Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(3):443-449. doi:10.7150/ijms.28353 This issue Cite
Research Paper
Decreased Ambient Oxygen Tension Alters the Expression of Endothelin-1, iNOS and cGMP in Rat Alveolar Macrophages
I-Chen Chen1,2,3, Yu-Tsai Lin4, Jhy-Shrian Huang1, Bin-Nan Wu5, Jong-Hau Hsu1,2, Mian-Shin Tan6*, Zen-Kong Dai1,2* ![]()
1. Department of Pediatrics, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
2. Department of Pediatrics, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
3. Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
4. Department of Otolaryngology, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan
5. Department of Pharmacology, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
6. Department of Biomedical Science and Environmental Biology, College of Life Sciences, Kaohsiung Medical University, Kaohsiung, Taiwan
*These two authors contributed equally to this work.
Received 2018-7-6; Accepted 2018-12-28; Published 2019-2-28
Abstract
Background: Hypoxia plays an important role in the vascular tone of pulmonary circulation via the vasculature and parenchymal tissue. Endothelin-1 (ET-1), a potent vasoconstrictive peptide, plays a role in inflammation in mononuclear cells. Nitric oxide synthase (NOS), which generates nitric oxide (NO)/cyclic 3', 5'-monophosphate (cGMP), is coexpressed with ET-1 in many cell types. The aim of this study was to assess whether hypoxia induces the production of ET-1 and associated expression of NOS, NO/cGMP and chemokines in rat alveolar macrophages (AMs).
Methods: NR8383 cells were cultured under hypoxic (1% oxygen) conditions for 0, 2, 4, 8 and 12 hours. Levels of ET-1, inducible NOS (iNOS), phosphorylated iNOS (p-iNOS), nitrite/nitrate (NOx), cGMP and monocyte chemoattractant protein-1 (MCP-1) were measured.
Results: ET-1, p-iNOS, NOx, and cGMP increased significantly in AMs after 4 hours of hypoxia (p < 0.05). ET-1 and MCP-1 mRNA increased after 8 hours (p < 0.05). The protein expression of ET-1, MCP-1, and p-iNOS increased in a time-dependent manner, while iNOS expression decreased with time.
Conclusions: The changes in ET-1, p-iNOS, and the NO/cGMP pathway in AMs may help elucidate the mechanisms in the hypoxic lung. Understanding changes in the endothelin axis in hypoxic AMs is a crucial first step to unravel its role in pulmonary circulation.
Keywords: hypoxia, endothelin-1, alveolar macrophage, nitric oxide synthase
Introduction
Acute hypoxia causes pulmonary vasoconstriction, whereas chronic hypoxia leads to the development of pulmonary hypertension and pulmonary vascular remodeling [1]. Alveolar hypoxia, which is present in many lung diseases such as acute respiratory distress syndrome, asthma, bronchopulmonary dysplasia, and obstructive sleep apnea syndrome may lead to severe cardiopulmonary diseases such as pulmonary hypertension [2-4]. Growing evidence has indicated that hypoxia alters the expression of various vasoactivators and inflammatory factors in pulmonary non-vascular cells such as macrophages, leukocytes, and dendritic cells [1]. Alveolar macrophages (AMs) respond rapidly to foreign material, play a central role in homeostasis, and initiate alveolar inflammation in response to hypoxia [5].
AMs are located at the air-tissue interface in alveoli and are among the first cells to contact external organisms or substances [2]. Although many different cells contribute to the inflammatory response to hypoxia, activated AMs are thought to play an important role in the lung, particularly in terms of the production of inflammatory cytokines. As such, AMs have become increasingly studied in multiple models of hypoxia [6].
AMs secrete many cytokines/chemokines in response to hypoxia and/or reoxygenation, including macrophage inflammatory protein 2, macrophage inflammatory protein 1 alpha, cytokine-induced neutrophil chemoattractant, monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-α, and RANTES [1, 6, 7]. However, the mechanism by which hypoxia prompts the production of vasoactivators or inflammatory substances by AMs is not entirely clear.
Endothelin-1 (ET-1) was first isolated from vascular endothelium in 1988 [8]. ET has three isoforms: ET-1, -2, and -3 [8, 9]. The pulmonary endothelial system has been implicated in the pathogenesis of pulmonary hypertension. Nitric oxide synthase (NOS), which is responsible for the generation of nitric oxide (NO)/cyclic 3', 5'-monophosphate (cGMP), is co-expressed with ET-1 in different diseases [10, 11]. ET-1 exerts vasodilatory effects by increasing NO/cGMP expression via activation of the ET-B receptor. Additionally, ET-1 is thought to be involved in bronchoconstriction and airway structural remodeling in the lung and has several pro-inflammatory properties of potential relevance to asthma [9]. Although ET-1 was originally isolated from endothelial cells, it is also synthesized and secreted by many other cells, including fibroblasts, mast cells, mononuclear cells (monocytes and macrophages), cardiomyocytes, and epithelial cells [12-16]. Several studies have demonstrated that ET-1 is secreted by macrophages under some circumstances, such as lipopolysaccharide, phorbol myristate acetate, or microbial challenges [14, 17, 18]. NO, catalyzed by inducible NOS (iNOS), is also secreted by macrophages. cGMP acts as an intracellular second messenger of NO, subsequently reducing blood vessel tone and blood pressure [19].
The NO/cGMP pathway is co-expressed with ET-1 in many different cells. However, no study has investigated whether ET-1 and iNOS/NO/cGMP are expressed simultaneously in AMs in response to oxidative stress. We hypothesized that hypoxic stimulation alters the expression of ET-1 and iNOS/NO/cGMP in AMs, which then show counteracting effects toward each other. Losing homeostasis in AMs may initiate the accumulation of vasoactivators or inflammatory substances, resulting in vasoconstriction, remodeling, and general inflammation in lung. Therefore, we investigated whether ET-1 - and the associated expression of NOS and NO/cGMP - can be induced by hypoxic AMs, similar to pulmonary endothelium.
Methods
All chemicals were purchased from Sigma Chemical Company (St. Louis, MO, USA). Fetal bovine serum (FBS) for cell culture was purchased from HyClone Laboratories (Logan, UT, USA). All media and antibiotics were purchased from GIBCO-BRL (Grand Island, NY, USA).
Cell preparation
NR8383 cells (Sprague-Dawley strain, ATCC# CL-2192) were obtained from the Taiwan Food Industry Research and Development Institute. The cells were initially maintained in Ham's F-12 medium with 15% heat-inactivated FBS, 2 mM L-glutamine, and 1.5 g/L sodium bicarbonate. The cells were grown at 37°C in 5% CO2, passaged after 3-4 days, removed from culture dishes with 5 mM EDTA in phosphate-buffered saline, washed, and resuspended in cell culture media before use.
Hypoxia exposure study and cell viability assay
To determine the ability of AMs to produce ET-1, the cells were cultured in the absence of hypoxic stimulation or 1% oxygenation for 2-8 hours. The cells were plated into 10-cm plates at a density of 2 × 106 cells/cm2. After 24 hours, the cells were treated with FBS-free media for 12 hours and placed into a hypoxia incubator with 1% O2, 5% CO2, and 94% N2. The cells were collected at 0, 2, 4, and 8 hours later. Cell viability for each experiment was determined by trypan-blue staining. At the different time points, cell lysates were collected and assayed for EDN1, iNOS, and MCP-1mRNA expression. The culture supernatants were collected and assayed for NO, cGMP, and ET-1 expression.
EDN1, iNOS, and MCP-1mRNA
Total RNA was isolated from the NR8383 cells using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). mRNA expression was quantified by reverse transcription-polymerase chain reaction (RT-PCR) using the specific primers listed in Table 1. The NCBI Reference Sequence ID for the primers are NM_012548.2 (EDN1), NM_012611.3 (iNOS), NM_031530.1(MCP-1), and NM_031144.3 (actin).
Oligonucleotide primers used in this study
| Gene | Forward sequence | Reverse sequence | |
|---|---|---|---|
| EDN1 | TGTAGTCAATGTGCTCG | CGAGCACATTGACTACA | |
| iNOS | GCTACACTTCCAACGCAACA | ACAATCCACAACTCGCTCCA | |
| MCP-1 | AGTATTCATGGAAGGGAATAG | CTATTCCCTTCCATGAATACT | |
| Actin | AGCCATGCCAAATGTCTCAT | ATGAGACATTTGGCATGGCT | |
RNA (2 μg) was then reverse-transcribed into cDNA using SuperScript II Reverse Transcriptase (Invitrogen). PCR was performed using 1 μL of the reverse transcription products in a total volume of 25 μL. Actin was used as an internal control to normalize the relative amount of cDNA in each reaction. The reaction mixture contained 10 mM Tris-HCl, pH 8.3, 1.5 mM MgCl2, 50 mM KCl, 200 mM dNTPs, 2 mM of each primer, and 1 U of Ex Taq Polymerase (Takara Bio, Shiga, Japan). After an initial enzyme activation step for 15 min at 95°C, PCR was carried out for ET-1 (30 s at 94°C, 30 s at 55°C, and 60 s at 72°C), iNOS (60 s at 94°C, 60 s at 60°C, and 60 s at 72°C), MCP-1 (60 s at 94°C, 60 s at 62°C, and 60 s at 72°C), and actin (1 min at 94°C, 30 s at 58°C, and 30 s at 72°C), followed by a 10 min extension at 72°C. The DNA fragments were separated by 2% agarose gel electrophoresis and visualized by ethidium bromide staining.
Cyclic GMP and ET-1 expression
Cell pellets (n=6) were acidified with 0.6% trifluoroacetic acid to give a 10% w/v homogenate, which was centrifuged at 2,000 × g for 15 min at 4°C. The supernatant was washed and dried under a stream of nitrogen at 60°C, and the dried extract was dissolved in a 20 µL assay buffer prior to analysis. cGMP level was determined using a cGMP enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI, USA), and the secreted ET-1 was measured using an enzyme immunoassay kit (Biomedica Group, Wien, Austria).
NO production
NO production was measured as nitrite/ nitrate (NOx) using Griess reagent (Sigma). Briefly, equal volumes of culture supernatant and Griess reagent (100 μL) were mixed for 10 min at room temperature, and absorbance at 540 nm was measured using a Labsystems Multiskan Ascent assay plate reader (Thermo Scientific, Rockford, IL, USA). A graded solution of NO2 was used to construct a standard curve, and the results represent the mean values from three separate samples.
Western blotting
A total of 50-75g μg protein was separated using 10-12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, Stafford, VA, USA) in a tank blotter (25 mM Tris, 0.192 M glycine, pH 8.3, 20% methanol) at 30 V overnight. The membranes were blocked in 5% skim milk in 10 mM Tris-HCl (pH 8.0, 150 mM NaCl, 0.05% Tween 20) overnight and incubated with anti-β-actin (1:20000, SC-47778; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti- endothelin-1 (1:500, NB300-526; Novus Biologicals, Littleton, CO, USA), p-iNOS (1:500, phosphorylated iNOS) (PA5-37667; Thermo Fisher Scientific, Waltham, MA, USA), anti-MCP1 (1:500, NBP1-07035; Novus Biologicals, Littleton, CO, USA), or iNOS/NOSII antibody (1:1000, sc-7271; Santa Cruz Biotechnology) for 2 h. The blots were washed twice with Tris-HCl (pH 8.0, 150 mM NaCl, 0.05% Tween-20) for 10 min and incubated with a second antibody (anti-rabbit or anti-mouse immunoglobulins) (IRDye; Odyssey Li-COR Biosciences, Lincoln, NE, USA) at a 1:20000 dilution for 1 hour. Then the signals were visualized and analyzed using the Odyssey infrared imaging system (Odyssey LI-COR).
Statistical analysis
One-way analysis of variance followed by Duncan's test was used for multiple comparisons using Instat-2 software (GraphPad, San Diego, CA, USA). Data are presented as means ± standard deviation. A p-value < 0.05 was considered significantly.
Results
Hypoxia upregulates EDN1 mRNA and ET-1 production
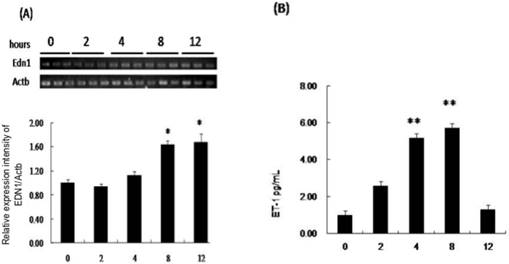
EDN1 mRNA increased significantly after 8 hours of hypoxia, but not at 2 or 4 hours compared to that in media from AMs that were not subjected to hypoxia (negative control) (Fig. 1A). The ratio of EDN1 mRNA to negative control was 1.62:1 after 8 hours of hypoxia. Rat AMs constitutively secreted ET-1, and the concentration increased significantly during 4-12 hours compared to that in media from AMs that were not subjected to hypoxia (Fig. 1B). The ratios of ET-1 production to the negative control after 4, 8, and 12 hours of hypoxia were 1.99:1, 3.51:1, and 4.70:1, respectively.
The production of EDN1 mRNA and secretion of ET-1 by NR8383 cells under a 1% O2 environment. NR8383 cells were cultured under hypoxia for 0, 2, 4, 8 and 12 hours. At the indicated times, cell lysates were collected and assayed for EDN1 mRNA (A), and culture supernatants were collected and assayed for ET-1 peptide (B). (A) EDN1 mRNA was increased significantly in the cell lysates of the AMs after hypoxia for 8 hours. (B) ET-1 was increased at 4 hours and continued to increase until 8 hours. (*p < 0.05 vs. 0 hour, **p < 0.01 vs. 0 hour, n = 6)
Hypoxia upregulates iNOS mRNA, NO, and cGMP expression
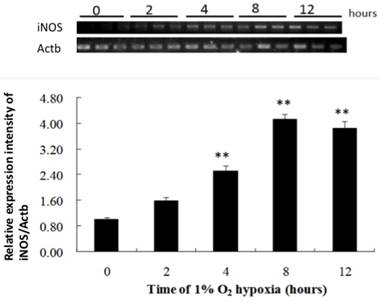
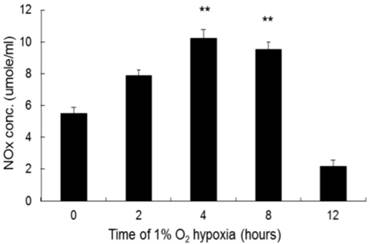
Hypoxia did not alter iNOS mRNA expression in the cell lysate until 4 hours after exposure, compared to that in the negative control. iNOS mRNA expression continued to increase throughout the incubation period (Fig. 2). The ratios of iNOS mRNA to negative control after 4 and 8 hours of hypoxia were 2.54: 1 and 4.18:1, respectively. NO level increased significantly after 4 hours of hypoxia compared to that in the negative control and continued up to 8 hours of hypoxia (Fig. 3). The ratios of NO expression to the negative control after 4 and 8 hours of hypoxia were 1.86:1 and 1.72:1, respectively.
The production of iNOS by NR8383 cells under a 1% O2 environment. NR8383 cells were cultured under hypoxia over 0, 2, 4, 8, and 12 hours. At the indicated times, cell lysates were collected and assayed for iNOS mRNA by RT-PCR. iNOS mRNA expression was significantly increased after 4 hours and continued to increase throughout the incubation period. (**p < 0.01 vs. 0 hour, n = 6) iNOS: inducible nitric oxide synthase; RT-PCR, reverse transcriptase polymerase chain reaction.
The production of NO by NR8383 cells under a 1% O2 environment. NR8383 cells were cultured under hypoxia over 0, 2, 4, 8, and 12 hours. At the indicated times, culture supernatants were collected and assayed for NO by using the Griess reagent. NO expression was increased significantly after 4 hours and continued to increase until 8 hours. (**p < 0.01 vs. 0 hour, n = 6)
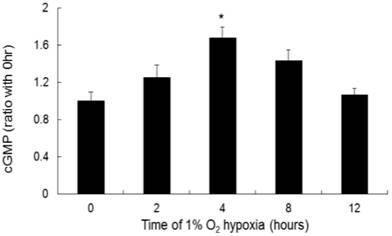
cGMP increased significantly at 4 hours of hypoxia compared to that in the negative control (Fig. 4). The ratios of cGMP expression to the negative control after 4 and 8 hours of hypoxia were 1.65:1 and 1.4:1, respectively.
The production of cGMP by NR8383 cells under a 1% O2 environment. NR8383 cells were cultured under hypoxia over 0, 2, 4, 8, and 12 hours. At the indicated times, cGMP (from 2 × 107 pellets/tube), the secondary messenger of NO, was collected and assayed. cGMP expression was significantly increased after 4 hours of hypoxia. (*p < 0.05 vs. 0 hour, n = 6)
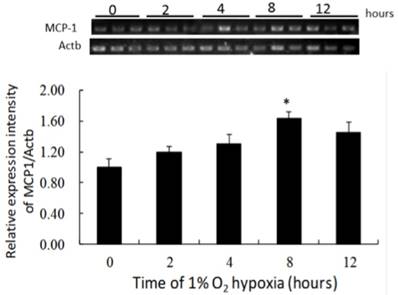
MCP-1 mRNA expression after hypoxia
MCP-1 mRNA expression increased gradually during incubation under 1% O2, and had increased significantly after 8 hours of hypoxia compared to that in the negative control (Fig. 5). The ratio of MCP-1 mRNA expression to the negative control was 1.59:1 after 8 hours of hypoxia.
The production of MCP-1 by NR8383 cells under a 1% O2 environment. NR8383 cells were cultured under hypoxia over 0, 2, 4, 8, and 12 hours. At the indicated times, cell extracts were analyzed for MCP-1 mRNA by RT-PCR. MCP-1 mRNA expression was increased significantly after 8 hours compared to 0 hours. (*p < 0.05 vs. 0 hour, n = 6) MCP-1: monocyte chemoattractant protein-1; RT-PCR, reverse transcriptase polymerase chain reaction.
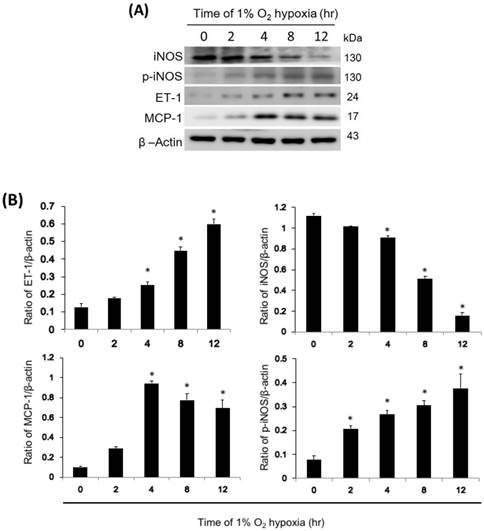
ET-1, iNOS, p-iNOS, and MCP-1 cellular protein expression
In NR8383 cells exposed to 1% hypoxia, ET-1 and MCP-1 expression increased after 4 hours of hypoxia and continued to increase for 12 hours, while iNOS expression decreased after 4 hours of hypoxia and continued to decrease for 12 hours (Fig. 6). In addition, active p-iNOS levels increased 2 hours after hypoxia and continued to increase for 12 hours. However, the level of iNOS, p-iNOS, ET-1, and MCP-1 didn't change with time in normoxic condition. (Fig. S1)
Effect of iNOS, p-iNOS, MCP-1, and ET-1 in NR8383 cells under a 1% O2 environment. (A) The cells were exposed to 1% O2 hypoxia for 0, 2, 4, 8, and 12 hours, and western blot analysis was subsequently performed to determine iNOS, p-iNOS, MCP-1, and ET-1 protein levels. Representative blots are from three independent experiments. (B) Quantification of band intensities. All data were reported as the means ± SD of at least three separate experiments. The t-test was used to determine statistical significance. * p < 0.05 vs. 0 hour control group. SD, standard deviation.
Discussion
In a hypoxic environment, pulmonary vasoconstriction plays an important role in the development of chronic pulmonary hypertension and pulmonary vascular remodeling [1]. Growing evidence has indicated that hypoxia increases the expression of lung inflammatory cytokines and chemokines, as well as the accumulation of AMs [20]. On the other hand, upregulation or an imbalance in the endothelin-NOS axis and influx of monocyte/ macrophages are implicated in hypoxia-induced injuries [1]. Nevertheless, a possible mechanism underlying macrophage-derived ET-1 in hypoxic injury has never been formally addressed; thus, our results may fill this gap.
ETs are peptides originally isolated from endothelial cells that have potent vasoactive and mitogenic properties and are also found in human and murine macrophages [17, 18]. In the current study, the increased ET-1 mRNA and protein expression levels both in the AM supernatant and cell pellets suggest that hypoxia triggers AMs to secrete ET-1 by modulating the gene. The more rapid increase in the ET-1 protein level than that of ET-1 mRNA may be because hypoxia accelerates the conversion of the ET-1 precursors to the active form, hypoxia stimulates AMs to secrete the active form of ET-1, or the half-life of ET-1 mRNA is shorter than the ET-1 protein. ET-1 mRNA decomposes or is suppressed more easily than the ET-1 protein under hypoxia [21].
ET-1 is a strong vasoconstricting hormone, and NOS, which generates NO, is a strong vasorelaxant. ET-1 vasodilates via type B (ETB) receptors and vasoconstricts via type A (ETA) receptors [22]. ET-1 and ET-3 are equipotent in their ability to stimulate ETB receptors and induce release of NO to relax the vasculature [23]. ET-1 and iNOS are co-expressed in various cells, such as endothelial, glial, nerve, and smooth muscle cells, as well as macrophages [11]. A close topographical relationship has also been reported between cells that synthesize NOS and ET-1 [24]. When the ET-1 expression is upregulated, NOS-NO cGMP is also upregulated to overcome the vasoconstrictive effect of ET-1 completely or incompletely. Interestingly, our study revealed that ET-1, NO, and iNOS mRNA expression increased simultaneously after 4-8 hours of hypoxia, but the ratio of the ET-1 production was much higher than the cGMP ratio, indicating an imbalance between ET-1 and cGMP in AMs, like endothelial cells, under hypoxic conditions, inferring that hypoxia can result in vasoconstriction by AMs.
Exhaled NO and iNOS increase in patients with sleep apnea [25-27]. Blood ET-1 concentrations also increase significantly in these patients, accompanied by increases in cAMP, cGMP, thromboxane B2, and angiotensin II [28, 29]. These findings suggest that sleep apnea is associated with a unique cardiopulmonary interaction in which respiratory inflammation and endothelial dysfunction occur. In addition, exhaled breath markers including peroxide (H2O2), oxidative stress index (8-isoprostane), inflammatory substances (leukotriene B4), and nitrates of patients with sleep apnea, are positively correlated with disease severity [27].The findings of the current study showed the increases in ET-1, iNOS, and NO/cGMP from AMs, suggesting that AMs may also play an important role in endothelin system dysfunction in response to hypoxia. AMs rapidly secrete ET-1 as an autocrine or paracrine factor, which positively feeds back to AMs and stimulates secretion of iNOS, NO, and cGMP [30, 31].
MCP-1 is an important chemokine related to AMs that initiates systemic microvascular inflammatory responses to alveolar hypoxia [32, 33]. In this study, we also demonstrated that MCP-1 mRNA expression increased after 8 hours of hypoxia, suggesting that a simple hypoxic challenge without lipopolysaccharide stimulation caused AMs to generate inflammatory chemokines and induce systemic inflammation. Many respiratory diseases lead to hypoxia without an infectious process, such as obstructive sleep apnea syndrome, asthma, and bronchopulmonary dysplasia. Of these, simple alterations in ambient O2 tension without a lung infection suggest the possible mechanisms occurring in AMs.
Furthermore, several studies have suggested that ET-1 is one of the factors regulating chemokine production [34, 35]. Increased ET-1 expression stimulates MCP-1 production [34, 35]. In addition, ET-1 transcriptionally regulates MCP-1 via nuclear factor κB and activator protein 1 in primary cultures of human airway smooth muscle cells [36]. The current study also revealed the time sequence of ET-1 and MCP-1 expression by AMs. Earlier expression of the ET protein than MCP-1 mRNA may imply that ET-1 regulates the expression of MCP-1. Activation of ET-1, iNOS, the NO/cGMP pathway, and MCP-1 may play a central role in pathological conditions in which alveolar hypoxia triggers pulmonary vasoconstriction and subsequent systemic inflammatory response.
In conclusion, we observed hypoxia-induced pulmonary endothelial system dysfunction and activation of iNOS and the NO/cGMP pathway by AMs. The endothelial axis has attracted increased attention as a potential target for therapies designed to treat respiratory distress disease, cardiovascular disease, and inflammatory pathologies. The important role of AMs as a source of ET-1 during hypoxia is being increasingly recognized. Here, we unraveled the endothelial dysfunction caused by AMs under conditions of pulmonary hypoxia and provide preliminary data for the continued analysis of the induction and regulation of ET-1 production by AMs under hypoxic conditions.
Supplementary Material
Supplementary figures and tables.
Abbreviations
AMs: Alveolar macrophages; MCP-1: monocyte chemoattractant protein-1; ET-1: Endothelin-1; NOS: Nitric oxide synthase; NO: nitric oxide; cGMP: cyclic 3', 5'-monophosphate; iNOS: Inducible NOS; p-iNOS: phosphorylated iNOS.
Acknowledgements
This research was supported by the grant from the Kaohsiung Medical University Hospital, Taiwan (KMUH100-0M22) and Ministry of Science and Technology, R.O.C. (MOST 106-2314-B-037-078- and MOST 107-2314-B-037-068 -MY2). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We also thank Chao-Hsiung Chen for his technical support in revising the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stenmark KR, Davie NJ, Reeves JT, Frid MG. Hypoxia, leukocytes, and the pulmonary circulation. Journal of applied physiology. 2005;98:715-21
2. Madjdpour C, Jewell UR, Kneller S, Ziegler U, Schwendener R, Booy C. et al. Decreased alveolar oxygen induces lung inflammation. American journal of physiology Lung cellular and molecular physiology. 2003;284:L360-7
3. Chao J, Wood JG, Gonzalez NC. Alveolar hypoxia, alveolar macrophages, and systemic inflammation. Respiratory research. 2009;10:54
4. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A. et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34-41
5. Lambrecht BN. Alveolar macrophage in the driver's seat. Immunity. 2006;24:366-8
6. Naidu BV, Krishnadasan B, Byrne K, Farr AL, Rosengart M, Verrier ED. et al. Regulation of chemokine expression by cyclosporine A in alveolar macrophages exposed to hypoxia and reoxygenation. Ann Thorac Surg. 2002;74:899-905 discussion 905
7. Sharma AK, Fernandez LG, Awad AS, Kron IL, Laubach VE. Proinflammatory response of alveolar epithelial cells is enhanced by alveolar macrophage-produced TNF-alpha during pulmonary ischemia-reperfusion injury. American journal of physiology Lung cellular and molecular physiology. 2007;293:L105-13
8. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y. et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411-5
9. Xu J, Zhong NS. Mechanisms of bronchial hyperresponsiveness: the interaction of endothelin-1 and other cytokines. Respirology. 1999;4:413-7
10. Dai ZK, Wu BN, Chen IC, Chai CY, Wu JR, Chou SH. et al. Attenuation of pulmonary hypertension secondary to left ventricular dysfunction in the rat by Rho-kinase inhibitor fasudil. Pediatric pulmonology. 2011;46:45-59
11. Rafols D, Steiner J, Rafols JA, Petrov T. Intracellular coexpression of endothelin-1 and inducible nitric oxide synthase underlies hypoperfusion after traumatic brain injury in the rat. Neuroscience letters. 2004;362:154-7
12. Boscoe MJ, Goodwin AT, Amrani M, Yacoub MH. Endothelins and the lung. The international journal of biochemistry & cell biology. 2000;32:41-62
13. Wanecek M, Weitzberg E, Rudehill A, Oldner A. The endothelin system in septic and endotoxin shock. European journal of pharmacology. 2000;407:1-15
14. Divino JN, Chawla KS, da Silva CM, Bjorge AM, Brittingham A. Endothelin-1 production by the canine macrophage cell line DH82: enhanced production in response to microbial challenge. Veterinary immunology and immunopathology. 2010;136:127-32
15. Coulombe M, Battistini B, Stankova J, Pouliot P, Bissonnette EY. Endothelins regulate mediator production of rat tissue-cultured mucosal mast cells. Up-regulation of Th1 and inhibition of Th2 cytokines. Journal of leukocyte biology. 2002;71:829-36
16. Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annual review of pharmacology and toxicology. 2001;41:851-76
17. Ehrenreich H, Anderson RW, Fox CH, Rieckmann P, Hoffman GS, Travis WD. et al. Endothelins, peptides with potent vasoactive properties, are produced by human macrophages. The Journal of experimental medicine. 1990;172:1741-8
18. Wahl JR, Goetsch NJ, Young HJ, Van Maanen RJ, Johnson JD, Pea AS. et al. Murine macrophages produce endothelin-1 after microbial stimulation. Experimental biology and medicine. 2005;230:652-8
19. Krumenacker JS, Hanafy KA, Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain research bulletin. 2004;62:505-15
20. Adegunsoye A, Balachandran J. Inflammatory response mechanisms exacerbating hypoxemia in coexistent pulmonary fibrosis and sleep apnea. Mediators Inflamm. 2015;2015:510105
21. McClellan G, Weisberg A, Rose D, Winegrad S. Endothelial cell storage and release of endothelin as a cardioregulatory mechanism. Circ Res. 1994;75:85-96
22. Schneider MP, Boesen EI, Pollock DM. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annual review of pharmacology and toxicology. 2007;47:731-59
23. Schiffrin EL. Endothelin: potential role in hypertension and vascular hypertrophy. Hypertension. 1995;25:1135-43
24. Vanhoutte PM. Say NO to ET. J Auton Nerv Syst. 2000;81:271-7
25. Depalo A, Carpagnano GE, Spanevello A, Sabato R, Cagnazzo MG, Gramiccioni C. et al. Exhaled NO and iNOS expression in sputum cells of healthy, obese and OSA subjects. Journal of internal medicine. 2008;263:70-8
26. Fortuna AM, Miralda R, Calaf N, Gonzalez M, Casan P, Mayos M. Airway and alveolar nitric oxide measurements in obstructive sleep apnea syndrome. Respiratory medicine. 2011;105:630-6
27. Petrosyan M, Perraki E, Simoes D, Koutsourelakis I, Vagiakis E, Roussos C. et al. Exhaled breath markers in patients with obstructive sleep apnoea. Sleep & breathing = Schlaf & Atmung. 2008;12:207-15
28. Hu X, Kang J, Xiao D, Wang W, Hou X, Yu R. The influence of nasal continual positive airway pressure on some vasoactive substances in patients with obstructive sleep apnea syndrome. Chinese journal of tuberculosis and respiratory diseases. 2002;25:36-8
29. Lurie A. Endothelial dysfunction in adults with obstructive sleep apnea. Advances in cardiology. 2011;46:139-70
30. Karkoulias K, Lykouras D, Sampsonas F, Drakatos P, Canova S, Tsoukalas G. et al. The role of Endothelin-1 in obstructive sleep apnea syndrome and pulmonary arterial hypertension: pathogenesis and Endothelin-1 antagonists. Current medicinal chemistry. 2010;17:1059-66
31. Kelly-Cobbs AI, Harris AK, Elgebaly MM, Li W, Sachidanandam K, Portik-Dobos V. et al. Endothelial endothelin B receptor-mediated prevention of cerebrovascular remodeling is attenuated in diabetes because of up-regulation of smooth muscle endothelin receptors. The Journal of pharmacology and experimental therapeutics. 2011;337:9-15
32. Chao J, Wood JG, Gonzalez NC. Alveolar macrophages initiate the systemic microvascular inflammatory response to alveolar hypoxia. Respiratory physiology & neurobiology. 2011;178:439-48
33. Chao J, Wood JG, Blanco VG, Gonzalez NC. The systemic inflammation of alveolar hypoxia is initiated by alveolar macrophage-borne mediator(s). American journal of respiratory cell and molecular biology. 2009;41:573-82
34. Chen P, Shibata M, Zidovetzki R, Fisher M, Zlokovic BV, Hofman FM. Endothelin-1 and monocyte chemoattractant protein-1 modulation in ischemia and human brain-derived endothelial cell cultures. Journal of neuroimmunology. 2001;116:62-73
35. Koyama Y, Kotani M, Sawamura T, Kuribayashi M, Konishi R, Michinaga S. Different actions of endothelin-1 on chemokine production in rat cultured astrocytes: reduction of CX3CL1/fractalkine and an increase in CCL2/MCP-1 and CXCL1/CINC-1. Journal of neuroinflammation. 2013;10:51
36. Sutcliffe AM, Clarke DL, Bradbury DA, Corbett LM, Patel JA, Knox AJ. Transcriptional regulation of monocyte chemotactic protein-1 release by endothelin-1 in human airway smooth muscle cells involves NF-kappaB and AP-1. Br J Pharmacol. 2009;157:436-50
Author contact
![]() Corresponding author: Zen-Kong Dai, Department of Pediatrics, Kaohsiung Medical University, 100 Shih-Chuan 1st Road, San-Ming District, Kaohsiung 807, Taiwan. Tel: 886-73121101-6507; Fax: 886-3208316; E-mail: zenkongedu.tw
Corresponding author: Zen-Kong Dai, Department of Pediatrics, Kaohsiung Medical University, 100 Shih-Chuan 1st Road, San-Ming District, Kaohsiung 807, Taiwan. Tel: 886-73121101-6507; Fax: 886-3208316; E-mail: zenkongedu.tw