: hepatocyte growth factor, Hgfa: hepatocyte growth factor activator, Hprt1: hypoxanthine phosphoribosyltransferase 1, Met: mesenchymal-epithelial transition, Nppa: natriuretic peptide A, Nppb: natriuretic peptide B.
Statistical analysis
Data are reported as mean ± standard error of the mean. Data distribution was determined by Shapiro-Wilk normality test. Means were compared by unpaired Student's t-test for normally distributed data or non-parametric Mann-Whitney U test for nonparametric data, with P<0.05 considered statistically significant. Mean PCR efficiency estimates (E) per amplicon and quantification cycle (Cq) values per sample were determined with LinRegPCR software, version 2015.0 (Heart Failure Research Center, NL). Relative gene expression ratios were calculated using Pfaffl method [23]. Statistical evaluations and correlations were performed using GraphPad Prism, version 6 (GraphPad Software, USA).
Results
Progressive increase of RV weight and RV pressure after MCT
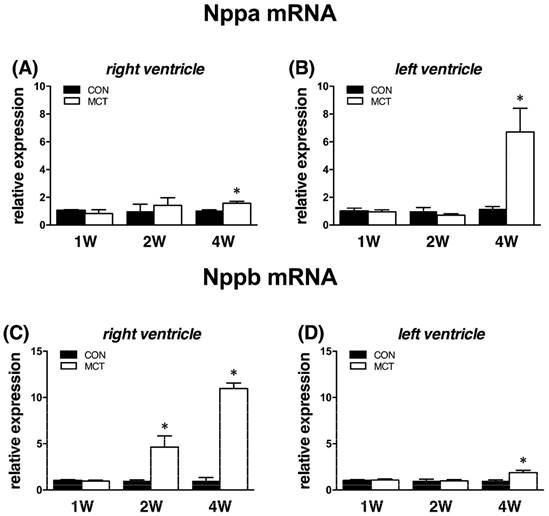
Body weight in MCT groups decreased from week 2 (-7%) to week 4 (-10%), both P<0.05. This was accompanied by increased relative weights of RVs and lungs at weeks 1, 2 and 4, respectively (all P<0.05; see Table 2). For comparison, relative weights of LVs were significantly increased in MCT groups solely at week 4 (P<0.05). Relative liver weight increased at week 2 and at week 4, both P<0.05 vs CON (Table 2). This was accompanied by significant increase of RVP in MCT-treated rats exclusively at week 4 (MCT 4W: 51±5 mmHg; CON 4W: 25±2 mmHg; P<0.05), while RV function in MCT groups remained unaltered at week 1 and 2. Natriuretic peptides genes, Nppa and Nppb gene (natriuretic peptide A and B), were used as markers of cardiac damage. We observed unchanged ventricular expression of these genes at week 1, but Nppb mRNA in RV was significantly elevated 4.7-fold already at week 2, i.e. in the stage with stable RV pressure. At the final week, expression of both natriuretic peptides was significantly increased in both heart ventricles (P<0.05, Figure 1), though it was more pronounced in RV.
Relative (when related to B2m and Hprt1) mRNA expression of Nppa gene after 1, 2 and 4 weeks after MCT injection in (A) right ventricle, (B) left ventricle, and Nppb gene in (C) right ventricle (D) left ventricle samples (n=6-12 per group; mean±SEM; *P<0.05 vs. CON).
Relative organ weights when related to their absolute body weights (BW) of right ventricle (RV), left ventricle (LV), lung and liver samples after 1, 2 and 4 weeks after MCT injection (n=6-12 per group; mean±SEM; *P<0.05 vs. CON).
| 1W | 2W | 4W | ||||
|---|---|---|---|---|---|---|
| CON | MCT | CON | MCT | CON | MCT | |
| RV/BW [mg/g] | 0.44±0.03 | 0.53±0.03* | 0.52±0.02 | 0.62±0.02* | 0.53±0.02 | 1.02±0.06* |
| LV/BW [mg/g] | 2.16±0.05 | 2.14±0.06 | 2.01±0.07 | 2.00±0.03 | 2.18±0.04 | 2.38±0.07* |
| lung/BW [mg/g] | 3.85±0.08 | 4.28±0.13* | 4.91±0.21 | 6.97±0.64* | 3.90±0.15 | 8.32±0.44* |
| liver/BW [mg/g] | 30.08±0.58 | 31.09±0.57 | 28.76±0.55 | 33.07±0.97* | 30.51±0.50 | 33.44±1.01* |
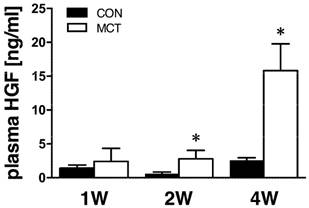
Progressive rise of plasma HGF concentrations after MCT
One week after MCT we observed no change in plasma HGF levels. At week 2, significant 3-fold elevation was detected and this trend further escalated to a 6-fold increase at week 4 (both P<0.05, Figure 2).
Relative plasma concentration of HGF protein after 1, 2 and 4 weeks after MCT injection (n=4-8 per group; mean±SEM; *P<0.05 vs. CON).
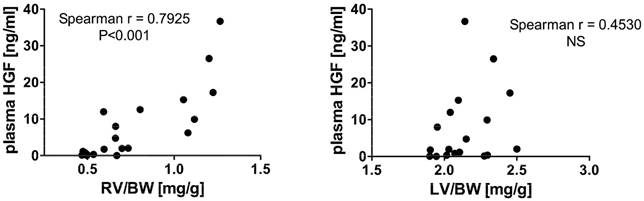
In the pooled group of MCT-treated rats, plasma HGF levels correlated positively with RV/BW (but not with LV/BW; see Figure 3). When divided to subgroups according to disease progression, this positive significant correlation between plasma HGF and RV/BW was present in the group of 4W MCT (Spearman r = 0.7857, P<0.05), but not in subgroups of 2W and 1W MCT (Spearman r = 0.1429, NS and Spearman r = 0.3143, NS, respectively). In healthy controls, we observed no relationship between plasma HGF and RV/BW neither LV/BW, respectively. We used RV/BW and LV/BW as a measures of organ remodeling, but all findings were valid also when using absolute RV and LV weights.
Relationship between plasma HGF concentrations and right and left ventricular weight to body weight ratio (RV/BW and LV/BW) in pooled group of all MCT-treated rats.
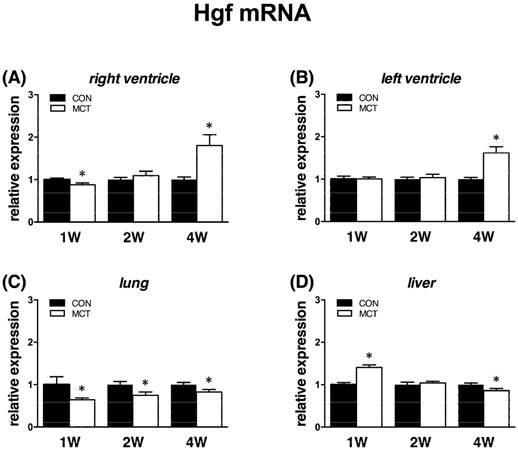
Increased cardiac Hgf mRNA expression at week 4
At week 1, we observed a decrease of Hgf mRNA expression in the RV (-11%, P<0.05) and lung (-33%, P<0.05), no change in the LV and a. pulmonalis and a significant increase (+46%, P<0.05) in liver. At week 2, we observed unaltered expression in all examined tissues except lung, where it remained decreased (-24%, P<0.05). At week 4, mRNA level of Hgf gene expression was increased in the left (+64%) and right (+76%) ventricle and decreased in a. pulmonalis (-29%), lungs (-19%) and liver (-16%), all P<0.05 vs CON (Figure 4).
Relative (when related to B2m and Hprt1) mRNA expression of Hgf gene after 1, 2 and 4 weeks after MCT injection in (A) right ventricle, (B) left ventricle, (C) lung and (D) liver samples (n=6-12 per group; mean±SEM; *P<0.05 vs. CON).
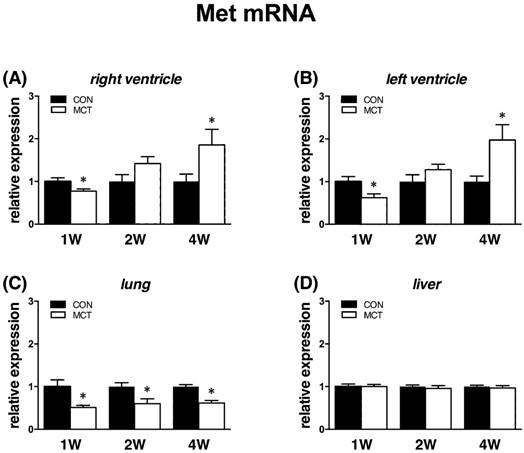
Increased cardiac Met mRNA expression at week 4
Met receptor gene expressions followed an almost identical pattern as Hgf. When compared to controls, at week 1 we observed decreases of Met expression in left (‑28%) and right (-15%) ventricle, lung (-48%), all P<0.05, but no change in a. pulmonalis and liver. At week 2, expressions of Met were similar in all tissues except for lung, where it remained significantly decreased (-35%, P<0.05). Finally, at week 4 we observed an increased expression of Met in both left (+119%) and right (+81%) ventricle, decreased in lung (‑40%), all P<0.05, and unchanged in a. pulmonalis and liver (Figure 5).
Relative (when related to B2m and Hprt1) mRNA expression of Met gene after 1, 2 and 4 weeks after MCT injection in (A) right ventricle, (B) left ventricle, (C) lung and (D) liver samples (n=6-12 per group; mean±SEM; *P<0.05 vs. CON).
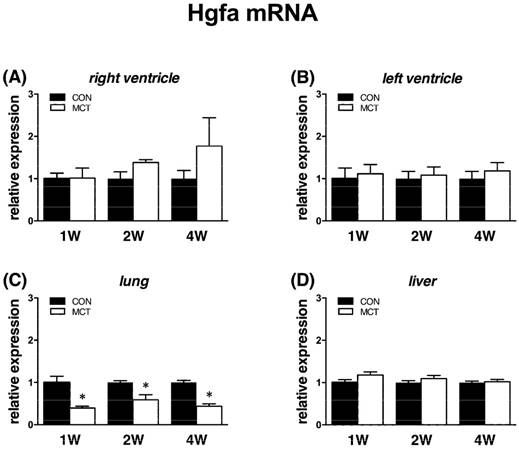
Reflecting mRNA expression of HGF regulators
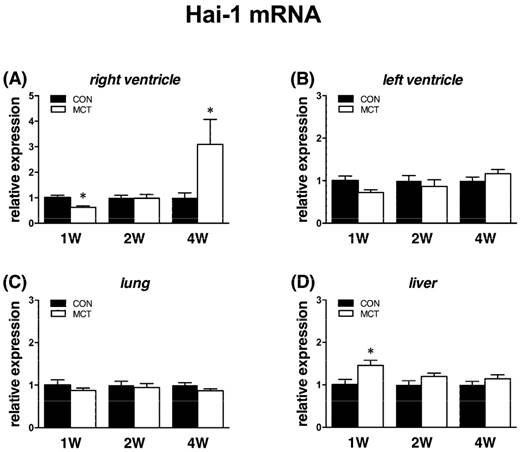
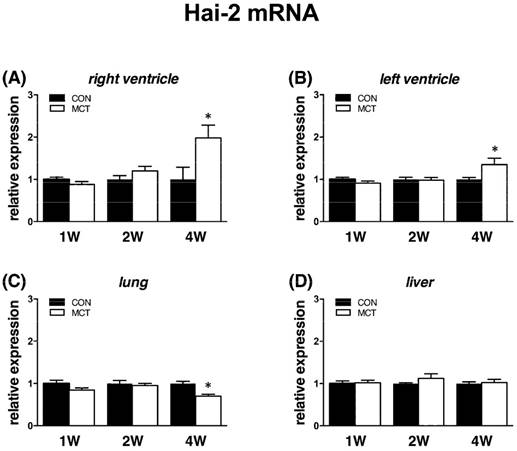
Expression of Hgfa was significantly (P<0.05) supressed in lung tissue from week 1 to week 4 as Hgf and Met, no change was observed in RV/LV and liver tissues. In case of HGFA inhibitors, Hai-1 gene showed the same significant changes (P<0.05) as Hgf gene in RV and in liver only at week 1, no changes were observed in LV and lung tissue. Hai-2 gene expression was similar to Hai-1, however significant (P<0.05) increase was seen at week 4 in both heart ventricles together with a decrease in lung tissue (Figures 6, 7, 8).
Relative (when related to B2m and Hprt1) mRNA expression of Hgfa gene after 1, 2 and 4 weeks after MCT injection in (A) right ventricle, (B) left ventricle, (C) lung and (D) liver samples (n=6-12 per group; mean±SEM; *P<0.05 vs. CON).
Relative (when related to B2m and Hprt1) mRNA expression of Hai-1 gene after 1, 2 and 4 weeks after MCT injection in (A) right ventricle, (B) left ventricle, (C) lung and (D) liver samples (n=6-12 per group; mean±SEM; *P<0.05 vs. CON).
Relative (when related to B2m and Hprt1) mRNA expression of Hai-2 gene after 1, 2 and 4 weeks after MCT injection in (A) right ventricle, (B) left ventricle, (C) lung and (D) liver samples (n=6-12 per group; mean±SEM; *P<0.05 vs. CON).
Discussion
The main finding of our study are the elevated plasma levels of HGF and increased mRNA levels solely in cardiac ventricles in the monocrotaline model of PH in rats. HGF could be an indicator of cardiac damage and, along with simultaneous upregulation of Met, also suggest the role of HGF/MET pathway in the progression cardiac damage in advanced PH.
Progression of the MCT-induced PH was accompanied by gradual rise of plasma HGF concentrations with robust elevations mainly at the late stage of PH. Plasma HGF positively correlated with RV/BW, but not with LV/BW ratio, further indicating a causal relationship with sustained pressure overload-related RV damage. Interestingly, we detected a significant increase of plasma HGF already two weeks after MCT administration, i.e. at a stage with as yet stable RV performance and only a moderately increased RV/BW ratio along with significantly upregulated mRNA expression for Nppb. These alterations collectively suggest very early cardiac damage reflected in plasma HGF levels, which could be a result of starting pressure overload from damaged pulmonary arteries, although we cannot exclude also direct monocrotaline cardiotoxicity [24]. Increased plasma HGF were reported in patients with various cardiovascular and pulmonary disorders [16, 17, 25, 26], as well as in patients with PH [19]. In patients with PAH, plasma levels of HGF correlated with PAH severity, although a relation between HGF and duration of PAH was not studied [19]. Our results confirm these findings and reveal that a rise of plasma HGF precedes the deterioration of RV function during the disease development and is in line with significantly rising Nppb expression in RV even in settings of stable RV pressure.
Although a link between increased plasma HGF and tissue damage was suggested [16, 17, 19, 25, 26], the source of detected HGF is mostly unknown. The cytokine is produced by cells of mesenchymal origin [9], while MET receptor is expressed mainly by epithelial cells, but is also found in other cell types including cardiomyocytes [27]. We showed that elevated plasma HGF mirrored, at least partially, the increased cardiac Hgf mRNA expression exclusively at week 4, the advanced stage of PH. Hgf gene in cardiomyocytes is normally silent, but can be enhanced [28] and this is known to have cardioprotective effects: anti-apoptosis [29], angiogenesis [30] and cell regeneration [31]. In cardiac ischemia/reperfusion injury, increased cardiac mRNA expression of Hgf is in line with elevated plasma levels of HGF [32]. The found correlation of cardiac Hgf expression and plasma HGF in our study is a novel finding. It suggests a role of HGF in cardiac compensatory processes and also, the possibility that cardiac tissue might contribute to increased HGF plasma levels in PH. One of possible alternative mechanisms of plasma HGF elevation may involve influence of erythropoietin, another prognostic indicator of cardiovascular mortality [33], which plays a role in PH development [34] and it can increase Hgf expression in stem cells [35]. Importantly, plasma levels of erythropoietin are known to be increased in PH patients [36] and MCT-injected rats as we reported previously [37]. Whether this is relevant also for the increased cardiac expression of Hgf remains to be elucidated, but it might provide an explanation for the surprising Hgf mRNA increase in both cardiac ventricles, i.e. in damaged RV as well as in “less affected” LV. This further supports the use of plasma HGF as a biomarker of cardiac damage in PH.
Contrariwise, we detected a sustained decrease of Hgf gene expression in lungs and stable or depressed (at week 4) Hgf mRNA in a. pulmonalis in MCT rats. Decreased HGF protein and mRNA levels in lungs in PH were reported [38, 39]. Interestingly, the finding of suppressed Hgf mRNA expression in lungs already one week after MCT injection is novel and it indicates that Hgf expression is impaired at early stages of PH. We speculate that reduced expression of Hgf, which is important in lung repair [39], mirrors its repressed protective activity [40] and therefore the organ is more susceptible to damage. MCT causes predominantly interstitial pulmonary fibrosis [20] and pulmonary arterial medial hypertrophy [11]. Importance of HGF is suggested by reports that MCT-induced vascular and lung injuries are, at least partially, preventable by HGF treatment, since Hgf overexpression attenuates medial hyperplasia and matrix accumulation in the pulmonary arteries [39] and prevents lung inflammation in rat models of PH [38]. As decrease of Hgf mRNA is present early, i.e. after one week following MCT injection, we can only speculate about a possible predominant MCT-related damage of cell types with more pronounced HGF expression. Nevertheless, progressive lung damage that is caused by MCT to induce PH leads to suppressed expression of HGF since the early stages and remains that way as the condition further develops [39], therefore promotion of its potential regenerative effects is most likely suppressed as well.
HGF is secreted from liver mesenchymal cells and is an important mediator of liver regeneration [41]. Consequently, plasma levels of HGF in our study might be influenced by hepatic expression. However, we observed increased Hgf mRNA levels exclusively at an early stage while in advanced PH the hepatic Hgf expression was reduced. Initial upregulation could be a result of MCT hepatotoxicity [42] as MCT undergoes hepatic metabolism leading to generation of a reactive dehydromonocrotaline. Proposed mechanism of hepatotoxicity involves inhibition of NADH oxidase activity of respiratory chain complex I, which causes liver damage [43]. The probable mechanism of MCT lung toxicity lies in the metabolic activation in the liver, which plays a crucial role in the development of this model. After the injection, MCT is quickly metabolized in the liver with CYP450 enzyme family involved to produce dehydromonocrotaline, the main active metabolite of MCT responsible for its damaging effects in pulmonary endothelium [44] probably by targeting the extracellular calcium-sensing receptor [45]. After metabolization, the metabolite is bound to erythrocytes and delivered to the pulmonary vascular endothelium [46] to initiate endothelial cell injury probably by various mechanisms [47, 48]. Thus, the early upregulation of Hgf mRNA might be associated with hepatic self-repair processes [49] after MCT administration as HGF stimulates hepatocytes proliferation in settings of liver injury [47, 48], while the late downregulation could be similar to chronic liver damage [50]. Although the mechanism remains unclear, we can likely exclude the hepatic overproduction as a source of elevated plasma HGF in rats with advanced PH.
We also explored the expression of Met receptor, the only HGF receptor [4]. In addition to HGF, MET receptor tissue and plasma levels are known to be increased in cardiac [51], liver [52], or lung disorders [39]. The initial decrease of Met in both heart ventricles found in our study could be associated with decreased Hgf mRNA expressions, at least in the RV. In accordance with elevated cardiac Hgf expression, we also detected an increased Met expression in both heart ventricles in advanced PH what could result from increased cardiac Hgf expression. Similarly, also the significant decrease of Met expression in lungs is related to depressed Hgf expression. These findings also suggest that lungs are predominantly affected by damage in the model of MCT-induced PH because of lacking regenerative capacity of HGF/MET pathway.
In overall, little is known about the mechanisms of HGF regulatory system (HGFA, HAI-1, HAI-2). In our experiment we detected changes reflecting the HGF/MET pathway behaviour. Expression of Hgfa showed no significant differences in cardiac and liver tissues, however it was significantly supressed in lung tissue since week 1 to week 4 in the same way as Hgf/Met in this compartment. This result may indicate that Hgfa gene could be one of the reasons for supressed expression of HGF/MET axis in lung tissue. It is considered a major regulator of HGF active form availability to the MET receptor as a substrate [8], but probably does not play a role in heart and liver in this condition. In case of HGFA inhibitors, Hai-1 and Hai-2, we observed a similar pattern to Hgf/Met in RV at week 4, where they were both significantly upregulated. This could be interpreted also as a compensatory reaction to an excessive mRNA expression of Hgf in cardiac ventricles and respective amounts of HGF in plasma at this stage of the disease progression. Hai-1 has two isoforms with dual roles, where one does not possess the inhibitory properties and therefore could lead to an enhanced HGF activation [53]. We also observed that Hai-1 was significantly upregulated also in liver at week 1 and Hai-2 was significantly upregulated also in LV and downregulated in lungs at week 4 in the same way as Hgf in these tissues. These results support, at least partially, our compensatory mechanism hypothesis, which could be potentially specific for this condition. Remarkably, we observed no upregulation of the Hgf/Met pathway or the intrinsic regulators in any of the analysed tissues in the week 2 after MCT administration. Therefore, the question of potential plasma HGF source at this time point remains elusive. We can only speculate about alterations in HGF systemic clearance.
This study has certain limitations. We measured only mRNA levels of Hgf/Met in tissues while protein levels would be desirable. However, studies reported a tight relation between mRNA and protein expression of HGF and also MET [38, 39, 54].
We conclude that increased HGF plasma levels temporally coincided and correlated with development of RV hypertrophy in the monocrotaline model of PH. This was associated with increased cardiac expression of Hgf and Met as PH advanced indicating that increased plasma HGF may be of cardiac origin underlining its role in disease development and also supporting its use as a cardiac-specific biomarker in PH.
Acknowledgements
The authors gratefully acknowledge the excellent technical assistance of Ms. Alena Hnizdova, Ms. Lenka Svobodova and Ms. Elena Vilemova. This research was funded by the Slovak Research and Development Agency (APVV-15-0685) and Science Grant Agency (VEGA 1/0294/15).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Galiè N, Humbert M, Vachiery JL. et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67-119
2. Humbert M, Morrell NW, Archer SL. et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S-24S
3. Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121:2045-2066
4. Rubin JS, Bottaro DP, Aaronson SA. Hepatocyte growth factor/scatter factor and its receptor, the c-met proto-oncogene product. Biochim Biophys Acta. 1993;1155:357-371
5. Miyazawa K, Shimomura T, Kitamura A. et al. Molecular-cloning and sequence-analysis of the cDNA for a human serine protease responsible for activation of hepatocyte growth-factor - structural similarity of the protease precursor to blood-coagulation factor-xii. J Biol Chem. 1993;268:10024-10028
6. Shimomura T, Denda K, Kitamura A. et al. Hepatocyte growth factor activator inhibitor, a novel Kunitz-type serine protease inhibitor. J Biol Chem. 1997;272:6370-6376
7. Kawaguchi T, Qin L, Shimomura T. et al. Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. J Biol Chem. 1997;272:27558-27564
8. Shimomura T, Kondo J, Ochiai M. et al. Activation of the Zymogen of Hepatocyte Growth Factor Activator by Thrombin. J Biol Chem. 1993;268:22927-22932
9. Saito S, Sakakura S, Enomoto M. et al. Hepatocyte growth factor promotes the growth of cytotrophoblasts by the paracrine mechanism. J Biochem. 1995;117:671-676
10. Pang Y, Liang MT, Gong Y. et al. HGF Reduces Disease Severity and Inflammation by Attenuating the NF-κB Signaling in a Rat Model of Pulmonary Artery Hypertension. Inflammation. 2018;41:924-931
11. Panganiban RAM, Day RM. Hepatocyte growth factor in lung repair and pulmonary fibrosis. Acta Pharmacol Sin. 2011;32:12-20
12. Ueda H, Nakamura T, Matsumoto K. et al. A potential cardioprotective role of hepatocyte growth factor in myocardial infarction in rats. Cardiovasc Res. 2001;51:41-50
13. Jayasankar V, Woo YJ, Bish LT. et al. Gene Transfer of Hepatocyte Growth Factor Attenuates Postinfarction Heart Failure. Circulation. 2003;108:230-236
14. Li M, Jiang D, Wang Y. et al. Signal Mechanisms of Vascular Remodeling in the Development of Pulmonary Arterial Hypertension. J Cardiovasc Pharmacol. 2016;67:182-190
15. Hoeper MM, Bogaard HJ, Condliffe R. et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D42-D50
16. Rychli K, Richter B, Hohensinner PJ. et al. Hepatocyte growth factor is a strong predictor of mortality in patients with advanced heart failure. Heart. 2011;97:1158-1163
17. Morishita R, Moriguchi A, Higaki J, Ogihara T. Hepatocyte growth factor (HGF) as a potential index of severity of hypertension. Hypertens Res. 1999;22:161-167
18. Sung YK, Zamanian RT, Wagner CA. et al. Differential expression of hepatocyte growth factor in patients with systemic sclerosis-associated pulmonary arterial hypertension. J Scleroderma Relat Disord. 2017;2:225-230
19. Liang M, Pang Y, Zhang S, Zhang M. Utility of Hepatocyte Growth Factor as a Biomarker for Early Diagnosis of Pulmonary Artery Hypertension. Mol Diagn Ther. 2016;20:463-468
20. Hayashi S, Mitsumori K, Imaida K. et al. Establishment of an animal model for pulmonary fibrosis in mice using monocrotaline. Toxicol Pathol. 1995;23:63-71
21. Ratnoff OD, Mirick GS. Influence of sex upon the lethal effects of an hepatotoxic alkaloid, monocrotaline. Bull Johns Hopkins Hosp. 1949;84:507-525
22. Sun X, Ku DD. Rosuvastatin provides pleiotropic protection against pulmonary hypertension, right ventricular hypertrophy, and coronary endothelial dysfunction in rats. Am J Physiol Heart Circ Physiol. 2008;294:H801-H809
23. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45
24. Akhavein F, St-Michel EJ, Seifert E. et al. Decreased Left Ventricular Function, Myocarditis, and Coronary Arteriolar Medial Thickening Following Monocrotaline Administration in Adult Rats. J Appl Physiol. 2007;103:287-295
25. Jin H, Yang R, Li W. et al. Early Treatment with Hepatocyte Growth Factor Improves Cardiac Function in Experimental Heart Failure Induced by Myocardial Infarction. J Pharmacol Exp Ther. 2003;304:654-660
26. Ziora D, Adamek M, Czuba Z. et al. Increased Serum Hepatocyte Growth Factor (HGF) Levels in Patients with Idiopathic Pulmonary Fibrosis (IPF) or Progressive Sarcoidosis. J Mol Biomark Diagn. 2014;5:1-4
27. Arechederra M, Carmona R, González-Nuñez M. et al. Met signaling in cardiomyocytes is required for normal cardiac function in adult mice. Biochim Biophys Acta. 2013;1832:2204-2215
28. Riess I, Sala V, Leo C. et al. A mouse model for spatial and temporal expression of HGF in the heart. Transgenic Res. 2011;20:1203-1216
29. Wang Y, Liu J, Tao Z. et al. Exogenous HGF Prevents Cardiomyocytes from Apoptosis after Hypoxia via Up-Regulating Cell Autophagy. Cell Physiol Biochem. 2016;38:2401-2413
30. Ruvinov E, Leor J, Cohen S. The effects of controlled HGF delivery from an affinity-binding alginate biomaterial on angiogenesis and blood perfusion in a hindlimb ischemia model. Biomaterials. 2010;31:4573-4582
31. Deuse T, Peter C, Fedak PWM. et al. Hepatocyte Growth Factor or Vascular Endothelial Growth Factor Gene Transfer Maximizes Mesenchymal Stem Cell-Based Myocardial Salvage After Acute Myocardial Infarction. Circulation. 2009;120:S247-S254
32. Nakamura T, Mizuno S, Matsumoto K. et al. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J Clin Invest. 2000;106:1511-1519
33. van der Meer P, Voors AA, Lipsic E. et al. Prognostic value of plasma erythropoietin on mortality in patients with chronic heart failure. J Am Coll Cardiol. 2004;44:63-67
34. Onal EM, Sag AA, Sal O. et al. Erythropoietin mediates brain-vascular-kidney crosstalk and may be a treatment target for pulmonary and resistant essential hypertension. Clin Exp Hypertens. 2017;39:197-209
35. Tari K, Atashi A, Kaviani S. et al. Erythropoietin induces production of hepatocyte growth factor from bone marrow mesenchymal stem cells in vitro. Biologicals. 2017;45:15-19
36. Karamanian VA, Harhay M, Grant GR. et al. Erythropoietin Upregulation in Pulmonary Arterial Hypertension. Pulm Circ. 2014;4:269-279
37. Malikova E, Galkova K, Vavrinec P, Vavrincova-Yaghi D, Kmecova Z, Krenek P, Klimas J. Local and systemic renin-angiotensin system participates in cardiopulmonary-renal interactions in monocrotaline-induced pulmonary hypertension in the rat. Mol Cell Biochem. 2016;418:147-157
38. Chen J, Zhang H, Zhang R. et al. Transfer of human hepatocyte growth factor reduces inflammation and prevents pulmonary arterial remodeling in monocrotaline-induced pulmonary arterial hypertensive rats. Int J Clin Exp Pathol. 2014;7:8763-8769
39. Ono M, Sawa Y, Mizuno S. et al. Hepatocyte Growth Factor Suppresses Vascular Medial Hyperplasia and Matrix Accumulation in Advanced Pulmonary Hypertension of Rats. Circulation. 2004;110:2896-2902
40. Hiramine K, Sata N, Ido A. et al. Hepatocyte growth factor improves the survival of rats with pulmonary arterial hypertension via the amelioration of pulmonary hemodynamics. Int J Mol Med. 2011;27:497-502
41. Bottaro D, Rubin J, Faletto D. et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802-804
42. Schoental R, Head M. Pathological changes in rats as a result of treatment with monocrotaline. Br J Cancer. 1955;9:229-237
43. Mingatto FE, Dorta DJ, dos Santos AB. et al. Dehydromonocrotaline inhibits mitochondrial complex I. A potential mechanism accounting for hepatotoxicity of monocrotaline. Toxicon. 2007;50:724-730
44. Mattocks AR. Toxicity of pyrrolizidine alkaloids. Nature. 1968;217:723-728
45. Xiao R, Su Y, Feng T. et al. Monocrotaline Induces Endothelial Injury and Pulmonary Hypertension by Targeting the Extracellular Calcium-sensing Receptor. J Am Heart Assoc. 2017;6:e004865
46. Pan LC, Lame MW, Morin D. et al. Red blood cells augment transport of reactive metabolites of monocrotaline from liver to lung in isolated and tandem liver and lung preparations. Toxicol Appl Pharmacol. 1991;110:336-346
47. Wilson DW, Segall HJ, Pan LC. et al. Mechanisms and pathology of monocrotaline pulmonary toxicity. Crit Rev Toxicol. 1992;22:307-325
48. Lee J, Reich R, Xu F, Sehgal PB. Golgi, trafficking, and mitosis dysfunctions in pulmonary arterial endothelial cells exposed to monocrotaline pyrrole and NO scavenging. Am J Physiol Lung Cell Mol Physiol. 2009;297:L715-L728
49. Zarnegar R, De Frances MC, Kost DP. et al. Expression of hepatocyte growth factor mRNA in regenerating rat liver after partial hepatectomy. Biochem Biophys Res Commun. 1991;177:559-565
50. Masuhara M, Yasunaga M, Tanigawa K. et al. Expression of hepatocyte growth factor, transforming growth factor α, and transforming growth factor β 1 messenger RNA in various human liver diseases and correlation with hepatocyte proliferation. Hepatology. 1996;24:323-329
51. Ono K, Matsumori A, Shioi T. et al. Enhanced expression of hepatocyte growth factor/c-Met by myocardial ischemia and reperfusion in a rat model. Circulation. 1997;95:2552-2558
52. Noguchi O, Enomoto N, Ikeda T. et al. Gene expressions of c-met and hepatocyte growth factor in chronic liver disease and hepatocellular carcinoma. J Hepatol. 1996;24:286-292
53. Kataoka H, Shimomura T, Kawaguchi T. et al. Hepatocyte Growth Factor Activator Inhibitor Type 1 Is a Specific Cell Surface Binding Protein of Hepatocyte Growth Factor Activator (HGFA) and Regulates HGFA Activity in the Pericellular Microenvironment. J Biol Chem. 2000;275:40453-40462
54. Mesarosova L, Ochodnicky P, Leemans JC, Florquin S, Krenek P, Klimas J. High glucose induces HGF-independent activation of Met receptor in human renal tubular epithelium. J Recept Signal Transduct Res. 2017;37:535-542
Author contact
![]() Corresponding author: Peter Krenek, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University in Bratislava, Odbojarov 10, 83232 Bratislava, Slovak Republic. E-mail: krenekuniba.sk
Corresponding author: Peter Krenek, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University in Bratislava, Odbojarov 10, 83232 Bratislava, Slovak Republic. E-mail: krenekuniba.sk